
Necroptosis: a new link between cell death and inflammation


Abstract
Necroptosis is a type of newly identified cell death induced by apoptotic stimuli under conditions where apoptotic execution is prevented. Studies over the past 10 years have revealed the molecular mechanism of necroptosis and challenged the old conception that necrosis is un-programmed. Recently, more and more data have emerged suggesting a close association between necroptosis and inflammation. In this review, the authors summarized the current knowledge of the mechanism of necroptosis, focusing on tumour necrosis factor α induced necroptosis and the roles of necroptosis in regulating inflammation. In particular, we discussed the occurrence of necroptosis and its relation with inflammation in neurological diseases hoping to provide new insight for the research and treatment of neuroinflammatory disorders.
Keywords
Introduction
Death is the most common ultimate fate of cells when challenged by death signals. By ultrastructural morphology, cell death is mainly classified as apoptosis, autophagy, or necrosis.[1] During past decades, extensive studies have been performed on apoptosis and autophagy, and have pictured very elegant molecular mechanisms for apoptosis and autophagy. By utilizing these mechanisms, apoptosis and autophagy can be finely regulated. Therefore, these 2 types of cell death are regarded as “programmed cell death”. Necrosis, however, has long been thought as acute and uncontrollable, largely owning to its elusive molecular mechanism, and thus been thought as “un-programmed”.
In the year of 2005, Dr. Jun-Ying Yuan[2] at Harvard University reported a novel type of necrosis, which occurred in cells when the apoptosis machinery is inhibited, but extracellular apoptotic stimulation persisted. This type of necrosis can be inhibited by a chemical named Necrostatin-1 (Nec-1), which suppresses the activity of receptor-interacting protein 1 (RIP1), suggesting that the cell death is molecularly regulated. Because the dying cells the researchers originally identified showed a mixture of ultrastructural features of both apoptosis and necrosis, for example, condensation of chromatin, disruption of the cell membrane and lysis of cytoplasmic contents, it was termed as necroptosis (necrosis + apoptosis). Later on, it was found to show mostly the morphological features of unregulated necrotic death.[3]
Molecular mechanism of necroptosis
Since Dr. Yuan’s publication, many studies have been performed on the occurrence and molecular mechanism of necroptosis. Most of the current knowledge about necroptosis comes from tumour necrosis factor α (TNF-α) induced necroptosis. Necroptosis is initiated when death signals such as TNF-α and Fas bind to their membrane receptors. This ligation leads to the formation of a membrane associated protein complex, named complex I.[4] Complex I is composed by: (1) proteins which have a death domain such as tumor necrosis factor receptor (TNFR)-associated death domain (TRADD), Fas-associated death domain (FADD); (2) RIP1; (3) TNFR-associated factors (TRAF), such as TRAF2 or TRAF5, and (4) cellular inhibitor of apoptosis protein 1 (cIAP1) and cIAP2.[5] TRADD acts as an adaptor for recruiting RIP1 to TNFR1. Subsequently, TRAF2/3/5 and cIAPs are added into the protein complex.[6] If E3 ubiquitin ligase is activated, TRAF2/5 and cIAP1/2 can ubiquitinate RIP1, which results in stabilization of the RIP1-containing plasma membrane associated complex that activates nuclear factor kappa and mitogen-activated protein kinases, and thus promoting cell survival.[7] Therefore, protein complex I determines the fate of cells to either survival or death.[8]
Activation of necroptosis signalling starts with deubiquitination of RIP1 and other components by deubiquitinating enzyme cylindromatosis, which removes ubiquitin chains from RIP1, thus, destabilizing Complex I.[9] Deubiquitinated RIP1 is released from Complex I and combines with FADD, TRADD, receptor-interacting protein 3 (RIP3) and caspase-8 to form Complex II.[10] Active caspase-8 can cleave and inactivate RIP1 and RIP3, thereby promoting the exogenous apoptosis pathway.[11]
If caspase-8 is inhibited, RIP1 and RIP3 will remain active and combine together by the common RHM domain to take part in forming a necrosome, which initiates a downstream signal cascade resulting in necroptosis.[12] Although RIP1 and RIP3 are both essential in the process, over-expressed RIP3 can induce necroptosis without enough RIP1, but not vice versa.[13] It has been demonstrated that RIP3 activates downstream signalling pathways, in particular, the phosphorylated mixed lineage kinase domain-like protein (MLKL),[14] which plays a central role in the execution of necroptosis. Two models have been proposed for its function: (1) acts as a platform at the plasma membrane for the recruitment of Ca2+ or Na+ ion channels,[15] (2) as a direct pore-forming complex on cell membranes through binding of the amino-terminus of the 4-helical bundle domain to negatively charged phosphatidylinositol phosphates.[16] Previous studies have suggested that phosphoglycerate mutase 5 involved mitochondrial fragmentation might be the key downstream molecule of MLKL for necroptosis execution.[17] However, recent evidences have challenged this idea.[18]
The mechanism of necroptosis discussed above is outlined in Figure 1. Besides the TNF-α induced extrinsic necroptosis, an intrinsic necroptosis signalling initiated by intracellular reactive oxygen species has been proposed recently. Translocation of p53 has been suggested to play a role in ischemia induced intrinsic necroptosis.[19] More detailed mechanisms of intrinsic necroptosis remain to be elucidated.
