
Neuroinflammation and neurological alterations in chronic liver diseases


Abstract
Several million people with chronic liver diseases (cirrhosis, hepatitis) show neurological alterations, named hepatic encephalopathy (HE) with cognitive and motor alterations that impair quality of life and reduces life span. Inflammation acts synergistically with hyperammonemia to induce cognitive and motor alterations in patients with chronic liver disease and minimal hepatic encephalopathy (MHE). Previous studies in animal models have suggested that neuroinflammation is a major player in HE. This would also be the case in patients with liver cirrhosis or hepatitis C with HE. Rats with MHE show microglial activation and neuroinflammation that is associated with cognitive impairment and hypokinesia. The anti-inflammatory drug ibuprofen reduces microglial activation and neuroinflammation and restores cognitive and motor functions in rats with MHE. Chronic hyperammonemia per se induces neuroinflammation. Both peripheral inflammation and hyperammonemia would contribute to neuroinflammation in chronic liver failure. Therefore, neuroinflammation may be a key therapeutic target to improve the cognitive and motor alterations in MHE and overt HE. Identifying new targets to reduce neuroinflammation in MHE without inducing secondary effects would serve to develop new therapeutic tools to reverse the cognitive and motor alterations in patients with HE associated with chronic liver diseases
Keywords
Introduction
Current evidence suggests that chronic inflammatory diseases lead to neuroinflammation. Increased neuroinflamamtion can result in neurological impairment with deficits in cognition and motor function. For example, patients with diabetes, rheumatoid arthritis, obesity or chronic kidney disease can develop neurological deficits.[1-9] Inflammation and neuroinflammation are major contributing factors to cognitive and motor deficits in situations such as postoperative cognitive dysfunction, ageing and in some mental (e.g. schizophrenia) and neurodegenerative (e.g. Alzheimer’s disease) diseases.[10-16]
Patients suffering from chronic liver diseases (mainly cirrhosisand/orhepatitis) also show chronic inflammation which can led to hepatic encephalopathy (HE): any alteration in cerebral function which is a consequence of previous liver failure.[17] There are two main types of liver diseases which induce HE: acute liver failure and chronic liver diseases. The effects and mechanisms underlying the cerebral alterations in acute and chronic liver failure are completely different.[17] This review will focus only on the mechanisms involved in HE in chronic liver diseases, mainly in liver cirrhosis.
Chronic liver diseases affect more than 5 million people in USA and a similar number in the European Union.[18] Patients with the chronic liver disease do not show neurological alterations at the beginning of the disease. However, with the progression of liver failure, most of these patients will suffer from some grade of HE. There are two main forms of HE in chronic liver disease: (1) minimal hepatic encephalopathy (MHE), in which the symptoms are not evident but can be unveiled using psychometric tests; (2) clinical or overt HE, in which the symptoms are evident. Once the symptoms are evident, clinical HE is graded in four stages according to the West Haven criteria.[19]
Approximately, 40-50% of cirrhotic patients present with MHE with mild cognitive impairment, attention deficits, psychomotor slowing, reduced mental processing speed and bimanual and visuomotor incoordination.[20-22] This incidence means that more than two million people suffer from MHE in USA, in the European Union and even more in the rest of the world.
