
Hemodynamics, inflammation, vascular remodeling, and the development and rupture of intracranial aneurysms: a review


Abstract
The central nervous system is an immunologically active environment where several components of the immune and inflammatory response interact among them and with the constituents of nervous tissue and vasculature in a critically orchestrated manner, influencing physiologic and pathologic processes. In particular, inflammation takes a central role in the pathogenesis of intracranial aneurysms (IAs). The common pathway for aneurysm formation involves endothelial dysfunction and injury, a mounting inflammatory response, vascular smooth muscle cells (VSMCs) phenotypic modulation, extracellular matrix remodeling, and subsequent cell death and vessel wall degeneration. We conducted a literature review (1980-2014) by Medline and EMBASE databases using the searching terms “IA” and “cerebral aneurysm” and further search was performed to link the search terms with the following key words: inflammation, hemodynamic(s), remodeling, macrophages, neutrophils, lymphocytes, complement, VSMCs, mast cells, cytokines, and inflammatory biomarkers. The aim of this review was to summarize the most recent and pertinent evidences regarding the articulated processes of aneurysms formation, growth, and rupture. Knowledge of these processes may guide the diagnosis and treatment of these vascular malformations, the most common cause of subarachnoid hemorrhage, which prognosis remains dismal.
Keywords
Introduction
Saccular intracranial aneurysms (IAs) are the most frequent cause of subarachnoid hemorrhage (SAH), the stroke type with the higher morbidity and mortality.[1,2] A precise evaluation of their rupture risk is crucial to orient treatment of unruptured IAs relatively to the risk of endovascular or surgical treatment.[2-7] Statistic studies involved patients suffering from aneurysm rupture found that cigarette smoking, arterial hypertension, ethnic origin, age, previous SAH, size ≥ 7 mm, localization of IAs at the posterior circulation and aneurysm’s shape are the most important variables regarding the rupture risk.[2-7]
However, the majority of IAs diagnosed following their rupture is small and located on the anterior circulation,[8,9] which indicates that the statistical approach does not allow individualizing the risk of rupture. Elucidating the pathogenic pathways inherent to the development and rupture of IAs may allow identifying more reliable markers of rupture-prone IAs. A growing body of evidence supports the correlation between modification of hemodynamic factors and arterial wall alteration leading to IAs development and rupture.[10-13] Particularly, wall shear stress (WSS) gradient might be an important factor of vascular remodeling through multiple mechanisms involving endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) modification of gene expression triggered by local inflammatory reaction and leading to degenerative changes of arterial wall.[14] In this article, we summarize the existing data, extracted from a review of the pertinent literature, regarding inflammation and hemodynamic stress in the pathogenesis of IAs. Our endeavor is to explore the causative relationships that may link hemodynamics, inflammation, vascular remodeling and the development and rupture of IAs. This approach may provide effective tools to predict the individual risk of aneurysmal rupture more reliably than statistical methodology.
Methods of the review
The literature review was conducted using Medline and EMBASE searches that included works published between 1980 and 2014. The terms “IA” and “cerebral aneurysm” were used as text words and MESH headings with appropriate subheadings. A further search was performed to link cerebral and IAs and the following key words: inflammation, hemodynamic(s), remodeling, macrophages, neutrophils, lymphocytes, complement, VSMCs, mast cells, cytokines, and inflammatory biomarkers. Textbooks, journal bibliographies, and conference proceedings were also included. Language restrictions were not used.
Inflammation and intracranial aneurysms remodeling
The central nervous system is an immunologically active environment where a complex set of interactions links the various constituents of the immune and inflammatory system with the constituents of nervous tissue and vasculature.[15] Aneurysm formation begins with a hemodynamically triggered endothelial dysfunction where inflammation initiated by nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation and imbalance between nitric oxide (NO) and peroxynitrite anion (ONOO-) in favor of ONOO- seem to hold a key role.[16] The following mounting inflammatory response implicates several inflammatory cells and mediators and phenotypic modulation of VSMCs from a contractile to a pro-inflammatory/pro-matrix remodeling phenotype with myointimal hyperplasia, inflammation and wall degeneration.
Several in vitro and in vivo studies found evidences that infiltration of inflammatory cells in the arterial wall initiates aneurysm formation and promotes its rupture through production of inflammatory cytokines, adhesion molecules, immunoglobulins, reactive oxygen species (ROS), complement and inflammatory cell-induced upregulation of matrix-degrading proteinases, among which macrophages, neutrophils, and lymphocytes hold a central role.[17,18]
Macrophages
Monocytes originally produced in bone marrow enter circulation and infiltrate endothelium at the site of the hemodynamic injury, where they differentiate into macrophages.[18] Macrophage is one of the first cell types to respond to injury, but it also regulates the later immune response.[18] The role of these inflammatory cells in IAs formation is demonstrated by the fact that macrophage depleted mouse displays a lower risk of IAs.[19] Macrophage action on the arterial wall is mediated by secretion of cytokines and proteinases.