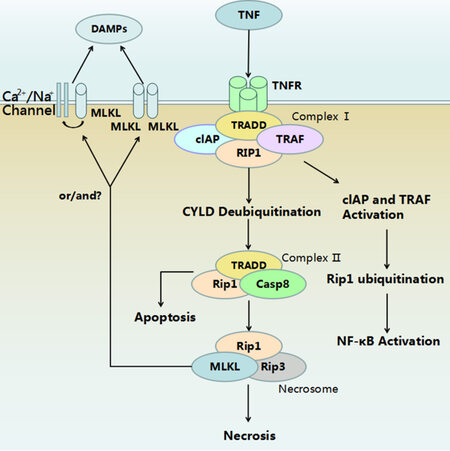
Figure 1. Mechanism of tumour necrosis factor-α (TNF-α) induced necroptosis. Binding of TNF-α to its receptor results in formation of Complex I. Activation of cIAP and tartrate resistant acid phosphatase activates downstream NF-κB signaling and subsequently promote cell survival. Complex II acts as a switch between apoptosis and necroptosis. Activation of caspase-8 guides the cells to apoptosis. Inhibition of caspase-8 leads to formation of a necrosome. Membrane translocation of phosphrylated MLKL disrupts cell membrane. The mechanisms underlying the lysis of cytoplasmic contents during necrosis are still unclear. DAMPs: damage associated molecular patterns; MLKL: mixed lineage kinase domain-like protein; TNF: tumour necrosis factor; TNFR: tumour necrosis factor receptor; cIAP: calf intestinal alkaline phosphatise; TRADD: tumor necrosis factor receptor associated death domain; TRAF: TNFR-associated factors; RIP1: receptor-interacting protein 1; CYLD: cylindromatosis; Casp8: caspase-8; RIP3: receptor-interacting protein 3; NF-kB: nuclear factor kappa.
Necroptosis and inflammation
It is known that apoptosis triggers minor or no inflammation, while necrosis induces inflammation via releasing damage associated molecular patterns (DAMPs), such as nuclear high mobility group box-1 proteins, mitochondrial DNA, and IL-1 family cytokines.[20] The insertion of MLKL into cell membranes immediately suggested a possible role of MLKL in the release of DAMPs. Because DAMPs stimulate pattern-recognition receptors such as toll-like receptors, necroptosis is thought to be beneficial in innate immune responses. For example, vaccinia virus encodes an inhibitor of caspase-1 and 8. In cases of vaccinia virus infection, the cells exhibit RIP3-dependent necroptosis, which mobilize immune cells against viruses.[21] Therefore, in certain virus-infected diseases, necroptosis seems to be an evolutionarily cellular anti-virus strategy.
In terms of bacterial infection, it has long been known that TNF is an important driver of bacterial sepsis,[22] suggesting that necroptosis may also be a pro-inflammatory factor in the bacterial infection-induced inflammation. In consistent with above mentioned role of RIP/MLKL-dependent necroptosis in the destructive inflammation after virus infection, RIP3 deficient mice are more resistant to TNF induced systematic inflammation.[23] However, some studies showed that RIP3-/- macrophages respond almost normally to liters per second stimulation, indicating that RIP3 may not be crucial for acute inflammation after bacterial infection.[24] In line with this idea, RIP3-dependent necroptosis and TNF expression was observed in tuberculosis infected tissue,[25] suggesting a role of necroptosis in the bacterial induced chronic inflammation.
In addition to its roles in infectious diseases, necroptosis has also been demonstrated to be involved in chronic sterile inflammation. For example, up-regulation of RIP3 and phosphorylated MLKL were detected in alcoholic and drug-induced liver injury. RIP3 depletion, or necrostatin-1 (Nec-1) administration can significantly protect liver cells from these injuries.[26] In ischemia-reperfusion conditions, necroptosis was reported in multiple tissues, including brain, heart, kidney and retina.[27-29] In other chronic inflammatory diseases such as atherosclerosis, receptor-interacting serine/threonine-protein kinase 3-dependent macrophage necroptosis has been thought of as a direct driver of atherosclerotic plaque formation.[30] Although it has been clearly demonstrated that necroptotic cells release DAMPs, how DAMPs mediate this necroptosis-triggered sterile inflammation remains to be experimentally validated.