Minimal hepatic encephalopathy has important consequences on daily life of patients with chronic liver diseases. Attention, mental processing speed, visuomotor and bimanual coordination are necessary for many daily tasks such as driving cars or dressing. Patients with MHE have impaired driving ability, which is associated with peripheral inflammation, with increased levels of interleukin-6 (IL-6) and IL-18.[23-25] They also have increased risk of traffic, home and work accidents and more falls, fractures, and hospitalizations, which pose a high economic burden to health systems. In addition, MHE predisposes patients to clinical HE with more serious neurological impairment that can lead to coma and death and reduces life span. Early diagnosis and treatment of MHE would improve the quality of life and survival of patients and economic burden.[26-29]
Inflammation and neuroinflammation in the neurological alterations in patients with chronic liver disease and MHE
Inflammation is a main contributor to the changes in cognitive and motor functions found in MHE patients suffering from chronic liver disease. For example, the serum levels of the pro-inflammatory cytokines IL-6 and IL-18 are increased in cirrhotic patients with MHE compared with cirrhotics without MHE. Moreover, the levels of IL-6 and IL-18 correlate with the grade of MHE evaluated using psychometric tests.[30]
Hyperammonemia plays a synergistic role with inflammation in the induction of the neurologic impairment [Figure 1]. Hyperammonemia developed in cirrhotic patients by ingestion of an amino acid mixture induces neuropsychological impairment during inflammation, but not after its resolution.[31] The contribution of inflammation and hyperammonemia to the neurological impairment in different pathological situations with different grades of inflammation and hyperammonemia was assessed by Felipo et al.,[32] who showed that the joint presence of inflammation and hyperammonemia, is enough to induce mild cognitive impairment, even in the absence of liver failure. The studies by Shawcross et al.[31] and Felipo et al.[32] support the idea that, as occurs in other chronic diseases, in chronic liver diseases, peripheral inflammation is involved in the induction of the cognitive and motor alterations associated to MHE and to overt HE.
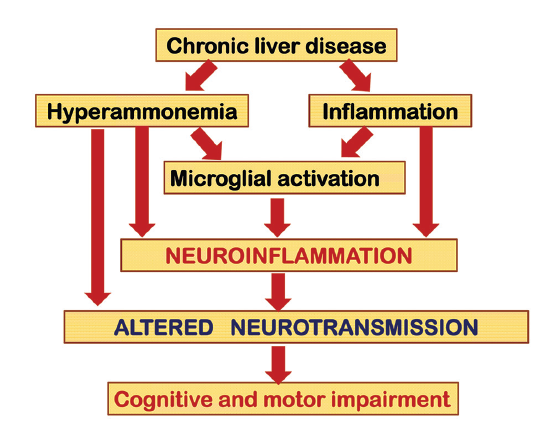
Figure 1. Hyperammonemia and inflammation act synergistically to induce neurological alterations in chronic liver disease. Chronic liver failure induces both hyperammonemia and peripheral inflammation, leading to microglial activation and neuroinflammation, which alters neurotransmission, leading to cognitive and motor impairment
Peripheral inflammation may lead to neuroinflammation by different mechanisms. The presence of neuroinflammation in patients with chronic liver diseases has not been thoroughly studied. One of the main methods to assess neuroinflammation is to assess by positron emission tomography (PET); the binding of ligands to the translocator protein (18 kDa) (TSPO), previously known as peripheral type benzodiazepine receptor.[33] Cagnin et al.[34] showed using PET techniques that cirrhotic patients with HE show increased binding of [11C](R)-PK11195 to TSPO in brain.[34] Moreover, the patient with the most severe cognitive impairment had the highest increases in regional [11C](R)-PK11195 binding. The authors suggested that an altered glial cell state might be causally related to impaired brain functioning in HE. These data suggest the presence of neuroinflammation in patients with HE, correlating with the grade of cognitive impairment.[34]
In support for the role of microglia, Dennis et al.[35] performed immunohistochemistry studies and cytokine assays in fixed postmortem brain tissue from chronic alcoholics with cirrhosis and HE, alcoholics without HE and controls. About half of the patients with HE showed what they called “proliferative HE (pHE)” and the other half did not.[35] The microglia in pHEs displayed an activated morphology with hypertrophied cell bodies and short, thickened processes. These patients also showed increased levels of IL-6. In contrast, the microglia in white matter regions of the non-proliferative HE cases were less activated and appeared dystrophic. The authors suggest that microglial proliferation may form part of an early neuroprotective response in HE that ultimately fails to halt the course of the disease because underlying etiological factors such as high cerebral ammonia and systemic inflammation remain.[35] In support, Zemtsova et al.[36] also analyzed markers of neuroinflammation in postmortem brain tissue from patients with cirrhosis that presented with and without HE and noncirrhotic controls. They found an up-regulation of the microglial activation marker ionized calcium-binding adaptor molecule-1 (Iba-1) in the cerebral cortex from patients with cirrhosis who have HE but not from patients with cirrhosis who do not have HE. In this study, mRNA and protein expression of iNOS and cyclooxygenase (COX-2) or mRNA expression of pro-inflammatory cytokines and chemokine monocyte chemoattractive protein-1 was analyzed in cerebral cortex. They were not different in patients with liver cirrhosis and HE than in patients with cirrhosis who did not have HE or control patients without cirrhosis. This report shows increased microglial activation in patients with HE but did not find increased levels of pro-inflammatory markers.