Cytokines are peptides, proteins, and glycoproteins that mediate inflammatory and immune response.[20-22] Several cytokines secreted by macrophages have been found involved in the pathogenesis of IA especially monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor alpha (TNF-α), stromal cells derived factor-1 (SDF-1/CXCL12). MCP-1 is a chemotactic cytokine, also known as chemokine, for monocytes, lymphocytes, and some granulocytes secreted by macrophages.[23] Its implication in the development of IAs is demonstrated by a decrease of IAs and arterial wall inflammation in MCP-1 knockout mice.[24] TNF-α is another proinflammatory cytokine secreted by macrophages.[18] Experimental data suggest a critical role of TNF-α in the formation and rupture of aneurysms in a murine model of cerebral aneurysm formation induced by hypertension and a single stereotactic injection of elastase into the basal cistern.[25] TNF-α knockout mice and those pre-treated with 3,6’-dithiothalidomide (DTH), a synthesized TNF-α inhibitor, had significantly decreased the incidence of aneurysm formation and rupture as compared to sham mice. Protein and mRNA expression of TNF-α in the cerebral vasculature were not significantly different in TNF-α knockout mice and in those pre-treated with DTH. However, TNF-α expression was higher in unruptured and the highest in ruptured aneurysms when compared to other conditions of aneurysms, where it co-localized to both smooth muscle cells and macrophages. SAH occurred between 7 and 21 days following aneurysm induction. Initiation of DTH treatment 6 days after aneurysm induction did not alter the incidence of aneurysm formation but resulted in aneurysmal stabilization and a significant decrease in rupture. Therefore, it can be inferred that inhibitors of TNF-α could be beneficial in preventing aneurysmal progression and rupture.[25] TNF-α upregulates the adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and E-selectin in ECs, fibroblasts, and SMCs.[26] These adhesion molecules attract and facilitate migration of leucocytes through the arterial wall, predisposing to atherosclerosis and IAs development.[26] Macrophages secrete also another potent chemoattractant cytokine, such as SDF-1/CXCL12.[27-29] Besides promoting angiogenesis by recruiting endothelial progenitor cells from the bone marrow through a CXCR4 dependent mechanism,[30] SDF-1 is associated with angiogenesis and migration and proliferation of macrophages in the walls of human and murine aneurysms, possibly playing a role in the development of IAs.[31] Macrophages also directly promote degradation of ECM by secreting a zinc and calcium-dependent family of endopeptidases known as matrix metalloproteinases (MMPs) and modulate their activity by producing tissue inhibitors of metalloproteinases (TIMPs), MCP-1 and TNF-α.[32-34] Beside from degrading all kinds of ECM proteins, these proteinases, particularly MMP-9 secreted also by VSMCs,[35,36] induce macrophage migration and infiltration across ECM.[37] Overexpression of MMP-9 in the wall of excised IA was first documented by Kim et al.,[38] followed by Takemura et al.[39] who later showed by immunohistochemical analysis of the overexpression of MMP-1, -2, and -9 in aneurysm walls. Moreover, the levels of MMP-2 and -9 were found to be higher in ruptured compared with unruptured aneurysms in a series of 30 patients, suggesting that MMP-mediated excessive breakdown of vessel ECM eventually leads to aneurysmal rupture.[40] The central role of MMPs in the pathogenesis of IAs is further demonstrated by the fact that inhibition of TIMPs promotes aneurysm formation in a rodent model.[41] Under physiological conditions, MMPs production is regulated at the level of transcription. An imbalance between the active MMPs and TIMPs leads to the accelerated destruction of connective tissue associated with several vascular diseases including IAs.[42]
Neutrophils
Other important inflammatory cells migrating to the site of arterial injury are neutrophils. As for macrophages, they secrete cytokines and MMPs. In addition, they produce peroxidases.[18] The array of neutrophil-derived cytokines is similar to that of macrophages, including TNF-α and cxc-chemokines such as interleukin (IL)-1β. However, unlike macrophages, they do not secrete cc-chemokines such as MCP-1.[39] IL-1β and cxc-chemokines are involved in various inflammatory cellular activities such as cell proliferation, differentiation, and apoptosis. Upregulation of IL-1β in animal models of IAs is associated with aneurysm formation and progression by reduced collagen biosynthesis in the aneurysm wall both at the transcriptional and post-transcriptional levels.[43,44] In addition, neutrophils secrete macrophage inflammatory protein 1-α, which reinforces cytokines’ action to promote inflammatory cell recruitment, migration, activation, and differentiation.[45]
Lymphocytes
Other first-responding inflammatory cells found in IAs wall are lymphocytes. They infiltrate arterial wall in early phases of aneurysm formation and rupture.[46] They are involved in the production of proinflammatory cytokines such as TNF-α, interferon-γ (IFN-γ) and IL-6.[47]
Complement
The role of complement in the mechanism of IA formation is not fully elucidated. In one study by a Helsinki group that compared ruptured with unruptured IA, the expression and activation of complement membrane attack complex were greater in ruptured samples and was associated significantly with aneurysm wall degeneration and inflammatory cell infiltration.[48] These authors showed in another study that complement activation occurs via the classical pathway as evidenced by the presence of classical pathway activators (IgG, IgM, C restive protein, oxidized low-density lipoprotein) in the IA wall.[49]
Vascular smooth muscle cells
As mentioned above, VSMCs are crucial in the process of IAs formation, and rupture. They are mostly found in the medial layer and synthesize the matrix for the structural integrity to the arterial wall. Thinning of this layer contributes to aneurysm formation and rupture.[50] In response to endothelial injury VSMCs first migrate into the intima where they multiply and give rise to myointimal hyperplasia.[51] Successively, VSMCs from a differentiated phenotype whose primary characteristic is contraction, dedifferentiation, losing capacity of expressing contractile genes, like myocardin, acquiring the capacity to express genes that may affect the rigidity or elasticity of the vascular wall such as collagen alfa2(I) gene (COL1A2) and upregulating proinflammatory genes, such as MMPs, MCP-1, VCAM 1, and IL.[52,53] This phenotypic modulation of VSMCs is induced by TNF-α and mediated by Kruppel-like transcription factor 4.[54] Phenotypically modulated VSMCs are no longer in spindle shape forming tightly compacted bands, but spider-like cells dissociated from each other, nonproliferating and noncontractile.[55] Likewise, aneurysmal rupture is associated with degeneration and caspase-mediated apoptotic loss of VSMCs of the aneurysm wall.[17,56,57]
Mast cells