It should be pointed out that many studies used Nec-1 to inhibit necroptosis. However, recent studies reported that Nec-1 has off-target effects. Besides inhibiting the kinase activity of RIP1, it inhibits the activity of endoleamine 2,3-oxygenase, which by itself, modulates inflammation.[31] Therefore, one should be sure to explain the results obtained solely by Nec-1 treatment.
Necroptosis and neurological diseases
Necroptosis was initially identified in ischemic brain. As the molecular mechanisms of necroptosis have been gradually discovered, necroptosis has been reported in more and more neurological diseases.
Spinal cord injury (SCI) is well known for its devastating effects on patients. One pathological feature of SCI is secondary injury characterized by chronic inflammation, astrogliosis and cavity formation.[32] Previous studies have demonstrated that application of Nec-1 can be protective for SCI, but which cells undergo necroptosis is unknown.[33,34] Our recent studies demonstrated that RIP3 and phosphorylated MLKL are up-regulated in reactive astrocytes and microglia after SCI.[35,36] Reactive astrocytes, which line the spinal cavity, die by M1 microglia/macrophage induced necroptosis partially through toll like receptor/myeloid differentiation 88 signalling.[35]Microglia, the major player of chronic inflammation post-SCI, die through endoplasmic reticulum stress involved necroptosis.[36] These researches raised the straightforward question of how necroptosis regulateschronic inflammation after SCI.
Multiple sclerosis is another neurodegenerative diseases characterized by demyelization and chronic inflammation. The link between inflammation and demyelination has long been recognized. A recent study from Prof. Jun-Ying Yuan’s group reported that TNF-α induces the death of oligodendrocytes in a RIP1/3 dependent manner.[37] In the mouse model of Gaucher’s disease, systemic TNF-α and IL-1β are elevated, and RIP3 is up-regulated in microglia and neurons. RIP3 knockout can significantly ameliorate the development of disease and prolong the survival of animals.[38] Amyotrophic lateral sclerosis (ALS) is the most adult onset motor neuron degenerative disease, in which inflammation is the most striking hallmark of pathological changes. Recently, it has been demonstrated that in the spinal cord of the ALS model, motor neurons also undergo necroptosis.[39] These studies suggested that necroptosis in different neurodegenerative diseases is cell type specific. The underlying mechanisms remain to be further investigated.
Conclusion
In summary, this progress brings us the new concept that necrosis can be chronic and controllable, although the mechanism of necroptosis remains far from fully revealed. The current evidences suggest that pro-inflammatory factors, e.g. TNF-α, can induce necroptosis, which in turn triggers inflammation. As we know, inflammation is complex and dynamic. The outcome of inflammation depends on the coordination of different types of immune cells. Even sub-populations of immune cells change their phenotypes in the course of inflammation, such as the M1-M2 switch of microglia/macrophages.[40] How necroptotic cells affect the different immune cell populations during the different time-phase of inflammation, or how necroptotic cells influence the phenotype of immune cells remain to be further investigated.
Recently, several studies showed that RIP1 and RIP3 might also be involved in inflammation independent of necroptosis.[41] For example, Inflammasome activation and release of IL-1 in smac-mimetic treated macrophages or in caspase-8 deficient dendrite cells are dependent on RIP3,[42] suggesting a cell-death independent role of RIP3 in inflammasome activation. This should be considered when evaluating results. Combined results from MLKL deficient cells or mice may be helpful for clarifying the point. In view of the importance of necroptosis and its roles in inflammation, better understanding of the interaction between necroptosis and inflammation will be helpful for treatment of inflammatory diseases.
Acknowledgments
The authors thank Dr. Hong Fan for his critical proofreading of the manuscript and suggestions for revision.
Financial support and sponsorship
This work was supported by National Natural Science Foundation of China to Dr. Ya-Zhou Wang (grant code: 81571224).
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nuñez, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012;19:107-20.
2. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005;1:112-9.
3. Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008;4:313-21.
4. Welz, PS, Wullaert, A, Vlantis, K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 2011;477:330-4.
5. Han, J, Zhong CQ, Zhang DW. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol 2011;12:1143-9.
6. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010;11:700-14.
7. Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 2004;24:1464-9.
8. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP (L) complex inhibits RIPK3-dependent necrosis. Nature 2011;471:363-7.
9. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009;137:1112-23.
10. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014;15:135-47.
11. Vanlangenakker N, Vanden Berghe T, Krysko DV, Festjens N, Vandenabeele P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med 2014;8:207-20.
12. Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep 2013;5:70-8.
13. Dondelinger Y, Aguileta M, Goossens V, Dubuisson C, Grootjans S, Dejardin E, Vandenabeele P, Bertrand MJ. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ 2013;20:1381-92.
14. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012;148:213-27.
15. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014;16:55-65.
16. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, and Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014;54:133-46.
