
Influence of chlorpyrifos oxon on the development and progression of Alzheimer's disease in amyloid precursor protein transgenic mice

Abstract
Aim: Alzheimer’s disease (AD) is a devastating neurological disorder and the most common form of dementia. Until date, the cause of AD eludes us, but a number of hypotheses have been put forward to try and understand the mechanisms involved. A series of studies have indicated that environmental factors, such as pesticides, heavy metals, and others can contribute to the development and progression of AD. Based on these data, we determined the impact of pesticides [chlorpyrifos oxon (CPO)] on AD-like pathogenesis in amyloid precursor protein (APP) transgenic mice.
Methods: APP mice were treated at various times with low-dose CPO (1 mg/kg/day), in utero (3-week of gestation), during lactation (3-week), or as young adults (continuous dosing).
Results: Exposure to CPO at all times enhanced neuro-inflammation and exacerbated oxidative stress in the brain prior to amyloid deposition. CPO-treated APP mice showed a decrease in memory and learning compared with untreated APP mice; furthermore, analyses of brain tissue sections and extracts showed an increase in Aβ levels and C-terminal fragment-β levels, a decrease in soluble APPα (sAPPα) levels, and an increase in plaque load. In addition, CPO-treated APP transgenic mice showed a significant decrease in neurotrophic factor levels (nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3) compared to vehicle-treated APP transgenic animals. Treatment with galantamine attenuated the effects of CPO by reducing amyloid β levels and amyloid load.
Conclusion: CPO accelerated and exacerbated the disease development and progression in the APP mice suggesting that pesticides may play a significant role in the pathogenesis of AD.
Keywords
Introduction
Amyloid β peptide (Aβ) containing senile plaques are one of the neuropathological hallmarks of Alzheimer’s disease (AD). Much of this work has focused on the biosynthesis of Aβ and factors that influence its deposition.[1] The Aβ peptides are generated via internal proteolysis of its precursor, the amyloid precursor protein (APP).[2,3] In addition, a variety of neuronal cytoskeletal alterations is prominent features of AD neuropathology.[4,5] Whether these abnormal features are the result or cause of neuronal loss is still controversial. [6-10] Early onset autosomal dominant AD is directly linked to mutations in one of the several genes: APP, presenilin 1 (PS1), or presenilin 2 (PS2).[11-13] In addition, several genes, most notably the apolipoprotein E (APOE) 4 allele, alter risk for later onset AD, and it is clear that mutation or polymorphism in several other genes can lead to similar AD phenotypes.[14]
Recent studies have suggested that early exposure of individuals to environmental toxins, drugs, and other agents appear to contribute to diseases that occur later in life.[15-18] Osmond and Barker have suggested (and this has been supported by others) that there are critical periods during fetal development where programming by a stimulus or insult has a lasting or lifelong effect.[19] We know that various tissues, including the brain, go through developmental programming to determine the number of cells (programmed cell death), types of cells, that participate to the final development of the brain.[20] It has been suggested that exposure of tissues to insults at critical times could result in reprogramming to give rise to diseases later in life.[18] It has been suggested that exposure of the brain to toxic agents triggers a process that will elicit the onset of AD, reprogram the brain to have AD start at an earlier age, or to exacerbate the process of AD.[21] This can be achieved by a number of ways. Metabolic, blood flow, endocrine, nutritional changes can affect blood lipids, plasma insulin, obesity, atherosclerosis, behavior and learning in small mammals and primates.[21-23] The application of this process to AD has been recently suggested and demonstrated in the latent early-life associated regulation model.[24,25]
This study evaluated the effects of the pesticide chlorpyrifos oxon (CPO) on the development of AD later in life. To test this hypothesis, we determined the influence of CPO on AD by treating APP transgenic mice during gestation, during lactation and after weaning. Our data showed that CPO exacerbated the pathogenesis of AD in the mouse model and that this process may be partially mediated by inflammation and by the effects of CPO on acetylcholinesterase (AChE) inhibition. These findings validate the influence of environmental toxins on the development and progression of AD.
Methods
Transgenic Alzheimer’s disease mice
The mice used for these studies expressed the mutant form of human presenilin-1 (DeltaE9) and the mutant form of the chimeric mouse/human APP695.[26] The mouse prion protein promoter directed the expression of both transgenes. The DeltaE9 mutation of the human presenilin-1 gene is a deletion of exon nine and corresponds to a form associated with early-onset AD. The APP695 gene harbors the K595N/M596L (Swedish) AD-causing mutations. The coding sequence of mouse Aβ peptide domain was humanized by replacing the three amino acids that differ between the two species with the human residues. These APP/ΔPS1-Tg (referred to as APP) mice start developing amyloid plaques at about 3-4 months of age. These mice were on a C57BL/6J background.
Polymerase chain reaction analysis was utilized to determine the genotype of the animals as previously described.[27] All experimental mice were male. Mice were given free access to food and water before and during the experiment.
Treatment of mice
Amyloid precursor protein transgenic mice were treated with CPO administered orally, dissolved in corn oil. During a preexperimental phase, all females were trained to drink corn oil from a syringe, to ensure proper administration of the vehicle and also to reduce stress associated with handling and exposure to a new stimulus. Following this training period, the females were paired with APP males in segregated cages, for up to a week, in order for mating to occur. The appearance of vaginal plugs was taken as evidence of successful insemination, at which time the females were separated from the males and placed in single housing in cages. Each female was randomly assigned to one of six CPO (Chem Service, West Chester, PA; 99.1% pure) dose groups: oil vehicle (at each time), 1 mg/kg bw/ day for 3-week during gestation, 1 mg/kg bw/day for 3-week during lactation, and 1 mg/kg bw/day starting at 2 months of age. For the gestation and lactation groups there were 6 females in each group; this was done to ensure that potential dose effects could not be confounded with individual variation associated with a single dam and to provide sufficient numbers of offspring for statistical analysis. There were no differences in weight or reduced locomotor activity following treatment (data not shown). Animal studies were conducted according to regulations by the National Institutes of Health and as approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina and Ralph H. Johnson VA Medical Center.
Age of mice for analysis of memory deficits and biomarkers
Memory function, amyloid plaque and brain biomarkers were evaluated after significant memory deficits developed in the AβPP mice.
Spatial memory deficit
The memory deficit in the animals was measured by the Morris water maze test as we have described previously.[27,28] Briefly, the spatial memory capability of each animal was assessed with the Morris water maze test [700-0718-4 W San Diego (SD) instruments] which evaluates memory in a swimming test. Mice were individually trained in a 1.2 m open field water maze in a pool filled with water to a depth of 30 cm and maintained at 25 °C. An escape platform (10 cm square) was placed 1 cm below the surface of the water. All animals underwent nonspatial pretraining for 4 consecutive days, which prepared the animals for the final behavioral test to determine the retention of memory to find the platform. Two days following the nonspatial pretraining, the hidden platform was placed in the center of one quadrant of the pool, the animal was released facing the pool wall in a random fashion, the time was recorded (latency period), and the distance traveled to reach the platform was measured using video recording (Smart Video Tracking System; SD Instruments).