Although best known for their role in allergy and anaphylaxis, mast cells play an important role in the inflammatory reaction leading to IAs formation and rupture mainly via cytokines release and expression and activation of MMPs.[58] Indeed, IAs formation is associated with the proliferation and degranulation of mastocytes, and ruptured aneurysms wall is richer in mast cells than unruptured IAs.[59] The finding that inhibitors of mast cell degranulation decrease the inflammatory reaction in aneurysm walls and block the progression of IAs in mice further support the participation of mast cells in the pathogenesis and rupture of IAs.[58]
Inflammatory cells interaction and arterial wall degeneration
Inflammation of arterial wall leading to formation of IAs is initiated by the infiltration of inflammatory cells (macrophages, neutrophils and lymphocytes), which release proinflammatory cytokines and proteinases as well as chemokines and chemoattractant cytokines, for the upregulating recruitment of inflammatory cells into the aneurysm wall.[60,61] In particular, levels of MCP-1, chemokine (C-C motif) ligand-5 (CCL5), monokine - induced - by - [gamma] - interferon, interferon-[gamma]-induced protein-10, Eotaxin, 2 other chemokines, IL-8 and IL-17 have been found to be higher in blood samples taken from the lumen of human IAs than blood samples from femoral arteries of the same patients.[61] Inflammatory cytokines lead to degradation and apoptosis of ECs and VSMCs through activation and upregulation of immune cells migration and activity.[21] Immune cells target not just cells but also ECM, the scaffolding structure that provides the arterial wall with tensile strength, elasticity, compressibility, adhesiveness as well as communicability between cells constituting the vessel wall.[17,18] In particular, macrophages secrete MMPs resulting in excessive proteolytic activity against connective tissue proteins, including collagens, elastin, and proteoglycans, which causes focal degradation of the vascular ECM and may contribute to aneurysm formation and growth.[31] Macrophages, in conjunction with lymphocytes, also act on VSMCs, for vessel wall remodeling.[21] Cytokines and growth factors secreted by macrophages and T-lymphocytes affect VSMCs phenotype changes[62] and promote their apoptosis.[48,63,64] One of the key initiators of apoptosis is interaction between the Fas receptor, which is expressed on the surface of both inflammatory cells and VSMCs, and its ligand (Fas-ligand, Fasl), which is expressed on the surface of macrophages and T-lymphocytes.[65] Their interaction induces VSMCs apoptosis through upregulation of cytokines such as TNF-α and interferons expressed by inflammatory cells.[17,54,56,57] These cytokines promote also the synthesis of NO, another factor inducing apoptosis.[66] Apoptotic loss of VSMCs induces arterial wall weakening by reducing matrix synthesis.[63,64]
Hemodynamic factors induce arterial wall injury and initiate wall inflammation
Endothelial dysfunction initially and vascular remodeling subsequently are triggered by shear stress.[67] This explains why IA is commonly found at arterial junctions, bifurcations or abrupt vascular angles where excessive hemodynamic stresses are exerted on arterial walls.[68] There is a close relation between WSS, endothelial dysfunction, and the downstream inflammatory reaction.[69,70]
Computational flow dynamic studies coupled with histological studies of the aneurysm wall demonstrated a correlation between hemodynamic conditions and inflammatory changes of intracranial arterial wall leading to aneurysm formation and rupture.[10-13,70] The most highlighted, even though, controversial factor studied is WSS,[12,71,72] which is the component of the forces coplanar with the cross-section of the artery, originating from blood circulation and acting on arterial walls.[73] WSS is related to dynamic viscosity of blood, flow velocity parallel to the arterial wall and distance of the vector to the wall.[73] Whether high or low WSS is involved in the arterial wall inflammatory damage, and development and rupture of IA are still matter of debate.[74] Hemodynamics in IAs is complex and includes areas of low and high WSS.[71,72] Several studies show that exposure to abnormal WSS drives endothelium-mediated proinflammatory reactions,[75] MMPs activation,[76-79] apoptosis of ECs and VSMCs,[80] ECM degradation, and arterial wall remodeling.[36,81] Spatial gradients and changes in WSS magnitude regulate ECs gene expression through the upregulation of transcription factors such as NF-κB under the conditions responding to cytokines, free radicals and other stimuli implicated in cell survival.[82-84] Oxidative stress in the arterial wall promotes IAs formation inducing direct endothelial injury, VSMCs phenotypic modulation and apoptosis, recruitment and invasion of inflammatory cells through upregulation of chemotactic cytokines and adhesion molecules, and MMPs activation.[85] Oxidative stress reflects an imbalance between the production of ROS and the arterial wall’s ability to readily detoxify the reactive intermediates or to repair the resulting damages. The ability of the arterial wall to counteract oxidative stress effects largely repose on NO action.[16] NO is an endothelium-derived relaxing factor that has several actions translating in anti-atherosclerotic properties: it modulates vasomotor tone, inhibits expression of MCP-1 and VCAM-1, prevents propagation of lipid oxidation, inhibits VSCMc proliferation, decreases platelet aggregation[86] and inhibits expression and activity of MMPs.[86] Practically, all risk factors for arterial wall damage (hypercholesterolemia, diabetes, insulin resistance, arterial hypertension, cigarette smoking) reduce production of endothelial NO through increased production of superoxide and other ROS.[16,86-90] These strong oxidants both disrupt NO-mediated arterial wall protection decreasing availability of NO and promote arterial wall inflammation increasing ONOO-production.[16,86-90] Under physiological conditions, unidirectional laminar shear stress increases NO availability through a biphasic action: within seconds after stress the endothelial nitric oxide synthase (eNOS) enzyme expressed in ECs is activated by a calcium-independent mechanisms.[91] Subsequently, eNOS expression is upregulated through NF-κB activation by a transient, one hour lasting, increase in eNOS mRNA transcription and a sustained increase in eNOS mRNA stability.[92] However, NF-κB activation leads also to an increased expression of pro-inflammatory genes encoding cytokines, VCAM-1, ICAM-1, tissue factor and MCP-1.[16,86-90] At a first glance, the NF-κB mediated protective and damaging effect appears to be contradictory. Nonetheless, the evidence of a negative feedback pathway on NF-κB activation mediated by NO production resolves this apparent paradox. If eNOS levels are relatively high, sufficient NO is produced to shut down NF-κB activation soon after shear is applied to endothelium through a classical negative feedback pathway.[16] Conversely, if eNOS levels are relatively low, NF-κB activation persists enough to restore eNOS to normal levels in order to ensure a long-lasting protection to ECs. The double effect of NF-κB activation also explains why shear stress may reveal both harmful and protective to endothelium. As a matter of fact, in case of alteration of the inhibitory limb of the NO-mediated negative feedback, the proinflammatory action of shear stress prevails, leading to endothelium damage, with elongation, migration, change in density, and loss of ECs and to VSMCs phenotype change with acquisition of pro-inflammatory/pro-matrix remodeling properties.