
Microglia and astroglia: the role of neuroinflammation in lead toxicity and neuronal injury in the brain

Abstract
Lead (Pb2+), a ubiquitous environmental toxicant, may widely affect the function of many organs or systems of human beings, especially the brain. Although lead is believed to transport into the brain through the blood-brain barrier (BBB) and cause direct neuronal injury, growing data have shown that lead exposure could induce brain dysfunction by triggering microglial and astroglial activation, pro-inflammatory cytokine production and inflammatory response, generation of reactive oxygen species and oxidative stress, and finally result in BBB dysfunction and neuronal damage. This review summarizes recent studies regarding microglial and astroglial reaction, neuroinflammation, and neuronal death in the brain following lead insult, suggesting that reactive glial cells may represent a potential target for manipulation of lead-induced neuroinflammatory injury of the brain.
Keywords
Introduction
Lead (Pb2+) is a widely distributed heavy metal and environmental toxicant, and overexposure to lead due to pollution or accident can impair the function of the nervous system, especially learning and memory abilities of developing brains during childhood.[1-4] Epidemiologic data indicate that learning impairment may be caused by moderate lead exposure in young individuals.[2,3] This impairment is largely related to neuronal injury caused by lead toxicity, but the detailed mechanism by which lead exposure induces neuronal injury, neuronal death, and brain dysfunction still remains elusive.[4,5] Several studies have indicated that lead exposure may interfere with calcium signaling, suppress neurogenesis and neuronal differentiation, inhibit formation of long-term potentiation (LTP), influence secretion of neurotransmitters, and even enhance production of amyloid protein.[6-13] Lead can also bind to key metabolic enzymes such as pyruvate kinase, induce reactive oxygen species (ROS), impede the supply of energy to neurons, and cause neuronal apoptosis.[14-17] Moreover, recent studies have shown a crucial involvement of microglial and astroglial cells in neuroinflammatory injury induced by lead exposure.[10,11] Microglia and astrocytes are two major types of glial cells involved in the regulation of the immune response to pathological processes in the brain.[18] Functional activation of microglia and astrocytes and the resulting neuroinflammation are associated with infection, autoimmunity, and pathogenesis of neurodegenerative diseases. In response to lead exposure, microglia and astrocytes can increase the production and release of inflammatory cytokines, enhance ROS generation, impede antioxidant activity, and result in neuronal injury or neuronal loss in the brain or other parts of the central nervous system (CNS).[10,11,19-22]
Microglial activation, pro-inflammatory cytokines, and neuroinflammation
It is generally regarded that microglial cells are derived from blood monocytes that reset in the CNS during embryonic development and are functionally involved in neuronal maintenance, injury, and repair in a manner similar to peripheral macrophages.[23] Microglial cells are a predominant source of various inflammatory cytokines, that is, interleukin-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), and interferon-gamma (IFN-γ), which can then induce a broad spectrum of inflammatory reactions. The activation of microglia and astrocytes in response to internal and external stimuli or insults might further increase the release of cytotoxic substances, pro-inflammatory cytokines, ROS, and excitatory amino acids, thus causing further neuronal injury in the brain.[22]
Lead-induced inflammatory cytokines in microglial cells
Obvious morphological change and higher synthesis of cytokines have been observed in activated microglial cells after lead exposure.[10,11,24] For instance, elevated expression of IL-1β and TNF-α is found in the cerebral cortex after lead exposure, as well as increased expression of IL-1β and IL-6 in the hippocampus.[25,26]In vitro experiments have also confirmed the elevation of TNF-α expression after lead exposure.[27] Gene expression analysis has shown that levels of the pro-inflammatory factors IL-6 and TNF-α are significantly perturbed by the lead insult in multiple brain regions.[19,20] These cytokines are co-expressed in glial cells in response to lead crossing the blood-brain barrier (BBB) and might also represent a mechanism for lead toxicity to the immature brain. Conversely, anti-inflammatory factors such as IL-10 and transforming growth factor beta (TGF-β) are decreased in the cortex in response to lead, as detected by real time-polymerase chain reaction.