On the day after the last training session, the platform was removed, and a spatial probe test conducted. Each mouse was allowed to search for the platform for 60 s (memory retention) and the percent time spent in quadrant where the platform was located [northeast (NE) quadrant] and in the outer annular area were determined.
Brain amyloid plaque
Amyloid plaque load was assessed in brain sections (10 from each mouse) as we have described previously, achieved by immunohistochemical staining for Aβ (Aβ antibody 10D5, Elan pharmaceuticals).[27,28] Brain tissues were fixed in 4% paraformaldehyde and then in 4% parformaldehyde and 30% sucrose for 24 h and each at 4 °C. Tissues were washed in buffered saline and transferred to an optimum cutting temperature medium. Cryosections were cut and blocked with normal serum, incubated with anti-Aβ and stained with diaminobenzoic acid (vector ABC Elite kit, vector laboratories). Bright field light microscopy imaged brain areas from which stained amyloid areas were quantitated using image analysis (NIH Image software, NIH, Washington, DC).
Brain amyloid β analysis
Brain Aβ analysis was conducted as previously described for transgenic AD mice.[27,28] Briefly, animals were sacrificed and brain extracts were homogenized (1:3 weight/volume of buffer) in buffer of 5 mol/L guanidine HCl in 50 mmol/L Tris-HCl, pH 7.6, 150 mmol/L NaCl, plus protease inhibitors (Sigma). Homogenates were diluted to 0.5 mol/L guanidine and centrifuged (200,000 g for 20 min), and supernatant and pellet fractions were collected. The pellet from the brain extract procedure was sonicated in 6 mol/L guanidine and centrifuged at 200,000 g for 20 min at 4 °C, and the supernatant was diluted to 0.5 mol/L guanidine. The two supernatants were combined, and Aβ (40) and Aβ (42) (Aβ1-40 and Aβ1-42, respectively) were determined using enzyme-linked immunosorbent assays (ELISAs) kits specific for each peptide (IBL, JP27718 and JP27711). ELISAs measured Aβ peptides by methods previously described.[29-33] Protein content was determined by the Bradford method.
C-terminal fragment-β and soluble amyloid precursor protein α analysis
C-terminal fragment-β (CTF-β) is generated from APP by β-secretase in the amyloidogenic pathway, and soluble APP (soluble amyloid precursor protein) is generated from APP by α-secretase in the nonamyloidogenic pathway. Western blots measured CTF-β and sAPPα brains of transgenic mice, using the same amount of protein per gel lane, performed as previously described.[27,28] CTF-β was determined in the pellet fraction from the brain extract (antibody 8717, sigma) and sAPPα was assessed in the supernatant fraction from the brain extract (antibody 6E10, signet laboratories). Relative amounts of CTF-β and sAPPα were measured by densitometry and results were expressed as a percentage of the mean levels of CTF-β and sAPPα of the control groups (without protease gene knockouts). Control β-actin western blots (antiβ-actin from Cell Signaling Technology) was conducted to monitor equal loading of the same amounts of samples (20 μg protein) in each gel lane.
Immunohistochemistry staining
Cryosections of the right brain hemispheres were washed 3 times (5 min/wash) with Tris-buffered saline (TBS) (pH 7.4) buffer, followed by washing 1 time with 0.1% Triton X-100-TBS buffer for 5 min. Sections were then incubated in 3% H2O2 and TBS buffer for 30 min at room temperature to eliminate endogenous peroxidase activity. After 1 h of blocking with 5.0% serum (horse or goat), the sections were incubated overnight with primary antibodies. Primary antibody and dilutions were: glial fibrillary acidic protein (GFAP)-positive astrocytes (1:200 dilution, 2E1; BD Biosciences, San Jose, CA). The next day, sections were washed 3 times (5 min/wash) with 0.1% Triton X-100 and TBS buffer to remove excess primary antibody. Thereafter, primary antibodies were detected using horseradish peroxidase (HRP)-conjugated mouse immunoglobulin G Vectastain ABC kit and DAB/substrate reagents (vector laboratories, Burlingame, CA) according to the manufacturer’s instructions.
Enzyme-linked immunosorbent assays for inflammatory markers
Brain hemispheres were weighted and homogenized with 4 volumes of phosphate buffered saline buffer (125 mg/mL) containing complete protease inhibitor cocktail (Sigma-Aldrich, Saint Louis, MO, USA). The supernatant was then collected, and total protein was determined by the BCA method (Pierce Biotechnology, Rockford, IL, USA). Tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6 levels were measured with a mouse TNF-α, IL-1β and IL-6 ELISA kits (R and D Systems, Minneapolis, MN, USA).
Protein carbonyl content
Protein carbonyl content was determined by the OxiSelect Protein Carbonyl Kit (Cell Biolabs, Inc.) and ELISA. Briefly, bovine serum albumin (BSA) standards or protein samples (10 μg/mL) were adsorbed onto a 96-well plate for 2 h at 37 °C. The protein carbonyls present in the sample or standard were derivatized to dinitrophenyl (DNP) hydrazone and probed with an anti-DNP antibody, followed by an HRP conjugated secondary antibody. The protein carbonyl content in an unknown sample was determined by comparing with a standard curve that was prepared from predetermined reduced and oxidized BSA standards.
Statistical evaluation
Experiments consisted of 10 mice in each group. Each biochemical analysis consisted of two or three replicates. Statistical analyzes and data display were conducted utilizing computer software designed for scientific data analysis (Prism 4 GraphPad, Prism, La Jolla, CA). Quantitative data are displayed as the mean and standard error of the mean. Differences between groups were determined by ANOVA analysis and Dunnett’s multiple comparison tests used to determine differences between transgenic control mice and treated animals.
Results
Experimental protocol
To investigate the influence of environmental toxins on AD, we used the APP transgenic mice expressing the mutant form of human presenilin-1 (DeltaE9) and the mutant chimeric mouse/human APP695 residue form (51). The mouse prion protein promoter directs the expression of both transgenes. APP mice start developing amyloid plaques around 3-4 months of age. This will allowed us to study the process in a relatively short period and to allow for the generation of sufficient animals for the studies. APP mice were exposed to either 1 mg/kg CPO via ingestion by suspending the toxin in corn oil (CPO) or corn oil alone. For these studies, three experimental paradigms were employed [Figure 1]. (1) Pregnant APP female mice were fed CPO daily for 3-week from the beginning of gestation; (2) female APP mice were fed CPO daily for 3-week from the beginning of lactation; and (3) weaned APP offspring were fed CPO daily for up to 6 months of age. CPO has been shown to cross the placental and is secreted into the milk following treatment of rodents.[34,35]
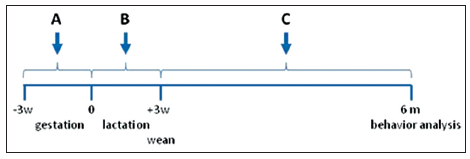
Figure 1. Experimental protocol for treatment of mice with chlorpyrifos oxon. This figure illustrates the protocol for the treatment of transgenic Alzheimer's disease mouse model utilized in this study. (a) Treatment of pregnant female amyloid precursor protein (APP) mice for 3-week during gestation; (b) treatment of female APP mice after birth and for 3-week during lactation; (c) treatment of APP mice following weaning until termination of the study at 6 months. All animals were assessed behaviorally prior to sacrifice. -3w, indicates time of plug following when male and female APP mice were placed together; 0, indicates time at birth; +3w, indicates time of weaning of APP mice
Administration of chlorpyrifos oxon exacerbates memory deficits in the amyloid precursor protein mice
The effect of the CPO (1 mg/kg) on the various paradigms was assessed for nonspatial pretraining for four consecutive days to learn the location of the hidden platform. Analyzes by the Morris water maze test on each day of the training period showed that the mice do learn, indicated by the reduced latency time for the mice to reach the hidden platform during the training period [Figure 2]. By the 4th day of training, the control APP in all groups showed the shortest latency period, representing enhanced learning, compared to the CPO APP mice. Mice administered CPO under each paradigm showed a worsening of the latency time suggesting that the treatment with CPO perturbs the learning process.