[17,50,61] ROS may also play a role in the pathogenesis of IAs.[93,94] Aoki et al.[95] demonstrated the upregulation of genes producing ROS and the downregulation of ROS-eliminating genes in a murine model of IAs. Moreover, the same study showed a similar effect of edaravone, a free radical scavenger, and of the deletion of ROS-producing gene: in two separate groups of animals both effectively inhibited IAs formation by suppressing inflammation in aneurysmal walls.[95] Moreover, low WSS may provide upregulation of proinflammatory cytokines and their receptors, such as IL-1α, IL-1 receptor, IL-6, and MCP-1.[96] In addition, an in vitro study showed that WSS on ECs causes a differential modulation of TNF-α-induced expression of adhesion molecules such as ICAM-1, VCAM-1, and E-selectin by reducing intracellular ECs ROS levels.[97] This may cause inhibition of TNF-α-induced VCAM-1 and E-selectin expression in ECs through inhibition of NF-κB activation.[97] However, the same authors showed that WSS-induced production of TNF-α stimulates the expression of another adhesion molecule, ICAM-1.[97] These apparently discordant findings indicate that a more thorough study of the cross-talk between these signaling molecules may shed further light onto the biological end-points produced by the WSS in modulating cytokine-induced adhesion molecule expression in ECs. Although the processes by which hemodynamic factors affect inflammation of the artery wall is incompletely known, vascular remodeling in response to abnormal WSS correlates with increased ECs and VSMCs apoptosis,[98,99] with upregulation of MMPs activity in both ECs[33] and VSMCs[36] and with upregulation of several transcription factors and inflammatory cytokines by inflammatory cells.[18] Along these lines, Wang et al.[82] showed in canine models that in areas of high shear stress (arterial bifurcations), aneurysm wall remodeling is associated with IL-1β and MMPs expression along with a loss of eNOS expression. In line with these findings, EC injury was found by Jamous et al.[100,101] to be the earliest change in aneurysm wall, followed by the formation of an inflammatory zone that leads to proteolytic destruction of the vascular ECM by MMPs and ultimately to aneurysm formation.
In summary, under physiologic condition shear stress promotes both endothelial NF-κB upregulation and immediate eNOS activation. In turn, NF-κB triggers both transient upregulation of eNOS and increased eNOS stability. This results in increased NO synthesis. NO protects arterial wall through a “direct” action on it as well as “indirectly” through a negative feedback on NF-κB activation. When the chain of events in red prevails, shear stress sustains arterial wall protection. Failure of NO-mediated direct and/or indirect arterial wall protection shifts the balance towards inflammation.
When the degeneration in the arterial wall, including loss of endothelial and smooth muscle cells and degradation of ECM are not healed, chronic remodeling of tissue takes place to alter the biomechanical properties of arterial wall and aneurysm formation, which eventually rupture [Figure 1].[63,64]
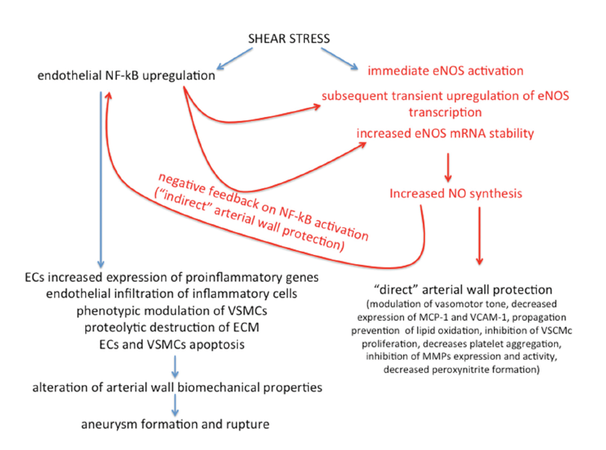
Figure 1. Impact on arterial wall condition of balance between shear stress‑mediated endothelial nuclear factor kappa B‑cell upregulation and nitric oxide synthesis
Inflammatory biomarkers and estimation of risk of rupture
Giving that considerable evidence suggest the involvement of inflammation in development and rupture of IAs,[19-25,31,35-41,48-64,70,76-81,93,94,99-101] preoperative noninvasive assessment of inflammatory status of the aneurysm wall may guide management of unruptured IAs.[102-105] One possible tool to identify rupture-prone IAs is ferumoxytol-enhanced magnetic resonance imaging (MRI).[102-104] Ferumoxytol is an ultrasmall superparamagnetic particle of iron oxide that reveals phagocytic activity of inflammatory cells because it is cleared by macrophages.[102-104] Ferumoxytol is hypointense on MRI T2*-weighted gradient echo sequences and hyperintense on T1-weighted spin echo sequences. It is detected inside blood vessels for ≤ 72 h and begins to clear within 24 h from injection. Its early uptake, within 24 h, is secondary to macrophage uptake and considering the crucial role of such inflammatory cells in development and rupture of IAs, it indicates active inflammation in aneurysm walls of unstable aneurysm, as suggested by one recent study.[102] These authors found that early ferumoxytol uptake was significantly higher in aneurysms with marked upregulation of inflammatory molecules such as cyclooxygenase-2 and microsomal prostaglandin E synthase-1 and macrophages, independently from site and size of the aneurysms.[101] Moreover, all three unruptured IAs of their series with early ferumoxytol uptake that were managed conservatively ruptured less than 6 months after diagnosis, which supports the hypothesis that inflammation is the cause but not the consequence of the rupture.[102] Another line of research aiming at differentiating ruptured and unruptured IAs on the basis of distinctive patterns of expression of inflammatory markers is the analysis of gene expression profile.[105] A recent study compared the gene expression arrays of ruptured and unruptured aneurysms and found a significant difference of expression of genes encoding macrophage-mediated inflammatory molecules according to the age of patients.[105] In particular, genes involved in vascular remodeling, inflammation, and atherosclerosis such as S100/calgranulin genes (S100A8, S100A9, and S100A12), cluster of differentiation 163, myeloperoxidase (MPO), were upregulated, while genes for Krüppel-like family of transcription factors (KLF2, KLF12, and KLF15) and CDKN2, which are respectively anti-inflammatory regulators and inhibitors of cellular proliferation, were downregulated, together with cell adhesion molecules and cytoskeletal proteins of arterial wall.[105] Therefore, the authors conclude that some of these identified genes may help identifying IAs at risk of rupture, which warrant early treatment.[105]
Conclusion
Aneurysm formation begins with a hemodynamically triggered endothelial inflammatory dysfunction, which is the cause rather than the consequence of aneurysms’ development and rupture. The proinflammatory action of shear stress prevails over its endothelium protective action when the balance between NF-κB-mediated production of NO and proinflammatory mediators (ROS, cytokines, adhesion molecules) shifts in favor of inflammation because of alteration of the inhibitory limb of the NO-mediated negative feedback on NF-κB activation. Targets of the inflammatory reaction are, besides ECs, ECM, and VSMCs. Endothelial injury, VSMCs phenotypic modulation with acquisition of pro-inflammatory/pro-matrix remodeling properties and subsequent Fas-mediated apoptotic cell death lead to the arterial wall weakening and aneurysm formation and rupture. Clarifying the causative relationships that link hemodynamics, inflammation, vascular remodeling, and the development and rupture of IAs may provide effective tools to predict the individual risk of aneurysmal rupture and aid the treatment decision-making process.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 1994;25:1342-7.