17. Wang Z, Jiang H, Chen S, Du F, and Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012;148:228-43.
18. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 2011;43:449-63.
19. Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012;149:1536-48.
20. Eigenbrod T, Park JH, Harder J, Iwakura Y, Núñez G. Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J Immunol 2008;181:8194-8.
21. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009;137:1112-23.
22. Clark IA. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev 2007;18:335-43.
23. Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK, Kelliher MA, Harris PA, Bertin J, Gough PJ, Shayakhmetov DM, Goguen JD, Fitzgerald KA, Silverman N, Lien E. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U S A 2014;111:7391-6.
24. Deaciuc IV, D'souza NB, de Villiers WJ, Burikhanov R, Sarphie TG, Hill DB, McClain CJ. Inhibition of caspases in vivo protects the rat liver against alcohol-induced sensitization to bacterial lipopolysaccharide. Alcohol Clin Exp Res 2001;25:935-43.
25. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 2013;153:521-34.
26. Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013;57:1773-83.
27. Koshinuma S, Miyamae M, Kaneda K, Kotani J, Figueredo VM. Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J Anesth 2014;28:235-41.
28. Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci U S A 2013;110:12024-9.
29. Gao S, Andreeva K, Cooper NG. Ischemia-reperfusion injury of the retina is linked to necroptosis via the ERK1/2-RIP3 pathway. Mol Vis 2014;20:1374-87.
30. Lin J, Li H, Yang M, Ren J, Huang Z, Han F, Huang J, Ma J, Zhang D, Zhang Z, Wu J, Huang D, Qiao M, Jin G, Wu Q, Huang Y, Du J, Han J. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep 2013;3:200-10.
31. Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis 2012;3:e437.
33. Wang Y, Wang H, Tao Y, Zhang S, Wang J, Feng X. Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience 2014;266:91-101.
34. Wang Y, Wang J, Yang H, Zhou J, Feng X, Wang H, Tao Y. Necrostatin-1 mitigates mitochondrial dysfunction post-spinal cord injury. Neuroscience 2015;289:224-32.
35. Fan H, Zhang K, Shan L, Kuang F, Chen K, Zhu K, Ma H, Ju G, Wang YZ. Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol Neurodegener 2016;11:14.
36. Fan H, Tang HB, Kang J, Shan L, Song H, Zhu K, Wang J, Ju G, Wang YZ. Involvement of endoplasmic reticulum stress in the necroptosis of microglia/macrophages after spinal cord injury. Neuroscience 2015;311:362-73.
37. Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, Geng J, Py B, Zhou W, Amin P, Berlink Lima J, Qi C, Yu Q, Trapp B, Yuan J. Activation of necroptosis in multiple sclerosis. Cell Rep 2015;10:1836-49.
38. Vitner EB, Salomon R, Farfel-Becker T, Meshcheriakova A, Ali M, Klein AD, Platt FM, Cox TM, Futerman AH. RIPK3 as a potential therapeutic target for Gaucher's disease. Nat Med 2014;20:204-8.
39. Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, Ikiz B, Hoffmann L, Koolen M, Nagata T, Papadimitriou D, Nagy P, Mitsumoto H, Kariya S, Wichterle H, Henderson CE, Przedborski S. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 2014;81:1001-8.
40. David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci 2011;12:388-99.
41. Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ 2015;22:225-36.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Pan YH, Liu XY, Liu JQ, Liu Q, Yang Y, Yang JL, Zhang XF, Wu Y, Wang YZ. Necroptosis: a new link between cell death and inflammation. Neurosciences 2016;3:156-60. http://dx.doi.org/10.20517/2347-8659.2015.59
AMA Style
Pan YH, Liu XY, Liu JQ, Liu Q, Yang Y, Yang JL, Zhang XF, Wu Y, Wang YZ. Necroptosis: a new link between cell death and inflammation. Neuroimmunology and Neuroinflammation. 2016; 3: 156-60. http://dx.doi.org/10.20517/2347-8659.2015.59
Chicago/Turabian Style
Pan, Yuan-Hang, Xun-Yuan Liu, Jia-Qiang Liu, Qian Liu, Yang Yang, Jia-Lei Yang, Xiu-Fen Zhang, Yin Wu, Ya-Zhou Wang. 2016. "Necroptosis: a new link between cell death and inflammation" Neuroimmunology and Neuroinflammation. 3: 156-60. http://dx.doi.org/10.20517/2347-8659.2015.59
ACS Style
Pan, Y.H.; Liu X.Y.; Liu J.Q.; Liu Q.; Yang Y.; Yang J.L.; Zhang X.F.; Wu Y.; Wang Y.Z. Necroptosis: a new link between cell death and inflammation. Neurosciences. 2016, 3, 156-60. http://dx.doi.org/10.20517/2347-8659.2015.59
About This Article
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 16
likes
Like This Article 16
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.