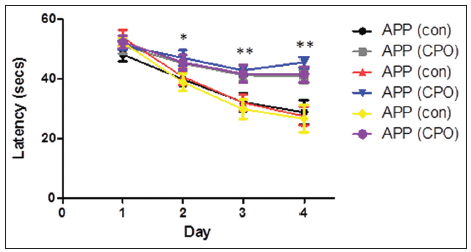
Figure 2. Treatment of amyloid precursor protein (APP) mice with chlorpyrifos oxon (CPO) results in diminished memory acquisition. APP mice (control, cornoil alone) and APP mice treated with CPO (CPO) from the different groups at 6 months of age were trained in the Morris water maze test on each of 4 consecutive days to learn the location of a submerged, invisible platform in a pool of water. The time that it took the mice to swim to the platform was recorded each day, measured as the latency period (in seconds, s), with shorter latency times indicating better memory acquisition. Latency (s) is shown as mean ± standard error of the mean (statistical significance, *P < 0.05, **P < 0.01, compared to APP control group; n = 12 per group)
Two days following training, mice were subjected to the final behavioral Morris water maze test to determine the memory deficits. CPO-treated mice exhibited substantial worsening of memory deficits, assessed by the latency period and distance traveled, which is the time and distance, respectively, that it took the animal to swim to the submerged platform [Figure 3]. The shorter time and distance traveled indicates better memory. The CPO treatment resulted in a 160% and 167% (gestation), 141% and 152% (lactation) and 140% and 142% (weanling) increase in the latency period and distance traveled, respectively [Figure 3a and b]. The 16 s latency period, and the 72 cm distance are the time and distance for wild-type mice (nontransgenic, same strain as APP mice) [Figure 3a and b, dotted lines].
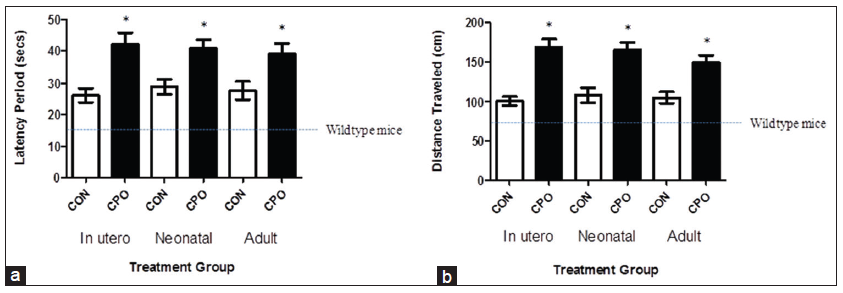
Figure 3. Treatment of amyloid precursor protein (APP) mice with chlorpyrifos oxon CPO results in exacerbated memory deficits. Memory deficits of APP mice were assessed 2 days after completion of the training in the Morris water maze test by measuring the latency period (a) and distance traveled (b) for animals to swim to the submerged, invisible platform. The shorter latency periods and shorter distances traveled indicate improved memory. APP mice (control, corn oil alone) compared to APP mice treated with CPO had shorter mean latency periods. Memory function of wild-type mice of the same strain and age is shown by the dotted line, as reported previously.[7] Values are expressed as mean ± standard error of the mean, and n = 12 per group. *Statistically significant (P < 0.05)
Administration of CPO resulted in substantial memory loss in the APP mice as illustrated by the reduced percent time spent in the NE quadrant (from which the submerged platform was removed), and the increased percent time spent in the outer annulus, compared to control APP mice [Figure 4]. The CPO treatment resulted in a 48% (gestation), 46% (lactation) and 35% (weanling) decrease in the percent time spent in the NE quadrant and a 215% (gestation), 213% (lactation) and 233% (weanling) increase in the percent time spent in the annulus. The APP mice (control) and APP CPO mice did not have a different swimming speed (data not shown). Thus, by the four parameters measured in the Morris water maze test, the CPO administration exacerbated the memory deficits that develop in the transgenic APP mice.
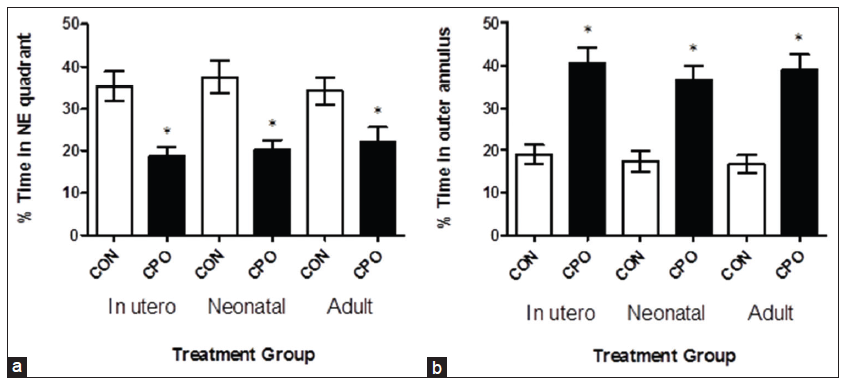
Figure 4. Treatment of amyloid precursor protein (APP) mice with chlorpyrifos oxon (CPO) results in diminished memory retention. The day after the last training session, the submerged platform was removed and the mice were allowed to swim in the pool for 60 s. The percent time each animal swam in the quadrant from which the platform had been removed (northeast [NE] quadrant) (a) and the percent time an animal swam in the annulus of the pool were recorded (b). Greater memory retention is reflected in a higher percent time in the northeast quadrant and lower percent time in the annulus. APP mice (control, corn oil alone) compared to APP mice treated with CPO had percent times in the quadrant of shorter duration. Values are expressed as the mean ± standard error of the mean, and n = 12 per group. *Statistically significant with P < 0.05
Administration of chlorpyrifos oxon to amyloid precursor protein mice exacerbates brain amyloid plaque load
Aβ immunohistochemistry of brain sections showed that the treatment with CPO increased brain amyloid plaques in the APP mice [Figure 5a and b]. Administration of CPO under all paradigms resulted in an increase in amyloid plaque load in the APP mice [Figure 5c]. Quantitative image analysis of the Aβ immunohistochemistry showed that the CPO resulted in a significant 214% (gestation), 234% (lactation) and 215% (weanling) increase in brain amyloid plaque load relative to control APP animals [Figure 5c].