Neuroinflammation has been also reported in patients with chronic hepatitis C (HC). However, in these patients a main contributor to neuroinflammation would be the presence of the virus into the brain. Postmortem studies of HC virus indicate that the viral replication might occur in the brain and microglia may be the locus for infection and subsequent neuroinflammatory activity.[37,38] Bokemeyer et al.[39] assessed the possible contribution of neuroinflammation to cognitive dysfunction in patients with HC who have mild liver disease. They performed an extensive neuropsychological examination and a magnetic resonance spectroscopy study in a group of patients with HC showing only mild liver disease but differing degrees of neuropsychiatric alterations. Choline, creatine and N-acetyl-aspartate and N-acetyl-aspartyl-glutamate concentrations in the basal ganglia were increased in the patients compared to controls. Fatigue correlated negatively with the metabolic changes.[39] As the increase of choline, creatine and myo-inositol are usually interpreted to indicate glial activation and macrophage infiltration in chronic inflammation they conclude that HC virus infection may induce neuroinflammation and brain dysfunction. The negative correlation with fatigue suggests a cerebral compensatory process after infection. Grover et al.[38] used two independent in vivo imaging techniques to assess the presence of neuroinflammation in patients with mild chronic HC and control subjects. Using magnetic resonance spectroscopy, they found that basal ganglia myo-inositol/creatine and choline/ creatine ratios were significantly elevated in patients with chronic HC compared to controls. Using PET with a ligand for microglial/brain macrophage activation, 11C-(R)-PK11195, Grover et al.[38] found evidence of microglial activation, which positively correlated with HC viremia and altered cerebral metabolism. This suggests that the HC virus induces neuroinflammation, which would contribute to the cognitive alterations in patients with HC. Although in patients with HC neuroinflammation seems to be caused mainly by the presence of the virus into the brain,[37,38] the studies from Cagnin et al.,[34] Dennis et al.[35] and Zemtsova et al.[36] suggest that neuroinflammation could also be present in patients with liver cirrhosis in the absence of virus infection.
Role of neuroinflammation in the cognitive and motor alterations in animal models of MHE
Neuroinflammation would be a main contributor to the cognitive and motor alterations in minimal and clinical HE associated to chronic liver disease. This possibility is clearly supported by studies in animal models. Cauli et al.[40] showed that rats with portacaval shunts (PCS), a main model of MHE,[41] show neuroinflammation. Following PCS, rats showed increased levels of inflammatory markers such as IL-6 levels or COX activity in the brain. PCS rats show impaired ability to learn a Y maze task due to reduced function of the glutamate-nitric oxide (NO)-cGMP pathway in cerebellum.[42] Cauli et al.[40] showed that chronic treatment with the anti-inflammatory ibuprofen reduced neuroinflammation and restored the ability to learn the Y maze task because it restores the function of the glutamate-NO-cGMP pathway in cerebellum in vivo. This report showed for the first time that neuroinflammation is a main contributor to the cognitive impairment in MHE [Figure 1].[40]
Neuroinflammation also contributes to other types of cognitive and motor alterations by altering other specific mechanisms modulating them. For example, neuroinflammation is a main contributor to hypokinesia in rats with MHE and chronic treatment with ibuprofen restored motor function.[43] The mechanisms and brain areas involved in neuroinflammation-induced hypokinesia are different from those involved in impairment of learning in the Y maze. Hypokinesia in PCS rats is due to the increased level of extracellular glutamate in substantia nigra pars reticulata (SNr). Glutamate activates metabotropic glutamate receptor 1 (mGluR1) and induces an increase extracellular gamma-aminobutyric acid (GABA) in ventromedial thalamus (VMT). This, in turn, reduces extracellular glutamate in motor cortex, which reduces motor activity. Blocking mGluR1 in SNr with an antagonist normalizes GABA in VMT, glutamate in motor cortex and motor activity.[44] A down-regulation of glutamate transporters contributes to the increased levels of extracellular glutamate in SNr.[44] Chronic treatment of rats with MHE with ibuprofen normalizes the amount of glutamate transporters, reduces extracellular glutamate in SNr and normalizes motor activity.[43] The presence of neuroinflammation and its contribution to cognitive and motor alterations have also been demonstrated in the other main animal model of MHE: rats with bile duct ligation (BDL). Rodrigo et al.[45] showed that BDL rats have microglial activation, mainly in cerebellum, as demonstrated by immunohistochemistry. This is associated with increased levels of inflammatory markers [inducible nitric oxide synthase (iNOS), IL-1β, prostaglandin E2] and with cognitive impairment and hypokinesia. Similar to the previous studies, chronic treatment with ibuprofen reduced microglial activation and inflammatory markers and restored cognitive and motor functions in the BDL rats.[45] The above reports clearly support the idea that MHE in chronic liver failure is associated with neuroinflammation that affects different cerebral processes resulting in different types of cognitive and motor alterations [Figure 1].