2. Bederson JB, Connolly ES Jr, Batjer HH, Dacey RG, Dion JE, Diringer MN, Duldner JE Jr, Harbaugh RE, Patel AB, Rosenwasser RH; American Heart Association. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke 2009;40:994-1025.
3. da Costa LB, Gunnarsson T, Wallace MC. Unruptured intracranial aneurysms: natural history and management decisions. Neurosurg Focus 2004;17:E6.
4. Greving JP, Wermer MJ, Brown RD Jr, Morita A, Juvela S, Yonekura M, Ishibashi T, Torner JC, Nakayama T, Rinkel GJ, Algra A. Development of the PHASES score for prediction of risk of rupture of intracranial aneurysms: a pooled analysis of six prospective cohort studies. Lancet Neurol 2014;13:59-66.
5. Nishioka H, Torner JC, Graf CJ, Kassell NF, Sahs AL, Goettler LC. Cooperative study of intracranial aneurysms and subarachnoid hemorrhage: a long-term prognostic study. II. Ruptured intracranial aneurysms managed conservatively. Arch Neurol 1984;41:1142-6.
7. Wiebers DO, Whisnant JP, Huston J 3rd, Meissner I, Brown RD Jr, Piepgras DG, Forbes GS, Thielen K, Nichols D, O'Fallon WM, Peacock J, Jaeger L, Kassell NF, Kongable-Beckman GL, Torner JC; International Study of Unruptured Intracranial Aneurysms Investigators. Unruptured intracranial aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet 2003;362:103-10.
8. Hernesniemi J, Dashti R, Lehecka M, Niemelä M, Rinne J, Lehto H, Ronkainen A, Koivisto T, Jaaskelainen JE. Microneurosurgical management of anterior communicating artery aneurysms. Surg Neurol 2008;70:8-28.
9. Roessler K, Cejna M, Zachenhofer I. Aneurysmatic subarachnoidal haemorrhage: incidence and location of small ruptured cerebral aneurysms - a retrospective population-based study. Wien Klin Wochenschr 2011;123:444-9.
10. Shojima M, Oshima M, Takagi K, Torii R, Hayakawa M, Katada K, Morita A, Kirino T. Magnitude and role of wall shear stress on cerebral aneurysm: computational fluid dynamic study of 20 middle cerebral artery aneurysms. Stroke 2004;35:2500-5.
11. Cebral JR, Mut F, Weir J, Putman C. Quantitative characterization of the hemodynamic environment in ruptured and unruptured brain aneurysms. AJNR Am J Neuroradiol 2011;32:145-51.
12. Xiang J, Natarajan SK, Tremmel M, Ma D, Mocco J, Hopkins LN, Siddiqui AH, Levy EI, Meng H. Hemodynamic-morphologic discriminants for intracranial aneurysm rupture. Stroke 2011;42:144-52.
13. Kono K, Fujimoto T, Shintani A, Terada T. Hemodynamic characteristics at the rupture site of cerebral aneurysms: a case study. Neurosurgery 2012;71:E1202-8.
14. Hashimoto T, Meng H, Young WL. Intracranial aneurysms: links among inflammation, hemodynamics and vascular remodeling. Neurol Res 2006;28:372-80.
15. Dumont AS, Dumont RJ, Chow MM, Lin CL, Calisaneller T, Ley KF, Kassell NF, Lee KS. Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery 2003;53:123-33.
16. Harrison DG, Widder J, Grumbach I, Chen W, Weber M, Searles C. Endothelial mechanotransduction, nitric oxide and vascular inflammation. J Intern Med 2006;259:351-63.
17. Chalouhi N, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Koch WJ, Dumont AS. Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab 2012;32:1659-76.
19. Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N, Lawton MT, Young WL, Liang EI, Nuki Y, Hashimoto T. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke 2011;42:173-8.
20. Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev 2006;86:515-81.
21. Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol 2009;78:539-52.
22. Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol 2011;31:969-79.
24. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Egashira K, Hashimoto N. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke 2009;40:942-51.
25. Starke RM, Chalouhi N, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Wada K, Shimada K, Hasan DM, Greig NH, Owens GK, Dumont AS. Critical role of TNF-α in cerebral aneurysm formation and progression to rupture. J Neuroinflammation 2014;11:77.
26. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993;362:801-9.
27. Peled A, Petit I, Kollet O, Magid M, Ponomaryov T, Byk T, Nagler A, Ben-Hur H, Many A, Shultz L, Lider O, Alon R, Zipori D, Lapidot T. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 1999;283:845-8.