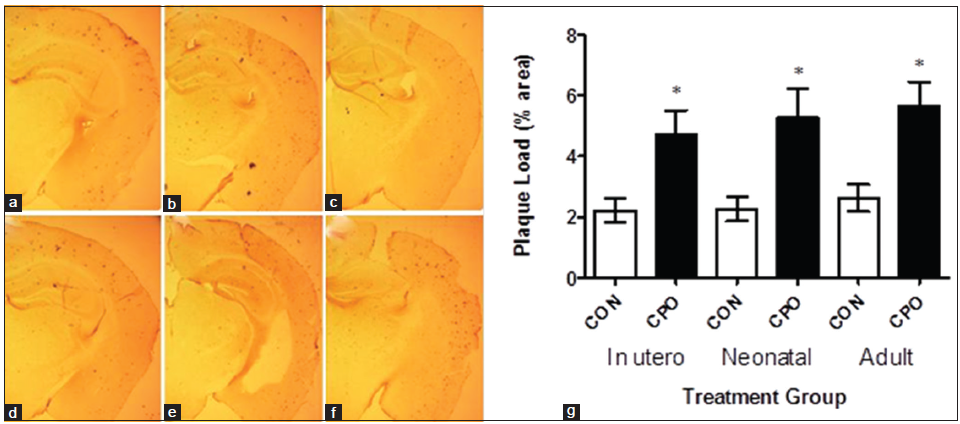
Figure 5. Treatment of amyloid precursor protein (APP) mice with chlorpyrifos oxon (CPO) results in increased brain amyloid plaque load. Amyloid plaque load was determined by immunohistochemistry and image analysis of brain sections from APP mice (control, corn oil alone) and CPO treated APP mice as shown in (a-c and d-f), respectively (representative images). Arrows indicate amyloid plaque deposits. (g) Quantitation showed that control APP mice had mean percent amyloid plaque loads lower than that of the CPO treated mice (n = 12 per group, values are expressed as mean ± standard error of the mean, *Statistically significant with P < 0.05)
Administration of chlorpyrifos oxon alters brain biomarkers in a manner characteristic of inflammation and increase in amyloid precursor protein processing by β-secretase
Amyloid precursor protein-derived Aβ peptides and APP-derived cleavage products resulting from amyloidogenic and nonamyloidogenic processing of APP were evaluated in the control and CPO treated mice. Amyloidogenic processing of APP by β-secretase produces the CTF-β fragment and Aβ peptides, and nonamyloidogenic processing of APP by α-secretase results in the sAPPα fragment.[27,31] Chlorpyrifos oxon administration to the transgenic APP mice at all-time points increased both brain Aβ1-40 and Aβ1-42 compared with control APP mice [Figure 6a and b]. The CPO treatment caused an increase in brain CTF-β levels relative to controls [Figure 6c and d]. The CPO treatment reduced sAPPα levels relative to controls [Figure 6e and f].Since CTF-β is a β-secretase cleavage product, an increase in CTF-β resulting from CPO treatment in the APP mice suggests that β-secretase activity is increased following administration of CPO. Increased production of CTF-β from APP in CPO-treated mice is likely to result in decreased APP available for α-secretase leading to a decrease in sAPPα [Table 1].
Aβ-related biomarker levels in the brains of APP mice
| In utero (nmol/L) | Neonatal (nmol/L) | Adult (nmol/L) | ||||
| Aβ1-40 | 81.2 ± 14.9 | 132 ± 16.3 | 85.3 ± 15.3 | 152 ± 30.4 | 68.2 ± 11.6 | 135 ± 26.1 |
| Aβ1-42 | 36.0 ± 6.34 | 65.2 ± 13.8 | 38.5 ± 8.38 | 76.6 ± 13.5 | 34.4 ± 8.54 | 69.8 ± 14.2 |
| In utero (% of control) | Neonatal (% of control) | Adult (% of control) | ||||
| sAPPα | 98.6 ± 13.6 | 58.4 ± 11.4 | 94.3 ± 11.0 | 63.4 ± 17.0 | 108 ± 11.4 | 46.2 ± 9.59 |
| CTFβ | 96.6 ± 10.9 | 145 ± 26.0 | 115 ± 20.9 | 165 ± 28.5 | 102 ± 16.2 | 157 ± 25.3 |
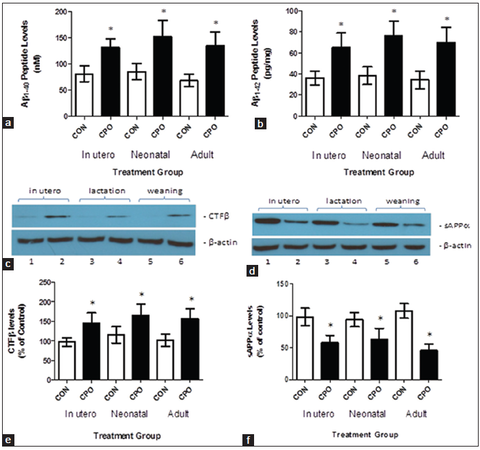
Figure 6. Treatment of amyloid precursor protein (APP) mice with chlorpyrifos oxon (CPO) results in changes in amyloid β (Aβ)-related biomarkers. Brain Aβ (40) and Aβ (42) (A-β1-40 and Aβ1-42, respectively) levels were determined by enzyme-linked immunosorbent assay. The APP mice (control, corn oil alone) compared to CPO-treated APP mice (CPO) had lower mean brain Aβ (40) levels (a) and mean Aβ (42) levels (b). Brain APP-derived CTF-β, generated by β-secretase, was assessed by western blot analysis (c). Relative quantitation by densitometry showed that the APP mice (control, corn oil alone [CON]) compared with CPO treated APP mice (CPO) had lower mean brain CTF-β levels (d). APP-derived sAPPβ was evaluated by western blot analysis (e). Quantitation by densitometry showed that the APP mice (control, CON) compared to CPO-treated APP mice (CPO) had higher mean brain sAPPβ levels (f) (n = 12 per group, values are expressed as mean ± standard error of the mean, *Statistically significant with P < 0.05)
Administration of chlorpyrifos oxon enhances inflammation and oxidative stress in the brain of amyloid precursor protein mice
In order to help determine the role of CPO on AD, we determined its impact on inflammatory markers in the brain following treatment. Brain samples from the control APP and CPO treated mice were examined for the cytokines tumor necrosis factor-α (TNF-α), interleukin 1β (IL-1β) and IL-6 levels at the termination of the study [Figure 7a-c]. All the treatment groups showed a significant elevation in the cytokine levels compared to the control APP mice [Table 2]. In addition, weaned APP mice were fed CPO (1 mg/kg) and then examined at various times up to 12 months for TNF-α levels [Figure 7d]. As seen in the figure, animals treated with CPO showed a significant increase in TNF-α levels early following treatment. However, while the TNF-α levels in the liver and blood returned to baseline within several days, the levels in the brain remained elevated for up to 12 months. Finally, brain sections from the APP mice treated with and without CPO during lactation were subject to immunohistochemistry for GFAP [Figure 7e]. As shown in figure, GFAP was significantly elevated in the CPO treated mice compared with the control animals.