Chronic hyperammonemia induces activation of microglia and neuroinflammation
Concerning the mechanisms by which chronic liver failure induces neuroinflammation, it seems that two main contributors would be involved: chronic hyperammonemia and peripheral inflammation.
In addition to PCS and BDL rats, a well-recognized rodent model of MHE are rats with “pure” hyperammonemia without liver failure induced by feeding an ammonium-containing diet.[41] As described above for rat model of MHE induced by PCS, rats with chronic hyperammonemia also show reduced function of the glutamate-NO-cGMP pathway in cerebellum[46] and reduced ability to learn the Y maze task,[47] which is restored when the function of the pathway is restored, for example, by treatment with sildenafil.[42] This suggests that hyperammonemia and neuroinflammation impair learning the ability by the same mechanism. This idea led Rodrigo et al.[45] to hypothesize that hyperammonemia would induce neuroinflamamtion in the brain, which would be responsible for impairment of the glutamate-NO-cGMP pathway and of cognitive function. To assess whether chronic hyperammonemia per se induces neuroinflammation, Rodrigo et al.[45] assessed in the brains of hyperammonemic rats without liver failure activation of microglia by immunohistochemistry and the levels of inflammatory markers [iNOS, IL-1β, and prostaglandin E 2 (PGE2)]. Hyperammonemic rats show microglial activation [Figure 1], mainly in cerebellum, and increased levels of inducible iNOS, IL-1β, and PGE2 which are associated with impaired function of the glutamate-NO -cGMP pathway and with cognitive impairment and hypokinesia.
Chronic treatment with ibuprofen reduced microglial activation and inflammatory markers and restored the function of the glutamate-NO-cGMP pathway in cerebellum and cognitive and motor functions in hyperammonemic rats.[44] In summary, this indicates that: (1) chronic hyperammonemia per se induces neuroinflammation; (2) neuroinflammation mediates the effects of chronic hyperammonemia on the function of the glutamate-NO-cGMP pathway in cerebellum and on cognitive and motor functions in hyperammonemic rats. As neurological alterations are mainly due to altered glutamatergic and GABAergic neurotransmission,[48,49] this suggests that neuroinflammation-mediated alterations in neurotransmission are mainly responsible for changes in cognitive and motor function in MHE and clinical HE [Figure 1].
Hyperammonemia may also alter neurotransmission by other mechanisms. For example, Thrane et al.[50] have shown that ammonia, at high concentrations, alters astrocyte potassium buffering, increasing extracellular potassium concentration and over-activating the Na+-K+-2Cl− co-transporter isoform 1 (NKCC1) in neurons. The consequent depolarization of the neuronal GABA reversal potential selectively impairs cortical inhibitory networks. This altered GABAergic neurotransmission may contribute to the neurological alterations in hyperammonemia. The alterations in glutamatergic and GABAergic neurotransmission in hyperammonemia and HE have been recently reviewed by Cauli et al.[48] In addition to the contribution of hyperammonemia, peripheral inflammation would also contribute to induction of neuroinflammation in chronic liver disease [Figure 1].