28. Ponomaryov T, Peled A, Petit I, Taichman RS, Habler L, Sandbank J, Arenzana-Seisdedos F, Magerus A, Caruz A, Fujii N, Nagler A, Lahav M, Szyper-Kravitz M, Zipori D, Lapidot T. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J Clin Invest 2000;106:1331-9.
29. Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med 2002;195:1145-54.
30. Zheng H, Fu G, Dai T, Huang H. Migration of endothelial progenitor cells mediated by stromal cell-derived factor-1alpha/CXCR4 via PI3K/Akt/eNOS signal transduction pathway. J Cardiovasc Pharmacol 2007;50:274-80.
31. Hoh BL, Hosaka K, Downes DP, Nowicki KW, Wilmer EN, Velat GJ, Scott EW. Stromal cell-derived factor-1 promoted angiogenesis and inflammatory cell infiltration in aneurysm walls. J Neurosurg 2014;120:73-86.
33. Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol 2008;75:346-59.
34. Thomson EM, Williams A, Yauk CL, Vincent R. Overexpression of tumor necrosis factor-α in the lungs alters immune response, matrix remodeling, and repair and maintenance pathways. Am J Pathol 2012;180:1413-30.
35. Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke 2007;38:162-9.
36. Kolega J, Gao L, Mandelbaum M, Mocco J, Siddiqui AH, Natarajan SK, Meng H. Cellular and molecular responses of the basilar terminus to hemodynamics during intracranial aneurysm initiation in a rabbit model. J Vasc Res 2011;48:429-42.
37. Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest 2008;118:3012-24.
38. Kim SC, Singh M, Huang J, Prestigiacomo CJ, Winfree CJ, Solomon RA, Connolly ES, Jr. Matrix metalloproteinase-9 in cerebral aneurysms. Neurosurgery 1997;41:642-66.
39. Takemura Y, Hirata Y, Sakata N, Nabeshima K, Takeshita M, Inoue T. Histopathologic characteristics of a saccular aneurysm arising in the non-branching segment of the distal middle cerebral artery. Pathol Res Pract 2010;206:391-6.
40. Jin D, Sheng J, Yang X, Gao B. Matrix metalloproteinases and tissue inhibitors of metalloproteinases expression in human cerebral ruptured and unruptured aneurysm. Surg Neurol 2007;68 Suppl 2:S11-6.
41. Aoki T, Kataoka H, Moriwaki T, Nozaki K, Hashimoto N. Role of TIMP-1 and TIMP-2 in the progression of cerebral aneurysms. Stroke 2007;38:2337-45.
42. Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest 2002;110:625-32.
43. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Morishita R, Hashimoto N. Reduced collagen biosynthesis is the hallmark of cerebral aneurysm: contribution of interleukin-1beta and nuclear factor-kappaB. Arterioscler Thromb Vasc Biol 2009;29:1080-6.
44. Johnston WF, Salmon M, Su G, Lu G, Stone ML, Zhao Y, Owens GK, Upchurch GR Jr, Ailawadi G. Genetic and pharmacologic disruption of interleukin-1ß signaling inhibits experimental aortic aneurysm formation. Arterioscler Thromb Vasc Biol 2013;33:294-304.
45. Kasama T, Miwa Y, Isozaki T, Odai T, Adachi M, Kunkel SL. Neutrophil-derived cytokines: potential therapeutic targets in inflammation. Curr Drug Targets Inflamm Allergy 2005;4:273-9.
46. Zhou HF, Yan H, Cannon JL, Springer LE, Green JM, Pham CT. CD43-mediated IFN-γ production by CD8+T cells promotes abdominal aortic aneurysm in mice. J Immunol 2013;190:5078-85.
47. Zheng S, Su A, Sun H, You C. The association between interleukin-6 gene polymorphisms and intracranial aneurysms: a meta-analysis. Hum Immunol 2013;74:1679-83.
48. Tulamo R, Frösen J, Junnikkala S, Paetau A, Pitkäniemi J, Kangasniemi M, Niemela M, Jaaskelainen J, Jokitalo E, Karatas A, Hernesniemi J, Meri S. Complement activation associates with saccular cerebral artery aneurysm wall degeneration and rupture. Neurosurgery 2006;59:1069-76.
49. Tulamo R, Frösen J, Junnikkala S, Paetau A, Kangasniemi M, Peláez J, Hernesniemi J, Niemela M, Meri S. Complement system becomes activated by the classical pathway in intracranial aneurysm walls. Lab Invest 2010;90:168-79.
50. Chalouhi N, Hoh BL, Hasan D. Review of cerebral aneurysm formation, growth, and rupture. Stroke 2013;44:3613-22.
51. Kosierkiewicz TA, Factor SM, Dickson DW. Immunocytochemical studies of atherosclerotic lesions of cerebral berry aneurysms. J Neuropathol Exp Neurol 1994;53:399-406.
52. Nakajima N, Nagahiro S, Sano T, Satomi J, Satoh K. Phenotypic modulation of smooth muscle cells in human cerebral aneurysmal walls. Acta Neuropathol 2000;100:475-80.
53. Sibon I, Mercier N, Darret D, Lacolley P, Lamazière JM. Association between semicarbazide-sensitive amine oxidase, a regulator of the glucose transporter, and elastic lamellae thinning during experimental cerebral aneurysm development: laboratory investigation. J Neurosurg 2008;108:558-66.
54. Ali MS, Starke RM, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Owens GK, Koch WJ, Greig NH, Dumont AS. TNF-α induces phenotypic modulation in cerebral vascular smooth muscle cells: implications for cerebral aneurysm pathology. J Cereb Blood Flow Metab 2013;33:1564-73.
55. Mérei FT, Gallyas F. Role of the structural elements of the arterial wall in the formation and growth of intracranial saccular aneurysms. Neurol Res 1980;2:283-303.
56. Sakaki T, Kohmura E, Kishiguchi T, Yuguchi T, Yamashita T, Hayakawa T. Loss and apoptosis of smooth muscle cells in intracranial aneurysms. Studies with in situ DNA end labeling and antibody against single-stranded DNA. Acta Neurochir (Wien) 1997;139:469-74.