Lead-induced reactive oxygen species generation in microglial cells
Lead exposure might destroy the glial support of neuronal cells by increasing ROS and other toxins in microglial cells.[28] The microglial inflammatory response is also associated with the production of ROS and nitric oxide (NO)-dependent reactive nitrogen species (RNS).[19] Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), which is ubiquitously expressed in microglia, contributes much to the production of superoxide and the induction of ROS.[29] Furthermore, NOX could be activated in monocytes and microglial cells by IL-1β, TNF-α, IFN-γ, and other pro-inflammatory cytokines.[19] The inducible NO synthase (iNOS) is also prevalent in microglia, and microglial NO generation regulates vascular relaxation and initiates rapidly induced, transiently regulated signaling events.[30] On the other hand, lead also increases NOX, which causes superoxide production and inhibits antioxidant production, and increases the accumulation of ROS in the brain.[22] It is well-known that neurons in the brain are vulnerable to excess ROS and RNS. Oxidative stress could result in the death of newly-born neurons by disrupting signaling processes, dysfunction of ion homeostasis, and protein misfolding.[29]
The signaling pathways involved in lead-induced microglial activation, however, need more investigation. In response to various environmental toxins including lead, microglia could enter the activated state and release ROS.[31] Pattern recognition receptors expressed on microglia might be one common signaling pathway. For example, toll-like receptors act as initiators of the nuclear factor kappa B (NF-κB) pathway when exposed to several toxins, such as lipopolysaccharide (LPS), resulting in the release of pro-inflammatory cytokines.[32] However, it is still not clear how lead could induce microglial activation and trigger inflammatory cytokine production, which remains a critical question to be answered.
Astrocytic activation and the neuroinflammatory response
The neurovascular unit in the brain comprises of neurons, blood vessels and their adjacent astrocytes.[33,34] The concept of a functional unit is a new one, and emphasizes the interaction between neurons and astrocytes under both normal and pathological physiological conditions. Astrocytes play a critical role in neuron function, including energy support, metabolism, and synapse formation.[35,36] Astrocytes maintain the trans-endothelial electric resistance (TEER) of the BBB.[37] Under pathological conditions, astrocytes might remove toxic substances and balance electrolyte and water levels.[33] It has been found that lead interferes with astrocyte functions such as energy metabolism, immune response, and ROS removal. Furthermore, astrocytes could collaborate with microglia to switch on neuroinflammatory reactions in the brain, and each of these effects can result in BBB dysfunction and injury to neurons.
Lead exposure leads to the insufficient supply of energy from astrocytes to neurons. Astrocytes contain a large number of mitochondria for energy and glutamate metabolism. Neurons in the brain show a preference for lactose and glutamine provided by astrocytes via shuttle routes.[38] Glycogen is exclusively localized in astrocytes in the adult brain[39] and can be metabolized to pyruvate, which is converted to lactate by lactate dehydrogenase mainly in astrocytes and then transported to neurons. When energy is insufficient, astrocytes can also use glycolysis from stored glycogen for the use of neurons.[40,41] Glycogen metabolism in astrocytes is also required for long-termmemory formation in the brain.[42] Lead exposure causes morphological and functional changes in astrocytic mitochondria.[9] Creatine kinase and pyruvate kinase are two key enzymes in astrocytes that are involved in the production of pyruvate and lactate, and lead can bind to their sulfhydryl groups and decrease activity, resulting in an insufficient supply of pyruvate and lactate from astrocytes to neurons.[16] Lead can also act on cytochrome C and adenosine triphosphate synthase to cause dysfunction of the electron transport chain in mitochondria and generation of free radicals.[14,43] Mitochondrial dysfunction in astrocytes affects the survival of motor neurons.[44] Accumulation of free radicals and ROS might enhance the lack of energy and glutamine, eventually causing neuronal apoptosis.[15]
Lead triggers inflammation through a collaboration of astrocytes with microglia. The functional collaboration between astrocytes and microglia might play an important role in neuroinflammation and BBB dysfunction in the brain.[45] Overexpression of inflammatory stimuli in the neurovascular unit may start a response to clear antigenic material, leading to destruction of the BBB as well as neuronal damage. Following lead exposure, astrocytes secrete a number of inflammatory cytokines such as TNF-α, IL-6, and IL-10 into surrounding tissues.[45] These cytokines further mediate the immune response, including activation of microglia and macrophages, and induce other adverse reactions, which might eventually result in the destruction of BBB tight junctions. Matrix metalloproteinases (MMPs) are an important family of proteins composed of a variety of zinc-dependent enzymes that are capable of degrading extracellular matrix proteins such as collagen, gelatin, viscous protein, fibronectin, and proteoglycans.[46] It has been hypothesized that inflammatory cytokines induce production of MMP-2 and MMP-9, two proteinases that degrade the extracellular matrix and basement membrane, in astrocytes, resulting in increased permeability of the BBB.[47] Other studies have shown that low concentrations of pro-inflammatory cytokines (such as TNF-α or IL-1β) or lead did not influence MMP-9 expression when administered separately, but combined administration of lead and cytokines could induce a marked synergistic elevation in MMP-9 expression.[48]
Functional crosstalk between microglia and astrocytes in neuroinflammation
The start of an inflammatory reaction to lead exposure depends on the interaction between the inflammatory responses of astrocytes and microglial cells. Following lead exposure, activation of astrocytes surrounding blood vessels is indicated by increased expression of glial fibrillary acidic protein (GFAP).[49,50] Therefore, the response to lead in astrocytes may affect the BBB. It has been shown that lead in the brain accumulates predominantly in astrocytes, as opposed to neurons.[51,52] Another culture experiment has shown that younger astrocytes accumulate and retain more lead than older astrocytes.[53] To protect neurons against lead, astrocytes serve as a lead pool in the process of neurogenesis. However, because astrocytes are not able to remove lead from their own cytoplasm effectively, the accumulated lead will finally cause progressive damage of astrocytes, the BBB, and nearby neurons.