The mechanisms by which hyperammonemia induces microglial activation and neuroinflammation have been poorly studied for the moment. Zemtsova et al.[36] studied the effects of large ammonia concentrations on rat microglia in vitro and in vivo. In cultured rat microglia, ammonia-stimulated cell migration and induced oxidative stress and an up-regulation of the microglial activation marker ionized calcium-binding adaptor molecule-1 (Iba-1). Up-regulation of Iba-1 was also found in the cerebral cortex from acutely ammonia-intoxicated rats. However, ammonia had no effect on microglial glutamate release, prostaglandin synthesis, and messenger RNA (mRNA) levels of iNOS, COX-2, and IL-1α/β, tumor necrosis factor α (TNFα), or IL-6, whereas in cultured astrocytes ammonia induced the release of glutamate, prostaglandins, and increased levels of IL-1β mRNA. Although this study was performed using ammonia levels much higher than those that can be found in chronic liver disease in vivo, these data suggest that ammonia per se may activate microglia. Additional factors would act synergistically with hyperammonemia to induce pro-inflammatory factors and neuroinflammation in chronic liver failure.
Therapeutic implications
The in vivo studies reported above in animal models show that neuroinflammation would be a key therapeutic target to improve the cognitive and motor alterations in MHE and overt HE. However, different approaches can be used to reduce neuroinflammation in chronic liver disease, but it is important to take into account the possible secondary effects of each treatment, which may depend on the type and grade of liver disease in each patient. For example, several studies show that ibuprofen ameliorates cognitive and motor function in rats with MHE.[40,43,45] However, ibuprofen is a non-steroidal anti-inflammatory drug (NSAID), and this type of anti-inflammatory is not recommended in cirrhotic patients because they may induce kidney damage and, in patients prone to portal hypertension (a common liver disease complication), the risk of gastrointestinal bleeding is intensified with NSAIDS.[51,52] Therefore, alternative therapeutic approaches are needed to reduce neuroinflammation that would present no secondary side-effects. As activation of microglia is a main contributor to neuroinflammation in animal models of HE, Agusti et al.[53] hypothesized that reducing directly microglial activation would be useful to improve cognitive and motor function in HE without inducing secondary effects in the kidney. Inhibiting the p38 MAP kinase antagonizes microglial activation[54] and inhibitors of p38 are being tested as therapeutic tools in different inflammatory and autoimmune diseases.[55] Taking this into account, Agusti et al.[53] assessed whether inhibiting p38 with SB239063 reduces neuroinflammation and improves cognitive and motor function in rats with MHE due to PCS without affecting the kidney. This group showed that treatment with SB239063 reduced microglial activation and inflammatory markers (PGE2, COX activity, iNOS, IL-1β, TNFα) in brain and completely restored learning ability, motor activity and coordination in PCS rats without inducing kidney damage. This suggests that inhibitors of p38 would improve the neurological status in HE without inducing secondary effects in the kidney. This type of inhibitors is being developed by different pharmaceutical companies to treat different chronic inflammatory diseases. It is likely that they can be developed further and following extensive pre-clinical and clinical evaluation become available to patients suffering from MHE.
Additional studies to identify and test new targets to reduce neuroinflammation in MHE without inducing secondary effects would be very useful to develop new therapeutic tools to reverse the cognitive and motor alterations in patients with MHE and clinical HE associated with chronic liver diseases.
Financial support and sponsorship
Supported by grants from Ministerio de Ciencia e Innovación (SAF2011-23051, CSD2008-00005), Consellería Educación Generalitat Valenciana (PROMETEO-2009-027, PROMETEOII/2014/033, ACOMP/2012/066, and ACOMP/2013/101).
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Diaz-Gerevini GT, Repossi G, Dain A, Tarres MC, Das UN, Eynard AR. Cognitive and motor perturbations in elderly with longstanding diabetes mellitus. Nutrition 2014;30:628-35.
2. Umemura T, Kawamura T, Umegaki H, Mashita S, Kanai A, Sakakibara T, Hotta N, Sobue G. Endothelial and inflammatory markers in relation to progression of ischaemic cerebral small-vessel disease and cognitive impairment: a 6-year longitudinal study in patients with type 2 diabetes mellitus. J Neurol Neurosurg Psychiatry 2011;82:1186-94.