Inflammatory marker levels in the brains of APP mice
| In utero (pg/mg) | Neonatal (pg/mg) | Adult (pg/mg) | ||||
|---|---|---|---|---|---|---|
| TNF-α | 100 ± 8.29 | 254 ± 35.9 | 109 ± 9.43 | 271 ± 38.2 | 112 ± 15.2 | 358 ± 43.9 |
| IL-1β | 99.7 ± 10.0 | 164 ± 16.8 | 97.3 ± 6.74 | 161 ± 19.0 | 105 ± 9.23 | 206 ± 25.4 |
| IL-6 | 95.6 ± 8.59 | 212 ± 41.8 | 91.7 ± 8.61 | 183 ± 26.6 | 107 ± 15.8 | 262 ± 39.4 |
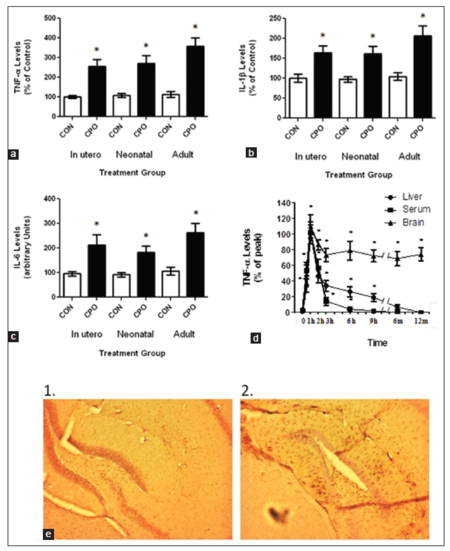
Figure 7. Treatment of amyloid precursor protein (APP) mice with chlorpyrifos oxon (CPO) results in exaggerated inflammation. Control APP mice and mice treated with CPO were evaluated for inflammatory markers. (a) Treatment of APP mice (3-week-old) with CPO showed constitutively elevated levels of tumor necrosis factor-α (TNF-α) in the brain compared to liver and serum levels. (b) Comparison of CPO treated APP mice to control APP mice showed a significant difference in the levels of TNF-α at all-time points. (c) Comparison of TNF-α (1), interleukin-1α (IL-1α) (2) and IL-6 (3) in the various treated groups. (d) APP mice were fed CPO (1 mg/kg) and then livers, blood and brains were examined at the indicated time points for TNF-α levels. Values are expressed as mean ± standard error of the mean, n = 12 per group. *Statistically significant (P < 0.05). (e) Immunostaining for glial fibrillary acidic protein (GFAP) in control (1) and CPO treated (2) animals (neonates). Brain sections were stained with anti-GFAP antibody
Furthermore, we analyzed the protein carbonyl content (a biomarker of reactive oxygen species) in the brain of the APP mice, and found that the level of oxidized proteins was significantly increased (> 50%) in mice treated with CPO than in the control APP mice [Figure 8].
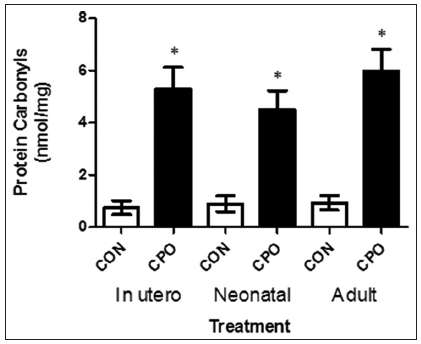
Figure 8. Protein carbonyl content in the brains of amyloid precursor protein (APP) transgenic mice treated with and without chlorpyrifos oxon. Extracts from the brains of APP transgenic mice were analyzed for the level of oxidized proteins by OxiSelect and enzyme-linked immunosorbent assay. Values are expressed as mean ± standard error of the mean, n = 12 per group. *Statistically significant (P < 0.05)
Administration of chlorpyrifos oxon decreases brain neurotrophic factors
To further characterize the changes in brain following treatment with CPO, APP mice were evaluated for neurotrophic factor levels [Figure 9]. Following treatment with CPO, mice were examined for nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 levels in the brain [Table 3]. As shown in figure, all treatments significantly reduced the neurotrophic factor levels in the brain.
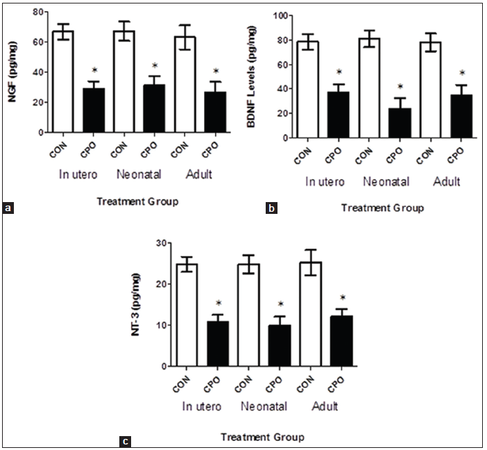
Figure 9. Treatment of amyloid precursor protein (APP) mice with chlorpyrifos oxon (CPO) results in a decrease in neurotrophic factors. Brain nerve growth factor, brain-derived neurotrophic factor and neurotrophin-3 levels (panels a, b and c, respectively) were determined by enzyme-linked immunosorbent assay in APP mice (control) and APP mice treated with CPO (CPO). Values are shown as the mean ± standard error of the mean, and n = 12. *Statistically significant (P < 0.05)
Neurotrophic factor levels in the brains of APP mice
| In utero (pg/mg) | Neonatal (pg/mg) | Adult (pg/mg) | ||||
|---|---|---|---|---|---|---|
| NGF | 66.7 ± 5.09 | 29.0 ± 4.91 | 67.1 ± 6.42 | 31.0 ± 6.12 | 63.0 ± 7.95 | 26.6 ± 6.79 |
| BDNF | 78.4 ± 6.56 | 37.4 ± 6.24 | 80.9 ± 6.80 | 23.9 ± 8.53 | 78.0 ± 7.23 | 35.1 ± 8.20 |
| NT-3 | 24.8 ± 1.82 | 10.8 ± 1.72 | 24.8 ± 2.23 | 9.96 ± 2.13 | 25.2 ± 3.09 | 12.1 ± 1.88 |
Administration of galantamine partially attenuates the effects of chlorpyrifos oxon on Aβ peptide levels and amyloid plaque load
As a potential therapeutic approach, we determined the impact of galantamine (a competitive and reversible cholinesterase inhibitor) that can protect against organophosphorus insecticides [Figure 10].