The response of microglia and astrocytes to neuroinflammation
Liu et al.[18] has proposed that activation of microglia in response to pathological conditions such as trauma, stroke, or neurodegenerative disorders occurs before activation of astrocytes. For instance, the activation of astrocytes occurs subsequently to microglial activation in respect to the cytokine expression sequence in Alzheimer’s disease.[54,55] A study with trimethyltin (TMT) treated rats, a model of neurodegenerative disease, revealed that GFAP significantly increases following microglial activation and that microglial activation requires lower concentrations of TMT than activation of astrocytes.[56] Considering that astrocytes are closer to the peripheral environment anatomically and more easily store toxic substances like lead, it may also be an imperceptible inflammatory signal released from astrocytes such as low amounts of TNF-α, free radicals, or ROS/NO that further initiates activation of microglial cells, leading to an inflammatory response.
The role of inflammatory cytokines and receptors in microglial-astrocytic interactions
Reciprocal activation of microglia and astrocytes mainly depends on inflammatory cytokines or their receptors.[57] Previous studies have shown that cytokines secreted from activated microglia also promote activation of astrocytes. Among those cytokines, IL-1 is a key mediator. IL-1β, mainly from microglia, can be rapidly expressed and may work to increase the secretion other cytokines such as IL-6, mainly from astrocytes, in order to promote inflammation.[35] Moreover, IL-1 might decrease the ability of astrocytes to reabsorb glutamic acid and promote the release of free radicals.[58,59] Experiments have shown that IL-1 receptor antagonists prevent pathological damage to astrocytes,[60] indicating that microglia might indirectly affect the function of astrocytes. In addition, microglial activation also promotes astrocytes to secrete TGF-β1 and IL-10.[61] When the severity of the immune response reaches a certain extent, however, TGF-β initiates a feedback loop to reduce the level of IL-1, inhibiting microglial activation and resulting in suppression of inflammation in the CNS.[62]
BBB dysfunction resulting from lead insult and neuroinflammation
Inflammatory cytokines and the inflammatory response are critical in the neurovascular unit and may result in alteration of BBB function. Brain microvascular endothelial cells (BMECs) are considered to be the anatomical and functional basis of the BBB.[63] As they are in direct contact with the circulating blood, BMECs are highly vulnerable to the impact of the blood environment. Studies have revealed that lead toxicity in the BBB or BMECs might influence tight junction proteins.[64] Tight junctions are key functional structures that bond BMECs together. Adhesion proteins are a component of tight junctions, and the zonula occludens (ZO) family plays a key role in connecting transmembrane proteins with actins inside the ECs to complete the structure of tight junctions.[65] In the cultured brain microvessel endothelial cell line RBE4, lead reduces the expression of tight junction proteins and lowers TEER, causing changes in ion permeability at the BBB and brain interstitial fluid ion regulation. As ZO-1 and ZO-2 are intracellular proteins, this suggests that cytoplasmic mechanisms may be associated with this process. Increased permeability of endothelial cells along with a decrease in occludin proteins has been detected following lead exposure.[65] The ZO family also seems to be susceptible to oxidative stress, and tight junctions are destroyed by lead-induced inflammation and ROS, leading to long-term BBB damage. However, other in vitro and in vivo experiments have revealed that claudin-1 mRNA and protein levels are downregulated without significant changes to ZO-1 and atresia proteins.[65,66]
The divalent iron ion channel [divalent metal transporter (DMT)] is a key element for the transport of iron across the BBB.[67] Many experiments have indicated that lead could also pass through DMT in a competitive way and may occupy this transporter when iron is deficient. Lead affects the offset of iron-regulated proteins, which allows it to more easily access endothelial cells.[68,69] When the concentration of iron is elevated, the transport of lead is effectively inhibited.[70] Interestingly, expression of fractalkine (CX3CL1), a mediator of neuron-glial signaling, is also enhanced after exposure to lead, especially in the hippocampus and forebrain.[10] In addition, lead also passes through and interferes with calcium channels, suggesting that lead might be able to cross the BBB in multiple or unknown other ways.