3. Hamed SA, Selim ZI, Elattar AM, Elserogy YM, Ahmed EA, Mohamed HO. Assessment of biocorrelates for brain involvement in female patients with rheumatoid arthritis. Clin Rheumatol 2012;31:123-32.
4. Shin SY, Julian L, Katz P. The relationship between cognitive function and physical function in rheumatoid arthritis. J Rheumatol 2013;40:236-43.
5. Nguyen JC, Killcross AS, Jenkins TA. Obesity and cognitive decline: role of inflammation and vascular changes. Front Neurosci 2014;8:375.
6. Spyridaki EC, Simos P, Avgoustinaki PD, Der mitzaki E, Venihaki M, Bardos AN, Margioris AN. The association between obesity and fluid intelligence impairment is mediated by chronic low-grade inflammation. Br J Nutr 2014;112:1724-34.
7. Miller AA, Spencer SJ. Obesity and neuroinflammation: a pathway to cognitive impairment. Brain Behav Immun 2014;42:10-21.
8. da Matta SM, Janaina Matos M, Kummer AM, Barbosa IG, Teixeira AL, Silva AC. Cognitive alterations in chronic kidney disease: an update. J Bras Nefrol 2014;36:241-5.
9. Silverwood RJ, Richards M, Pierce M, Hardy R, Sattar N, Ferro C, Savage C, Kuh D, Nitsch D, NSHD Scientific and Data Collection Teams. Cognitive and kidney function: results from a British birth cohort reaching retirement age. PLoS One 2014;9:e86743.
10. Boyer L, Richieri R, Dassa D, Boucekine M, Fernandez J, Vaillant F, Padovani R, Auquier P, Lancon C. Association of metabolic syndrome and inflammation with neurocognition in patients with schizophrenia. Psychiatry Res 2013;210:381-6.
11. Takeda S, Sato N, Morishita R. Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: relevance to pathogenesis and therapy. Front Aging Neurosci 2014;6:171.
12. Cherniack EP. A berry thought-provoking idea: the potential role of plant polyphenols in the treatment of age-related cognitive disorders. Br J Nutr 2012;108:794-800.
13. Bilbo SD, Smith SH, Schwarz JM. A lifespan approach to neuroinflammatory and cognitive disorders: a critical role for glia. J Neuroimmune Pharmacol 2012;7:24-41.
14. Hawkins MA, Alosco ML, Spitznagel MB, Strain G, Devlin M, Cohen R, Crosby RD, Mitchell JE, Gunstad J. The association between reduced inflammation and cognitive gains after bariatricsurgery. Psychosom Med 2014;[Epub ahead of print].
15. Hovens IB, Schoemaker RG, van der Zee EA, Absalom AR, Heineman E, van Leeuwen BL. Postoperative cognitive dysfunction: involvement of neuroinflammation and neuronal functioning. Brain Behav Immun 2014;38:202-10.
16. Barrientos RM, Hein AM, Frank MG, Watkins LR, Maier SF. Intracisternal interleukin-1 receptor antagonist prevents postoperative cognitive decline and neuroinflammatory response in aged rats. J Neurosci 2012;32:14641-8.
17. Felipo V. Hepatic encephalopathy: effects of liver failure on brain function. Nat Rev Neurosci 2013;14:851-8.
18. Leevy CB, Phillips JA. Hospitalizations during the use of rifaximin versus lactulose for the treatment of hepatic encephalopathy. Dig Dis Sci 2007;52:737-41.
19. Conn HO, Leevy CM, Vlahcevic ZR, Rodgers JB, Maddrey WC, Seeff L, Levy LL. Comparison of lactulose and neomycin in the treatment of chronic portal-systemic encephalopathy. A double blind controlled trial. Gastroenterology 1977;72:573-83.
20. Weissenborn K, Giewekemeyer K, Heidenreich S, Bokemeyer M, Berding G, Ahl B. Attention, memory, and cognitive function in hepatic encephalopathy. Metab Brain Dis 2005;20:359-67.