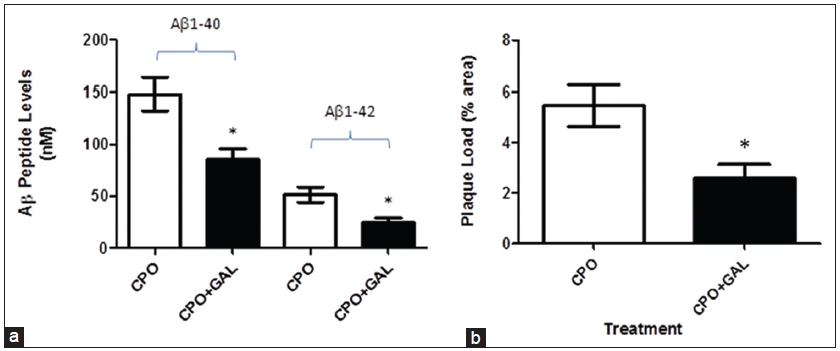
Figure 10. Treatment of amyloid precursor protein (APP) mice with galantamine partially attenuates the effects of chlorpyrifos oxon (CPO) on amyloid β (Aβ peptide levels and plaque load. Weaned APP mice (3-week of age) were treated with CPO (1 mg/kg) or CPO + galantamine (2 mg/kg/day, i.m.) and examined for Aβ peptide levels (a) and amyloid load (b) values are shown as the mean ± standard error of the mean, and n = 12. *Statistically significant (P < 0.05)
Treatment of weaned APP mice with CPO or CPO plus galantamine (2 mg/kg/day) showed a significant reduction in Aβ peptide levels (Aβ1-40-148.2 ± 15.87 nmol/L vs. 85.30 ± 10.43 nmol/L; Aβ1-42-51.6 ± 7.37 nmol/L vs. 24.7 ± 4.37 nmol/L, respectively) and plaque load (5.46 ± 0.81% area vs. 2.57 ± 0.56% area, respectively).
Discussion
The major result of this study is that CPO (organophosphate pesticide) exacerbates amyloid pathology and enhances memory deficits in the APP transgenic mouse model. This is one of the first studies to demonstrate that pesticides result in increased AD pathology. Moreover, mice treated in utero, during neonatal development or during adulthood all developed AD-like pathogenesis to a greater extent than control animals. These significant findings suggest that environmental factors may influence AD development and progression and that developing preventatives or therapeutics to attenuate the effects of the pesticides may limit the extent of AD and improve memory deficits of AD.
The aging United States population is leading to a growing number of individuals with neurodegenerative disorders.[36] Since only a small proportion of the individuals with AD have a genetic predisposition to the disease and because the pathogenesis of the disease remains to be elucidated, we need to consider alternative hypotheses to determine the disease process. Over the years, a number of studies have focused on the role of environmental toxins in AD but have not been able to link the two.[37-42] Previous studies have examined the role of heavy metals in the AD.[43-46] For years, we have known that heavy metals affect brain development resulting in abnormalities that persist throughout life. Several studies indicated that aluminum (Al) found in antiperspirants, antacids and occupational exposure can contribute to the development of AD.[47,48] However, just as many studies have indicated the Al has no effect.[48-50] The same can be said for lead (Pb), mercury (Hg), methylmercury (MeHg), iron (Fe), zinc (Zn), etc.[51-54] The association between solvent exposure and AD is weak and in some cases contradictory.[55,56] In addition, electromagnetic fields have a tenuous relationship to AD.[57,58] Finally, various pesticides have been linked to AD as well as other neurological disorders especially Parkinson’s disease. The effects of specific pesticides (organophosphates and carbamates) on the brain are well known and contribute to a number of pathological features. Tyas et al.[38] and Baldi et al.[40] showed that occupational exposure to defoliants/fumigants or general pesticides as statistically limited to AD. Furthermore, epidemiological studies have demonstrated that specific organochlorides (dichlorodiphenyltrichloroethane, dichlorodiphenyldichloroethylene and dieldrin) were present in the brains of both AD and Parkinson’s disease patients suggesting an etiological relationship for these chemicals.[59]
The findings of this study address the impact of environmental factors on the which exacerbates brain biochemical processes and memory deficits upon exposure in a mouse model expressing the APP transgene that is relevant to sporadic AD patients, representing more than 90% of the AD population. Environmental factors have been viewed in the field as potential targets for therapeutic intervention to prevent or attenuate pathology and memory deficits associated with AD.[44] Identification of such pathways involved in enhanced memory deficits was accomplished in this study by using the APP mouse model of AD.[28]
Treatment of mice in utero, during neonatal development or as adults resulted in an altered biomarker pattern consistent with enhanced AD-like activity. Biomarker analyses showed that pesticide exposure of the APP mice increased brain Aβ and CTF-β derived from APP by β-secretase, and reduced sAβPPα; these changes represent an altered Aβ-related pattern characteristic of augmenting the processing of APP. These data demonstrate that pesticides may enhance β-secretase activity in a transgenic mouse model expressing APP or may reduce the clearance of Aβ peptides via proteases or other mechanisms.[36,60] The biomarker data supports the hypothesis that exposure of the APP mice to CPO enhances memory deficits by altering the presence of Aβ peptides. Moreover, the data suggest that the improvement in memory deficits occurring with administration of galantamine may be due to the reversible inhibition of AChE activity.[61]
Our data shows that certain environmental toxins regulate oxidative stress and inflammatory processes triggered by the Aβ peptide. The toxins enhance Aβ production and exacerbate oxidative stress. Thus, the aim of this study was to determine the impact of environmental toxins on the pathogenesis of AD. The overall hypothesis is that environmental influence occurring during brain development and beyond result in damage to mitochondria, and reprogramming of the brain resulting in increased oxidative stress and inflammation.[62,63] This process alters expression of various genes related to the development of AD (increased APP expression, increase APP processing), which further exacerbates the disease course.[64] This susceptibility early in life exacerbates the normal process of amyloidogenesis in the aging brain, accelerating the onset of AD.[65] Several studies have suggested in human studies or demonstrated in animal studies that environmental toxins do influence neurodegeneration and neurobehavioral function.[44,65-68] Stewart et al.[67] showed that APOE genotype and previous exposure to lead can alter behavioral aspects of aged individuals. In addition, they showed that magnetic resonance imaging analysis of these individuals showed increased neurodegeneration compared to individuals not exposed to lead.[66] Finally, Zawia has shown that exposure of rats and mice to lead early in life can exacerbate APP processing and amyloid formation in the brain.[44,65,68]
In conclusion, this study shows that exposure of APP transgenic mice to pesticides as different times during development results in enhanced memory deficits and altered brain APP metabolism. In addition, we show that the CPO exacerbates inflammation, oxidative stress and suppresses neurotrophic factor expression that may contribute to the disease process. Importantly, these data validate the impact of environmental toxins on the enhancement of AD pathology and suggest that this process may contribute to the development and progression of AD in people.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature 1999;399:A23-31.