In one model involving exposure to lead, increased β-amyloid (Aβ) levels were found in the choroid plexus.[2] On the choroid epithelial cell surface, a critical transporter known as lipoprotein receptor-related protein-1 (LRP-1) is responsible for transporting Aβ out of the brain. LRP-1 knockout mice show higher levels of amyloid protein following lead exposure.[71] Lead could induce a significant reduction in LRP-1 expression by interfering with the LRP-1 gene promoter. These studies, therefore, suggest that lead neurotoxicity might also be related to memory deficits in the pathogenesis of Alzheimer’s disease.[72]
Neuronal damage induced by lead exposure and neuroinflammation
Lead-induced inflammatory reaction cascades within the neurovascular unit may cause neuronal damage.[21] It has been hypothesized that TNF-α, IL-1β, and IL-6 could cause neuronal apoptosis through glial activation.[73] Possible mechanisms of injury might be ROS production due to the pro-inflammatory cytokine IL-1β or increased glycogen consumption in astrocytes due to TNF-α and IL-1, thereby causing increased levels of toxic substances and affecting the metabolism of the cells.[74] TNF might also be involved in the expression of NO, suggesting another way by which could inactivate LTP. Furthermore, IL-1β acts on endothelial cell tight junction proteins, reducing the amount and location of occludin and increasing the permeability of the BBB.[75] Inflammatory reactions could also change the transport of multiple substances by affecting the role of glutamate receptors.[76] Lead-induced chemokines, mainly secreted from neurons, have been shown to act on microglial receptors and participate in the interactions between neurons and glial cells, resulting in changes in microglial and astrocyte morphology.[77]
Oxidative damage is fatal to brain neurons. In pathological conditions such as hypoxia, traumatic injury, and lead insult, these toxic free radicals might be over-generated and cause secondary injuries to neurons. Compared with neurons, astrocytes have higher levels of antioxidants such as glutathione (GSH), heme-oxygenase 1 and GSH S-transferase.[78] Neurons may maintain their antioxidant capacity by transporting and utilizing these substances, among which the GSH shuttle pathway is likely to be paramount.[79,80] GSH, the most abundant antioxidant in the brain, is mainly generated in astrocytes. Astrocytes store a much higher content of GSH-related enzymes in order to guarantee a supply to neurons.[78] GSH-depleted astrocytes display a reduced ability to protect neurons against oxidative injury.[78]
When lead enters astrocytes, it could directly deplete NADPH. More importantly, it affects glucose 6 phosphate dehydrogenase, a key enzyme of the pentose phosphate pathway, reducing the production of NADPH.[15] Both effects might result in a lack of GSH support from astrocytes to neurons. Lead is able to bind to GSH sulfhydryl groups and disable its ability as a ROS scavenger. Lead exposure results in an accumulation of ROS and a decrease in antioxidants. Increased levels of ROS contribute to higher BBB permeability, inducing oxidative damage to cellular molecules, activation of inflammatory mediators, and the destruction of tight junctions.[81] ROS also inhibit glutamate transporters and cause a secondary glutamate metabolism exception,[82] increasing the role of lead in the destruction of neurons. In addition, studies have shown that lead reduces many antioxidant molecules such as superoxide dismutase and catalase in adult mouse and rat brain.[68,83]
Finally, the phosphorylated cyclic-AMP response element binding (pCREB) is an important transcription factor for long-term memory, and lead could block the cAMP-CREB pathway by reducing pCREB, resulting in a decline in long-term memory.[6,7,13] The effect of lead exposure on CREB protein expression and phosphorylation in the cerebral cortex and hippocampus during postnatal development has been studied. Lead exposure did not affect total CREB levels, but decreased pCREB levels by about 30-38% in both cortex and hippocampus.[13] Disruptions in pCREB expression levels and the binding activity of CREB proteins may decipher intracellular mechanisms of lead neurotoxicity in developing brains.[12,13] In addition, the protein kinase C (PKC)/NF-κB pathway might be involved in lead-induced neuroinflammatory injury to brain neurons, as it represents a key stress response signal to inflammation.[84] The PKC/NF-κB pathway might also play a critical role in cell defense reactions and cell apoptosis. The PKC/NF-κB pathway has been shown to be involved in the regulation of NO and pro-inflammatory cytokine production in the LPS model of inflammation.[85] PKC/NF-κB pathway downstream products such as tumor necrosis factor-related apoptosis inducing ligand, caspase-1, and NOS2 are enhanced in animal models after lead exposure.[86]
Conclusion
In summary, microglial and astroglial responses might be critically involved in neuroinflammation and lead neurotoxicity in the brain. Microglia and astrocytes may have crosstalk or mutual activation by inflammatory cytokines and receptors. Lead (Pb2+) has been shown to contact and interfere with microglia and astrocytes, which may trigger microglial and astrocytic activation, enhance inflammatory cytokine generation and release, increase ROS and oxidative stress, and finally result in BBB dysfunction and neuronal injury [Figure 1]. Further extensive studies are still needed, however, to elucidate the specific signaling pathways for microglia and astrocytes partaking in neuroinflammation in the brain and to find new targets of manipulation for the prevention and treatment of lead neurotoxicity in human beings.