57. Guo F, Li Z, Song L, Han T, Feng Q, Guo Y, Xu J, He M, You C. Increased apoptosis and cysteinyl aspartate specific protease-3 gene expression in human intracranial aneurysm. J Clin Neurosci 2007;14:550-5.
58. Ishibashi R, Aoki T, Nishimura M, Hashimoto N, Miyamoto S. Contribution of mast cells to cerebral aneurysm formation. Curr Neurovasc Res 2010;7:113-24.
59. Hasan D, Chalouhi N, Jabbour P, Hashimoto T. Macrophage imbalance (M1 vs. M2) and upregulation of mast cells in wall of ruptured human cerebral aneurysms: preliminary results. J Neuroinflammation 2012;9:222.
60. Krischek B, Kasuya H, Tajima A, Akagawa H, Sasaki T, Yoneyama T, Ujiie H, Kubo O, Bonin M, Takakura K, Hori T, Inoue I. Network-based gene expression analysis of intracranial aneurysm tissue reveals role of antigen presenting cells. Neuroscience 2008;154:1398-407.
61. Chalouhi N, Points L, Pierce GL, Ballas Z, Jabbour P, Hasan D. Localized increase of chemokines in the lumen of human cerebral aneurysms. Stroke 2013;44:2594-7.
62. Kilic T, Sohrabifar M, Kurtkaya O, Yildirim O, Elmaci I, Günel M, Pamir MN. Expression of structural proteins and angiogenic factors in normal arterial and unruptured and ruptured aneurysm walls. Neurosurgery 2005;57:997-1007.
63. Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke 1999;30:1396-401.
64. Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, Jaaskelainen J. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke 2004;35:2287-93.
65. Schaub FJ, Han DK, Liles WC, Adams LD, Coats SA, Ramachandran RK, Seifert RA, Schwartz SM, Bowen-Pope DF. Fas/FADD-mediated activation of a specific program of inflammatory gene expression in vascular smooth muscle cells. Nat Med 2000;6:790-6.
66. Brüne B, von Knethen A, Sandau KB. Nitric oxide (NO): an effector of apoptosis. Cell Death Differ 1999;6:969-75.
67. Sho E, Sho M, Singh TM, Nanjo H, Komatsu M, Xu C, Masuda H, Zarins CK. Arterial enlargement in response to high flow requires early expression of matrix metalloproteinases to degrade extracellular matrix. Exp Mol Pathol 2002;73:142-53.
68. Kondo S, Hashimoto N, Kikuchi H, Hazama F, Nagata I, Kataoka H. Cerebral aneurysms arising at nonbranching sites. An experimental Study. Stroke 1997;28:398-403.
69. Nixon AM, Gunel M, Sumpio BE. The critical role of hemodynamics in the development of cerebral vascular disease. J Neurosurg 2010;112:1240-53.
70. Turjman AS, Turjman F, Edelman ER. Role of fluid dynamics and inflammation in intracranial aneurysm formation. Circulation 2014;129:373-82.
71. Boussel L, Rayz V, McCulloch C, Martin A, Acevedo-Bolton G, Lawton M, Higashida R, Smith WS, Young WL, Saloner D. Aneurysm growth occurs at region of low wall shear stress: patient-specific correlation of hemodynamics and growth in a longitudinal study. Stroke 2008;39:2997-3002.
72. Kadasi LM, Dent WC, Malek AM. Colocalization of thin-walled dome regions with low hemodynamic wall shear stress in unruptured cerebral aneurysms. J Neurosurg 2013;119:172-9.
73. Perktold K, Thurner E, Kenner T. Flow and stress characteristics in rigid walled and compliant carotid artery bifurcation models. Med Biol Eng Comput 1994;32:19-26.
74. Meng H, Tutino VM, Xiang J, Siddiqui A. High WSS or low WSS? Complex interactions of hemodynamics with intracranial aneurysm initiation, growth, and rupture: toward a unifying hypothesis. AJNR Am J Neuroradiol 2014;35:1254-62.
75. Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA 1999;282:2035-42.
76. Sforza DM, Putman CM, Cebral JR. Hemodynamics of Cerebral Aneurysms. Annu Rev Fluid Mech 2009;41:91-107.
77. Kim SC, Singh M, Huang J, Prestigiacomo CJ, Winfree CJ, Solomon RA, Connolly ES, Jr. Matrix metalloproteinase-9 in cerebral aneurysms. Neurosurgery 1997;41:642-66.
78. Bruno G, Todor R, Lewis I, Chyatte D. Vascular extracellular matrix remodeling in cerebral aneurysms. J Neurosurg 1998;89:431-40.
79. Kadirvel R, Ding YH, Dai D, Zakaria H, Robertson AM, Danielson MA, Lewis DA, Cloft HJ, Kallmes DF. The influence of hemodynamic forces on biomarkers in the walls of elastase-induced aneurysms in rabbits. Neuroradiology 2007;49:1041-53.
80. Ota R, Kurihara C, Tsou TL, Young WL, Yeghiazarians Y, Chang M, Mobashery S, Sakamoto A, Hashimoto T. Roles of matrix metalloproteinases in flow-induced outward vascular remodeling. J Cereb Blood Flow Metab 2009;29:1547-58.
81. Frösen J, Tulamo R, Paetau A, Laaksamo E, Korja M, Laakso A, Niemela M, Hernesniemi J. Saccular intracranial aneurysm: pathology and mechanisms. Acta Neuropathol 2012;123:773-86.
82. Wang Z, Kolega J, Hoi Y, Gao L, Swartz DD, Levy EI, Mocco J, Meng H. Molecular alterations associated with aneurysmal remodeling are localized in the high hemodynamic stress region of a created carotid bifurcation. Neurosurgery 2009;65:169-77.
83. Frangos JA, Eskin SG, McIntire LV, Ives CL. Flow effects on prostacyclin production by cultured human endothelial cells. Science 1985;227:1477-9.
84. Khachigian LM, Resnick N, Gimbrone MA Jr, Collins T. Nuclear factor-kappa B interacts functionally with the platelet-derived growth factor B-chain shear-stress response element in vascular endothelial cells exposed to fluid shear stress. J Clin Invest 1995;96:1169-75.