2. Checler F. Processing of the beta-amyloid precursor protein and its regulation in Alzheimer's disease. J Neurochem 1995;65:1431-44.
3. Wang R, Sweeney D, Gandy SE, Sisodia SS. The profile of soluble amyloid beta protein in cultured cell media. Detection and quantification of amyloid beta protein and variants by immunoprecipitation-mass spectrometry. J Biol Chem 1996;271:31894-902.
5. Galasko D, Hansen LA, Katzman R, Wiederholt W, Masliah E, Terry R, Hill LR, Lessin P, Thal LJ. Clinical-neuropathological correlations in Alzheimer's disease and related dementias. Arch Neurol 1994;51:888-95.
6. Selkoe DJ. Normal and abnormal biology of the beta-amyloid precursor protein. Annu Rev Neurosci 1994;17:489-517.
7. Roses AD. Apolipoprotein E affects the rate of Alzheimer disease expression: beta-amyloid burden is a secondary consequence dependent on APOE genotype and duration of disease. J Neuropathol Exp Neurol 1994;53:429-37.
8. Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 1996;2:864-70.
9. Hardy J. The Alzheimer family of diseases: many etiologies, one pathogenesis? Proc Natl Acad Sci U S A 1997;94:2095-7.
10. Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol 1998;8:447-53.
11. St. George-Hyslop PH. The molecular genetics of Alzheimer disease. In: Terry RD, Katzman R, Bick KL, editors. Alzheimer Disease. New York: Raven Press; 1994. pp. 345-52.
12. Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 1995;375:754-60.
13. Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 1995;269:973-7.
14. Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett 1992;135:235-8.
15. de la Fuente-Fernández R, Calne DB. Evidence for environmental causation of Parkinson's disease. Parkinsonism Relat Disord 2002;8:235-41.
16. Thiruchelvam M, Brockel BJ, Richfield EK, Baggs RB, Cory-Slechta DA. Potentiated and preferential effects of combined paraquat and maneb on nigrostriatal dopamine systems: environmental risk factors for Parkinson's disease? Brain Res 2000;873:225-34.
17. Thiruchelvam M, Richfield EK, Baggs RB, Tank AW, Cory-Slechta DA. The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson's disease. J Neurosci 2000;20:9207-14.
19. Osmond C, Barker DJ. Fetal, infant, and childhood growth are predictors of coronar y hear t disease, diabetes, and hypertension in adult men and women. Environ Health Perspect 2000;108 Suppl 3:545-53.
20. Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Front Behav Neurosci 2009;3:14.
21. Landrigan PJ, Sonawane B, Butler RN, Trasande L, Callan R, Droller D. Early environmental origins of neurodegenerative disease in later life. Environ Health Perspect 2005;113:1230-3.
22. Barker DJ, Lackland DT. Prenatal influences on stroke mortality in England and Wales. Stroke 2003;34:1598-602.
24. Lahiri DK, Maloney B. The "LEARn" (latent early-life associated regulation) model: an epigenetic pathway linking metabolic and cognitive disorders. J Alzheimers Dis 2012;30 Suppl 2:S15-30.
25. Lahiri DK, Maloney B, Zawia NH. The LEARn model: an epigenetic explanation for idiopathic neurobiological diseases. Mol Psychiatry 2009;14:992-1003.
26. Savonenko A, Xu GM, Melnikova T, Morton JL, Gonzales V, Wong MP, Price DL, Tang F, Markowska AL, Borchelt DR. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer's disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis 2005;18:602-17.
27. El-Amouri SS, Zhu H, Yu J, Marr R, Verma IM, Kindy MS. Neprilysin: an enzyme candidate to slow the progression of Alzheimer's disease. Am J Pathol 2008;172:1342-54.
28. Hook G, Hook V, Kindy M. The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer's disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J Alzheimers Dis 2011;26:387-408.
29. Hook V, Kindy M, Hook G. Cysteine protease inhibitors effectively reduce in vivo levels of brain beta-amyloid related to Alzheimer's disease. Biol Chem 2007;388:247-52.
30. Hook G, Hook VY, Kindy M. Cysteine protease inhibitors reduce brain beta-amyloid and beta-secretase activity in vivo and are potential Alzheimer's disease therapeutics. Biol Chem 2007;388:979-83.
31. Hook VY, Kindy M, Hook G. Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J Biol Chem 2008;283:7745-53.
32. Hook VY, Kindy M, Reinheckel T, Peters C, Hook G. Genetic cathepsin B deficiency reduces beta-amyloid in transgenic mice expressing human wild-type amyloid precursor protein. Biochem Biophys Res Commun 2009;386:284-8.
33. Kindy MS, Yu J, Zhu H, El-Amouri SS, Hook V, Hook GR. Deletion of the cathepsin B gene improves memory deficits in a transgenic ALZHeimer's disease mouse model expressing AßPP containing the wild-type ß-secretase site sequence. J Alzheimers Dis 2012;29:827-40.
34. Lassiter TL, Barone S Jr, Moser VC, Padilla S. Gestational exposure to chlorpyrifos: dose response profiles for cholinesterase and carboxylesterase activity. Toxicol Sci 1999;52:92-100.
35. Marty MS, Andrus AK, Bell MP, Passage JK, Perala AW, Brzak KA, Bartels MJ, Beck MJ, Juberg DR. Cholinesterase inhibition and toxicokinetics in immature and adult rats after acute or repeated exposures to chlorpyrifos or chlorpyrifos-oxon. Regul Toxicol Pharmacol 2012;63:209-24.
36. de Magalhães JP, Wuttke D, Wood SH, Plank M, Vora C. Genome-environment interactions that modulate aging: powerful targets for drug discovery. Pharmacol Rev 2012;64:88-101.
37. Rondeau V, Commenges D, Jacqmin-Gadda H, Dartigues JF. Relation between aluminum concentrations in drinking water and Alzheimer's disease: an 8-year follow-up study. Am J Epidemiol 2000;152:59-66.
38. Tyas SL, Manfreda J, Strain LA, Montgomery PR. Risk factors for Alzheimer's disease: a population-based, longitudinal study in Manitoba, Canada. Int J Epidemiol 2001;30:590-7.
39. Lindsay J, Laurin D, Verreault R, Hébert R, Helliwell B, Hill GB, McDowell I. Risk factors for Alzheimer's disease: a prospective analysis from the Canadian Study of Health and Aging. Am J Epidemiol 2002;156:445-53.
40. Baldi I, Lebailly P, Mohammed-Brahim B, Letenneur L, Dartigues JF, Brochard P. Neurodegenerative diseases and exposure to pesticides in the elderly. Am J Epidemiol 2003;157:409-14.
41. Kamel F, Hoppin JA. Association of pesticide exposure with neurologic dysfunction and disease. Environ Health Perspect 2004;112:950-8.
42. Kumar V, Kinsella LJ. Healthy brain aging:effect of head injury, alcohol and environmental toxins. Clin Geriatr Med 2010;26:29-44.