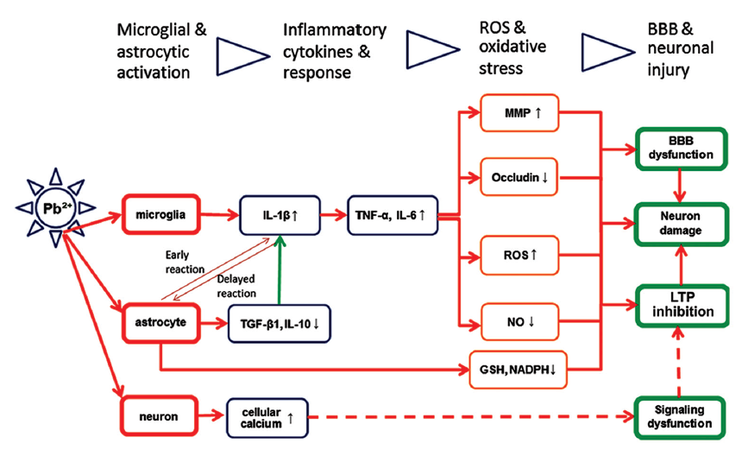
Figure 1. Correlative function of microglial and astroglial cells in the generation of inflammatory cytokines and neuronal injury. The lead (Pb2+) may contact and trigger microglial and astrocytic activation, enhance inflammatory cytokines and response, increase reactive oxygen species and oxidative stress, and finally result in blood‑brain barrier dysfunction, long‑term potentiation inhibition and inflammatory injury of various neurons in the brains
Financial support and sponsorship
The National Basic Research Program (2012CB525002, 2011CB504103) and National Natural Science Foundation of China (81272346, 31371374).
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Luo W, Ruan D, Yan C, Yin S, Chen J. Effects of chronic lead exposure on functions of nervous system in Chinese children and developmental rats. Neurotoxicology 2012;33:862-71.
2. Amato MS, Moore CF, Magzamen S, Imm P, Havlena JA, Anderson HA, Kanarek MS. Lead exposure and educational proficiency: moderate lead exposure and educational proficiency on end-of-grade examinations. Ann Epidemiol 2012;22:738-43.
3. Magzamen S, Imm P, Amato MS, Havlena JA, Anderson HA, Moore CF, Kanarek MS. Moderate lead exposure and elementary school end-of-grade examination performance. Ann Epidemiol 2013;23:700-7.
4. Senut MC, Cingolani P, Sen A, Kruger A, Shaik A, Hirsch H, Suhr ST, Ruden D. Epigenetics of early-life lead exposure and effects on brain development. Epigenomics 2012;4:665-74.
5. Mason LH, Harp JP, Han DY. Pb neurotoxicity: neuropsychological effects of lead toxicity. Biomed Res Int 2014;2014:840547.
6. Behl M, Zhang Y, Monnot AD, Jiang W, Zheng W. Increased beta-amyloid levels in the choroid plexus following lead exposure and the involvement of low-density lipoprotein receptor protein-1. Toxicol Appl Pharmacol 2009;240:245-54.
7. Toscano CD, Hashemzadeh-Gargari H, McGlothan JL, Guilarte TR. Developmental Pb2+exposure alters NMDAR subtypes and reduces CREB phosphorylation in the rat brain. Brain Res Dev Brain Res 2002;139:217-26.
8. Raunio S, Tähti H. Glutamate and calcium uptake in astrocytes after acute lead exposure. Chemosphere 2001;44:355-9.
9. Struzy-ska L, Bubko I, Walski M, Rafalowska U. Astroglial reaction during the early phase of acute lead toxicity in the adult rat brain. Toxicology 2001;165:121-31.
10. Struzynska L, Dabrowska-Bouta B, Koza K, Sulkowski G. Inflammation-like glial response in lead-exposed immature rat brain. Toxicol Sci 2007;95:156-62.
11. Liu MC, Liu XQ, Wang W, Shen XF, Che HL, Guo YY, Zhao MG, Chen JY, Luo WJ. Involvement of microglia activation in the lead induced long-term potentiation impairment. PLoS One 2012;7:e43924.
12. Struzynska L. A glutamatergic component of lead toxicity in adult brain: the role of astrocytic glutamate transporters. Neurochem Int 2009;55:151-6.
13. Toscano CD, McGlothan JL, Guilarte TR. Lead exposure alters cyclic-AMP response element binding protein phosphorylation and binding activity in the developing rat brain. Brain Res Dev Brain Res 2003;145:219-28.
14. Bressler J, Kim KA, Chakraborti T, Goldstein G. Molecular mechanisms of lead neurotoxicity. Neurochem Res 1999;24:595-600.