85. LaMack JA, Friedman MH. Individual and combined effects of shear stress magnitude and spatial gradient on endothelial cell gene expression. Am J Physiol Heart Circ Physiol 2007;293:H2853-9.
87. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000;87:840-4.
88. Inoue N, Kawashima S, Hirata KI, Rikitake Y, Takeshita S, Yamochi W, Akita H, Yokoyama M. Stretch force on vascular smooth muscle cells enhances oxidation of LDL via superoxide production. Am J Physiol 1998;274:H1928-32.
89. Lehoux S, Esposito B, Merval R, Loufrani L, Tedgui A. Pulsatile stretch-induced extracellular signal-regulated kinase 1/2 activation in organ culture of rabbit aorta involves reactive oxygen species. Arterioscler Thromb Vasc Biol 2000;20:2366-72.
90. Grote K, Flach I, Luchtefeld M, Akin E, Holland SM, Drexler H, Schieffer B. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase-derived reactive oxygen species. Circ Res 2003;92:e80-6.
91. Mata-Greenwood E, Grobe A, Kumar S, Noskina Y, Black SM. Cyclic stretch increases VEGF expression in pulmonary arterial smooth muscle cells via TGF-beta1 and reactive oxygen species: a requirement for NAD(P)H oxidase. Am J Physiol Lung Cell Mol Physiol 2005;289:L288-9.
92. Kuchan MJ, Frangos JA. Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am J Physiol 1994;266:C628-36.
93. Davis ME, Cai H, Drummond GR, Harrison DG. Shear stress regulates endothelial nitric oxide synthase expression through c-Src by divergent signaling pathways. Circ Res 2001;89:1073-80.
94. Starke RM, Chalouhi N, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Koch WJ, Dumont AS. The role of oxidative stress in cerebral aneurysm formation and rupture. Curr Neurovasc Res 2013;10:247-55.
95. Aoki T, Nishimura M, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Reactive oxygen species modulate growth of cerebral aneurysms: a study using the free radical scavenger edaravone and p47phox(-/-) mice. Lab Invest 2009;89:730-41.
96. Passerini AG, Polacek DC, Shi C, Francesco NM, Manduchi E, Grant GR, Pritchard WF, Powell S, Chang GY, Stoeckert CJ Jr, Davies PF. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci U S A 2004;101:2482-7.
97. Chiu JJ, Lee PL, Chen CN, Lee CI, Chang SF, Chen LJ, Lien SC, Ko YC, Usami S, Chien S. Shear stress increases ICAM-1 and decreases VCAM-1 and E-selectin expressions induced by tumor necrosis factor-[alpha] in endothelial cells. Arterioscler Thromb Vasc Biol 2004;24:73-9.
98. Cho A, Courtman DW, Langille BL. Apoptosis (programmed cell death) in arteries of the neonatal lamb. Circ Res 1995;76:168-75.
99. Cho A, Mitchell L, Koopmans D, Langille BL. Effects of changes in blood flow rate on cell death and cell proliferation in carotid arteries of immature rabbits. Circ Res 1997;81:328-37.
100. Jamous MA, Nagahiro S, Kitazato KT, Satoh K, Satomi J. Vascular corrosion casts mirroring early morphological changes that lead to the formation of saccular cerebral aneurysm: an experimental study in rats. J Neurosurg 2005;102:532-5.
101. Jamous MA, Nagahiro S, Kitazato KT, Tamura T, Aziz HA, Shono M, Satoh K. Endothelial injury and inflammatory response induced by hemodynamic changes preceding intracranial aneurysm formation: experimental study in rats. J Neurosurg 2007;107:405-11.
102. Hasan D, Chalouhi N, Jabbour P, Dumont AS, Kung DK, Magnotta VA, Young WL, Hashimoto T, Winn HR, Heistad D. Early change in ferumoxytol-enhanced magnetic resonance imaging signal suggests unstable human cerebral aneurysm: a pilot study. Stroke 2012;43:3258-65.
103. Neuwelt EA, Várallyay CG, Manninger S, Solymosi D, Haluska M, Hunt MA, Nesbit G, Stevens A, Jerosch-Herold M, Jacobs PM, Hoffman JM. The potential of ferumoxytol nanoparticle magnetic resonance imaging, perfusion, and angiography in central nervous system malignancy: a pilot study. Neurosurgery 2007;60:601-11; discussion 611-2.
104. Dósa E, Tuladhar S, Muldoon LL, Hamilton BE, Rooney WD, Neuwelt EA. MRI using ferumoxytol improves the visualization of central nervous system vascular malformations. Stroke 2011;42:1581-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Signorelli F, Gory B, Riva R, Labeyrie PE, Pelissou-Guyotat I, Turjman F. Hemodynamics, inflammation, vascular remodeling, and the development and rupture of intracranial aneurysms: a review. Neurosciences 2015;2:59-67. http://dx.doi.org/10.4103/2347-8659.154885
AMA Style
Signorelli F, Gory B, Riva R, Labeyrie PE, Pelissou-Guyotat I, Turjman F. Hemodynamics, inflammation, vascular remodeling, and the development and rupture of intracranial aneurysms: a review. Neuroimmunology and Neuroinflammation. 2015; 2: 59-67. http://dx.doi.org/10.4103/2347-8659.154885
Chicago/Turabian Style
Signorelli, Francesco, Benjamin Gory, Roberto Riva, Paul-Emile Labeyrie, Isabelle Pelissou-Guyotat, Francis Turjman. 2015. "Hemodynamics, inflammation, vascular remodeling, and the development and rupture of intracranial aneurysms: a review" Neuroimmunology and Neuroinflammation. 2: 59-67. http://dx.doi.org/10.4103/2347-8659.154885
ACS Style
Signorelli, F.; Gory B.; Riva R.; Labeyrie P.E.; Pelissou-Guyotat I.; Turjman F. Hemodynamics, inflammation, vascular remodeling, and the development and rupture of intracranial aneurysms: a review. Neurosciences. 2015, 2, 59-67. http://dx.doi.org/10.4103/2347-8659.154885
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.