43. Cornett CR, Markesbery WR, Ehmann WD. Imbalances of trace elements related to oxidative damage in Alzheimer's disease brain. Neurotoxicology 1998;19:339-45.
44. Zawia NH, Basha MR. Environmental risk factors and the developmental basis for Alzheimer's disease. Rev Neurosci 2005;16:325-37.
45. Liu G, Huang W, Moir RD, Vanderburg CR, Lai B, Peng Z, Tanzi RE, Rogers JT, Huang X. Metal exposure and Alzheimer's pathogenesis. J Struct Biol 2006;155:45-51.
46. Platt B. Experimental approaches to assess metallotoxicity and ageing in models of Alzheimer's disease. J Alzheimers Dis 2006;10:203-13.
47. Martyn CN, Barker DJ, Osmond C, Harris EC, Edwardson JA, Lacey RF. Geographical relation between Alzheimer's disease and aluminum in drinking water. Lancet 1989;1:59-62.
48. Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB. The association between aluminum-containing products and Alzheimer's disease. J Clin Epidemiol 1990;43:35-44.
49. Broe GA, Henderson AS, Creasey H, McCusker E, Korten AE, Jorm AF, Longley W, Anthony JC. A case-control study of Alzheimer's disease in Australia. Neurology 1990;40:1698-707.
50. Foster HD. How aluminum causes Alzheimer's disease: the implications for prevention and treatment of Foster's multiple antagonist hypothesis. J Orthomol Med 2000;15:21-51.
51. Lovell MA, Rober tson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci 1998;158:47-52.
52. Molina JA, Jiménez-Jiménez FJ, Aguilar MV, Meseguer I, Mateos-Vega CJ, González-Mu-oz MJ, de Bustos F, Porta J, Ortí-Pareja M, Zurdo M, Barrios E, Martínez-Para MC. Cerebrospinal fluid levels of transition metals in patients with Alzheimer's disease. J Neural Transm 1998;105:479-88.
53. Kowalik-Jankowska T, Ruta-Dolejsz M, Wisniewska K, Lankiewicz L, Kozlowski H. Possible involvement of copper (II) in Alzheimer disease. Environ Health Perspect 2002;110 Suppl 5:869-70.
54. Huang X, Moir RD, Tanzi RE, Bush AI, Rogers JT. Redox-active metals, oxidative stress, and Alzheimer's disease pathology. Ann N Y Acad Sci 2004;1012:153-63.
55. Graves AB, Rosner D, Echeverria D, Mortimer JA, Larson EB. Occupational exposures to solvents and aluminium and estimated risk of Alzheimer's disease. Occup Environ Med 1998;55:627-33.
56. Gun RT, Korten AE, Jorm AF, Henderson AS, Broe GA, Creasey H, McCusker E, Mylvaganam A. Occupational risk factors for Alzheimer disease: a case-control study. Alzheimer Dis Assoc Disord 1997;11:21-7.
57. Sobel E, Dunn M, Davanipour Z, Qian Z, Chui HC. Elevated risk of Alzheimer's disease among workers with likely electromagnetic field exposure. Neurology 1996;47:1477-81.
58. Graves AB, Rosner D, Echeverria D, Yost M, Larson EB. Occupational exposure to electromagnetic fields and Alzheimer disease. Alzheimer Dis Assoc Disord 1999;13:165-70.
59. Fleming L, Mann JB, Bean J, Briggle T, Sanchez-Ramos JR. Parkinson's disease and brain levels of organochlorine pesticides. Ann Neurol 1994;36:100-3.
60. El-Amouri SS, Zhu H, Yu J, Gage FH, Verma IM, Kindy MS. Neprilysin protects neurons against Abeta peptide toxicity. Brain Res 2007;1152:191-200.
61. Albuquerque EX, Pereira EF, Aracava Y, Fawcett WP, Oliveira M, Randall WR, Hamilton TA, Kan RK, Romano JA Jr, Adler M. Effective countermeasure against poisoning by organophosphorus insecticides and nerve agents. Proc Natl Acad Sci U S A 2006;103:13220-5.
62. Lahiri DK, Maloney B. The "LEARn" (Latent Early-life Associated Regulation) model integrates environmental risk factors and the developmental basis of Alzheimer's disease, and proposes remedial steps. Exp Gerontol 2010;45:291-6.
63. Fan LW, Tien LT, Lin RC, Simpson KL, Rhodes PG, Cai Z. Neonatal exposure to lipopolysaccharide enhances vulnerability of nigrostriatal dopaminergic neurons to rotenone neurotoxicity in later life. Neurobiol Dis 2011;44:304-16.
64. Zawia NH, Lahiri DK, Cardozo-Pelaez F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic Biol Med 2009;46:1241-9.
65. Wu J, Basha MR, Zawia NH. The environment, epigenetics and amyloidogenesis. J Mol Neurosci 2008;34:1-7.
66. Stewart WF, Schwartz BS, Davatzikos C, Shen D, Liu D, Wu X, Todd AC, Shi W, Bassett S, Youssem D. Past adult lead exposure is linked to neurodegeneration measured by brain MRI. Neurology 2006;66:1476-84.
67. Stewart WF, Schwartz BS, Simon D, Kelsey K, Todd AC. ApoE genotype, past adult lead exposure, and neurobehavioral function. Environ Health Perspect 2002;110:501-5.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Yu J, Zhu H, Bhat A, El-Sayed H, Gudz T, Gattoni-Celli S, Kindy MS. Influence of chlorpyrifos oxon on the development and progression of Alzheimer's disease in amyloid precursor protein transgenic mice. Neurosciences 2015;2:31-42. http://dx.doi.org/10.4103/2347-8659.149421
AMA Style
Yu J, Zhu H, Bhat A, El-Sayed H, Gudz T, Gattoni-Celli S, Kindy MS. Influence of chlorpyrifos oxon on the development and progression of Alzheimer's disease in amyloid precursor protein transgenic mice. Neuroimmunology and Neuroinflammation. 2015; 2: 31-42. http://dx.doi.org/10.4103/2347-8659.149421
Chicago/Turabian Style
Yu, Jin, Hong Zhu, Aruna Bhat, Hanaa El-Sayed, Tatyana Gudz, Sebastiano Gattoni-Celli, Mark S. Kindy. 2015. "Influence of chlorpyrifos oxon on the development and progression of Alzheimer's disease in amyloid precursor protein transgenic mice" Neuroimmunology and Neuroinflammation. 2: 31-42. http://dx.doi.org/10.4103/2347-8659.149421
ACS Style
Yu, J.; Zhu H.; Bhat A.; El-Sayed H.; Gudz T.; Gattoni-Celli S.; Kindy MS. Influence of chlorpyrifos oxon on the development and progression of Alzheimer's disease in amyloid precursor protein transgenic mice. Neurosciences. 2015, 2, 31-42. http://dx.doi.org/10.4103/2347-8659.149421
About This Article
Copyright

Data & Comments
Data


 Cite This Article 2 clicks
Cite This Article 2 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.