15. Flora SJ, Gautam P, Kushwaha P. Lead and ethanol co-exposure lead to blood oxidative stress and subsequent neuronal apoptosis in rats. Alcohol Alcohol 2012;47:92-101.
16. Lepper TW, Oliveira E, Koch GD, Berlese DB, Feksa LR. Lead inhibits in vitro creatine kinase and pyruvate kinase activity in brain cortex of rats. Toxicol In Vitro 2010;24:1045-51.
17. Mousa AM, Al-Fadhli AS, Rao MS, Kilarkaje N. Gestational lead exposure induces developmental abnormalities and up-regulates apoptosis of fetal cerebellar cells in rats. Drug Chem Toxicol 2015;38:73-83.
18. Liu W, Tang Y, Feng J. Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci 2011;89:141-6.
19. Kasten-Jolly J, Heo Y, Lawrence DA. Central nervous system cytokine gene expression: modulation by lead. J Biochem Mol Toxicol 2011;25:41-54.
20. Kasten-Jolly J, Pabello N, Bolivar VJ, Lawrence DA. Developmental lead effects on behavior and brain gene expression in male and female BALB/cAnNTac mice. Neurotoxicology 2012;33:1005-20.
21. Kumawat KL, Kaushik DK, Goswami P, Basu A. Acute exposure to lead acetate activates microglia and induces subsequent bystander neuronal death via caspase-3 activation. Neurotoxicology 2014;41:143-53.
22. Verstraeten SV, Aimo L, Oteiza PI. Aluminium and lead: molecular mechanisms of brain toxicity. Arch Toxicol 2008;82:789-802.
23. Kraft AD, Harry GJ. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int J Environ Res Public Health 2011;8:2980-3018.
24. Zurich MG, Eskes C, Honegger P, Bérode M, Monnet-Tschudi F. Maturation-dependent neurotoxicity of lead acetate in vitro: implication of glial reactions. J Neurosci Res 2002;70:108-16.
25. Li N, Liu F, Song L, Zhang P, Qiao M, Zhao Q, Li W. The effects of early life Pb exposure on the expression of IL1-β, TNF-α and Aβ in cerebral cortex of mouse pups. J Trace Elem Med Biol 2014;28:100-4.
26. Li N, Liu X, Zhang P, Qiao M, Li H, Li X, Zhang H, Yu Z. The effects of early life lead exposure on the expression of interleukin (IL) 1β, IL-6, and glial fibrillary acidic protein in the hippocampus of mouse pups. Hum Exp Toxicol 2015;34:357-63.
27. Cheng YJ, Liu MY, Wu TP, Yang BC. Regulation of tumor necrosis factor-alpha in glioma cells by lead and lipopolysaccharide: involvement of common signaling pathway. Toxicol Lett 2004;152:127-37.
28. Nava-Ruiz C, Méndez-Armenta M, Ríos C. Lead neurotoxicity: effects on brain nitric oxide synthase. J Mol Histol 2012;43:553-63.
29. Lipton SA, Gu Z, Nakamura T. Inflammatory mediators leading to protein misfolding and uncompetitive/fast off-rate drug therapy for neurodegenerative disorders. Int Rev Neurobiol 2007;82:1-27.
30. Choi SH, Lee DY, Kim SU, Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci 2005;25:4082-90.
31. Soltaninejad K, Kebriaeezadeh A, Minaiee B, Ostad SN, Hosseini R, Azizi E, Abdollahi M. Biochemical and ultrastructural evidences for toxicity of lead through free radicals in rat brain. Hum Exp Toxicol 2003;22:417-23.
32. Vogel SN, Fitzgerald KA, Fenton MJ. TLRs: differential adapter utilization by toll-like receptors mediates TLR-specific patterns of gene expression. Mol Interv 2003;3:466-77.
33. Sá-Pereira I, Brites D, Brito MA. Neurovascular unit: a focus on pericytes. Mol Neurobiol 2012;45:327-47.
34. Witthoft A, Em Karniadakis G. A bidirectional model for communication in the neurovascular unit. J Theor Biol 2012;311:80-93.
35. Rouach N, Avignone E, Même W, Koulakoff A, Venance L, Blomstrand F, Giaume C. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell 2002;94:457-75.
36. Stipursky J, Romão L, Tortelli V, Neto VM, Gomes FC. Neuron-glia signaling: implications for astrocyte differentiation and synapse formation. Life Sci 2011;89:524-31.
37. Siddharthan V, Kim YV, Liu S, Kim KS. Human astrocytes/ astrocyte-conditioned medium and shear stress enhance the barrier properties of human brain microvascular endothelial cells. Brain Res 2007;1147:39-50.
38. Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 2011;14:724-38.
41. Walls AB, Heimbürger CM, Bouman SD, Schousboe A, Waagepetersen HS. Robust glycogen shunt activity in astrocytes: effects of glutamatergic and adrenergic agents. Neuroscience 2009;158:284-92.
42. Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011;144:810-23.
43. Maiti AK, Saha NC, Paul G. Effect of lead on oxidative stress, Na+K+ATPase activity and mitochondrial electron transport chain activity of the brain of Clarias batrachus L. Bull Environ Contam Toxicol 2010;84:672-6.
44. Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de León A, Robinson KM, Mason RP, Beckman JS, Barbeito L, Radi R. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci 2008;28:4115-22.
45. Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 2006;7:41-53.
46. Hartmann C, Zozulya A, Wegener J, Galla HJ. The impact of glia-derived extracellular matrices on the barrier function of cerebral endothelial cells: an in vitro study. Exp Cell Res 2007;313:1318-25.
47. Rosenberg GA. Matrix metalloproteinases in multiple sclerosis: is it time for a treatment trial? Ann Neurol 2001;50:431-3.
48. Lahat N, Shapiro S, Froom P, Kristal-Boneh E, Inspector M, Miller A. Inorganic lead enhances cytokine-induced elevation of matrix metalloproteinase MMP-9 expression in glial cells. J Neuroimmunol 2002;132:123-8.
49. Sansar W, Ahboucha S, Gamrani H. Chronic lead intoxication affects glial and neural systems and induces hypoactivity in adult rat. Acta Histochem 2011;113:601-7.
50. Little AR, O'Callagha JP. Astrogliosis in the adult and developing CNS: is there a role for proinflammatory cytokines? Neurotoxicology 2001;22:607-18.
51. Lindahl LS, Bird L, Legare ME, Mikeska G, Bratton GR, Tiffany-Castiglioni E. Differential ability of astroglia and neuronal cells to accumulate lead: dependence on cell type and on degree of differentiation. Toxicol Sci 1999;50:236-43.
52. Tiffany-Castiglioni E, Sierra EM, Wu JN, Rowles TK. Lead toxicity in neuroglia. Neurotoxicology 1989;10:417-43.
53. Legare ME, Barhoumi R, Hebert E, Bratton GR, Burghardt RC, Tiffany-Castiglioni E. Analysis of Pb2+ entry into cultured astroglia. Toxicol Sci 1998;46:90-100.
54. Gahtan E, Overmier JB. Inflammatory pathogenesis in Alzheimer's disease: biological mechanisms and cognitive sequeli. Neurosci Biobehav Rev 1999;23:615-33.
55. Ii M, Sunamoto M, Ohnishi K, Ichimori Y. Beta-amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res 1996;720:93-100.
56. McCann MJ, O'Callaghan JP, Martin PM, Bertram T, Streit WJ. Differential activation of microglia and astrocytes following trimethyl tin-induced neurodegeneration. Neuroscience 1996;72:273-81.
57. Shao W, Zhang SZ, Tang M, Zhang XH, Zhou Z, Yin YQ, Zhou QB, Huang YY, Liu YJ, Wawrousek E, Chen T, Li SB, Xu M, Zhou JN, Hu G, Zhou JW. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via αB-crystallin. Nature 2013;494:90-4.
58. Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 2000;7:153-9.
59. Thornton P, Pinteaux E, Gibson RM, Allan SM, Rothwell NJ. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J Neurochem 2006;98:258-66.
60. Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci 2001;21:5528-34.
61. da Cunha A, Jefferson JA, Jackson RW, Vitkovic L. Glial cell-specific mechanisms of TGF-beta 1 induction by IL-1 in cerebral cortex. J Neuroimmunol 1993;42:71-85.
62. Herrera-Molina R, von Bernhardi R. Transforming growth factor-beta 1 produced by hippocampal cells modulates microglial reactivity in culture. Neurobiol Dis 2005;19:229-36.
63. Cardoso FL, Brites D, Brito MA. Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain Res Rev 2010;64:328-63.
64. Kim JH, Byun HM, Chung EC, Chung HY, Bae ON. Loss of integrity: impairment of the blood-brain barrier in heavy metal-associated ischemic stroke. Toxicol Res 2013;29:157-64.
65. Balbuena P, Li W, Ehrich M. Assessments of tight junction proteins occludin, claudin 5 and scaffold proteins ZO1 and ZO2 in endothelial cells of the rat blood-brain barrier: cellular responses to neurotoxicants malathion and lead acetate. Neurotoxicology 2011;32:58-67.
66. Shi LZ, Zheng W. Early lead exposure increases the leakage of the blood-cerebrospinal fluid barrier, in vitro. Hum Exp Toxicol 2007;26:159-67.
67. I Bannon D, Portnoy ME, Olivi L, Lees PS, Culotta VC, Bressler JP. Uptake of lead and iron by divalent metal transporter 1 in yeast and mammalian cells. Biochem Biophys Res Commun 2002;295:978-84.
68. Wang J, Wu J, Zhang Z. Oxidative stress in mouse brain exposed to lead. Ann Occup Hyg 2006;50:405-9.
69. Wang Q, Lin Y, Zhang W, Liu M, Chen Y, Chen J, Luo W. Lead induces dysregulation of iron regulatory protein 1 via the extracellular signal-regulated kinase pathway in human vascular endothelial cells. Brain Res 2012;1455:19-27.
70. Wang Q, Luo W, Zheng W, Liu Y, Xu H, Zheng G, Dai Z, Zhang W, Chen Y, Chen J. Iron supplement prevents lead-induced disruption of the blood-brain barrier during rat development. Toxicol Appl Pharmacol 2007;219:33-41.
71. Gu H, Wei X, Monnot AD, Fontanilla CV, Behl M, Farlow MR, Zheng W, Du Y. Lead exposure increases levels of β-amyloid in the brain and CSF and inhibits LRP1 expression in APP transgenic mice. Neurosci Lett 2011;490:16-20.
72. Stewart WF, Schwartz BS, Davatzikos C, Shen D, Liu D, Wu X, Todd AC, Shi W, Bassett S, Youssem D. Past adult lead exposure is linked to neurodegeneration measured by brain MRI. Neurology 2006;66:1476-84.
73. Liu JS, John GR, Sikora A, Lee SC, Brosnan CF. Modulation of interleukin-1beta and tumor necrosis factor alpha signaling by P2 purinergic receptors in human fetal astrocytes. J Neurosci 2000;20:5292-9.
74. Gavillet M, Allaman I, Magistretti PJ. Modulation of astrocytic metabolic phenotype by proinflammatory cytokines. Glia 2008;56:975-89.
75. Huber JD, Witt KA, Hom S, Egleton RD, Mark KS, Davis TP. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol 2001;280:H1241-8.
76. O'Shea RD. Roles and regulation of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol 2002;29:1018-23.
77. Biber K, Vinet J, Boddeke HW. Neuron-microglia signaling: chemokines as versatile messengers. J Neuroimmunol 2008;198:69-74.
78. Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 2003;23:3394-406.
79. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/ GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 2006;98:641-53.
80. McKenna MC. The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res 2007;85:3347-58.
81. Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res 2009;43:348-64.
82. Trotti D, Nussberger S, Volterra A, Hediger MA. Differential modulation of the uptake currents by redox interconversion of cysteine residues in the human neuronal glutamate transporter EAAC1. Eur J Neurosci 1997;9:2207-12.
83. Nehru B, Kanwar SS. N-acetylcysteine exposure on lead-induced lipid peroxidative damage and oxidative defense system in brain regions of rats. Biol Trace Elem Res 2004;101:257-64.
84. Xu SZ, Shan CJ, Bullock L, Baker L, Rajanna B. Pb2+reduces PKCs and NF-kappaB in vitro. Cell Biol Toxicol 2006;22:189-98.
85. Guo F, Lou Y, Feng N, Li G, Xie A, Huang X, Wang Y. Exposure to lanthanum compound diminishes LPS-induced inflammation-associated gene expression: involvements of PKC and NF-kappaB signaling pathways. Biometals 2010;23:669-80.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Liu JT, Dong MH, Zhang JQ, Bai Y, Kuang F, Chen LW. Microglia and astroglia: the role of neuroinflammation in lead toxicity and neuronal injury in the brain. Neurosciences 2015;2:131-7. http://dx.doi.org/10.4103/2347-8659.156980
AMA Style
Liu JT, Dong MH, Zhang JQ, Bai Y, Kuang F, Chen LW. Microglia and astroglia: the role of neuroinflammation in lead toxicity and neuronal injury in the brain. Neuroimmunology and Neuroinflammation. 2015; 2: 131-7. http://dx.doi.org/10.4103/2347-8659.156980
Chicago/Turabian Style
Liu, Jin Tao, Mo Han Dong, Jie Qiong Zhang, Ya Bai, Fang Kuang, Liang Wei Chen. 2015. "Microglia and astroglia: the role of neuroinflammation in lead toxicity and neuronal injury in the brain" Neuroimmunology and Neuroinflammation. 2: 131-7. http://dx.doi.org/10.4103/2347-8659.156980
ACS Style
Liu, JT.; Dong MH.; Zhang JQ.; Bai Y.; Kuang F.; Chen LW. Microglia and astroglia: the role of neuroinflammation in lead toxicity and neuronal injury in the brain. Neurosciences. 2015, 2, 131-7. http://dx.doi.org/10.4103/2347-8659.156980
About This Article
Copyright

Data & Comments
Data


 Cite This Article 26 clicks
Cite This Article 26 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.