21. Amodio P, Schiff S, Del Piccolo F, Mapelli D, Gatta A, Umilta C. Attention dysfunction in cirrhotic patients: an inquiry on the role of executive control, attention orienting and focusing. Metab Brain Dis 2005;20:115-27.
22. Felipo V, Ordono JF, Urios A, El Mlili N, Gimenez-Garzo C, Aguado C, Gonzalez-Lopez O, Giner-Duran R, Serra MA, Wassel A, Rodrigo JM, Salazar J, Montoliu C. Patients with minimal hepatic encephalopathy show impaired mismatch negativity correlating with reduced performance in attention tests. Hepatology 2012;55:530-9.
23. Bajaj JS. Minimal hepatic encephalopathy matters in daily life. World J Gastroenterol 2008;14:3609-15.
24. Wein C, Koch H, Popp B, Oehler G, Schauder P. Minimal hepatic encephalopathy impairs fitness to drive. Hepatology 2004;39:739-45.
25. Felipo V, Urios A, Valero P, Sanchez M, Serra MA, Pareja I, Rodriguez F, Gimenez-Garzo C, Sanmartin J, Montoliu C. Serum nitrotyrosine and psychometric tests as indicators of impaired fitness to drive in cirrhotic patients with minimal hepatic encephalopathy. Liver Int 2013;33:1478-89.
26. Romero-Gomez M, Boza F, Garcia-Valdecasas MS, Garcia E, Aguilar-Reina J. Subclinical hepatic encephalopathy predicts the development of overt hepatic encephalopathy. Am J Gastroenterol 2001;96:2718-23.
27. Roman E, Cordoba J, Torrens M, Torras X, Villanueva C, Vargas V, Guarner C, Soriano G. Minimal hepatic encephalopathy is associated with falls. Am J Gastroenterol 2011;106:476-82.
28. Stepanova M, Mishra A, Venkatesan C, Younossi ZM. In-hospital mor tality and economic burden associated with hepatic encephalopathy in the United States from 2005 to 2009. Clin Gastroenterol Hepatol 2012;10:1034-41.
29. Bajaj JS, Pinkerton SD, Sanyal AJ, Heuman DM. Diagnosis and treatment of minimal hepatic encephalopathy to prevent motor vehicle accidents: a cost-effectiveness analysis. Hepatology 2012;55:1164-71.
30. Montoliu C, Piedrafita B, Serra MA, del Olmo JA, Urios A, Rodrigo JM, Felipo V. IL-6 and IL-18 in blood may discriminate cirrhotic patients with and without minimal hepatic encephalopathy. J Clin Gastroenterol 2009;43:272-9.
31. Shawcross DL, Davies NA, Williams R, Jalan R. Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J Hepatol 2004;40:247-54.
32. Felipo V, Urios A, Montesinos E, Molina I, Garcia-Torres ML, Civera M, Olmo JA, Ortega J, Martinez-Valls J, Serra MA, Cassinello N, Wassel A, Jorda E, Montoliu C. Contribution of hyperammonemia and inflammatory factors to cognitive impairment in minimal hepatic encephalopathy. Metab Brain Dis 2012;27:51-8.
33. Kannan S, Balakrishnan B, Muzik O, Romero R, Chugani D. Positron emission tomography imaging of neuroinflammation. J Child Neurol 2009;24:1190-9.
34. Cagnin A, Taylor-Robinson SD, Forton DM, Banati RB. In vivo imaging of cerebral "peripheral benzodiazepine binding sites" in patients with hepatic encephalopathy. Gut 2006;55:547-53.
35. Dennis CV, Sheahan PJ, Graeber MB, Sheedy DL, Kril JJ, Sutherland GT. Microglial proliferation in the brain of chronic alcoholics with hepatic encephalopathy. Metab Brain Dis 2014;29:1027-39.
36. Zemtsova I, Gorg B, Keitel V, Bidmon HJ, Schror K, Haussinger D. Microglia activation in hepatic encephalopathy in rats and humans. Hepatology 2011;54:204-15.
37. Vivithanaporn P, Maingat F, Lin LT, Na H, Richardson CD, Agrawal B, Cohen EA, Jhamandas JH, Power C. Hepatitis C virus core protein induces neuroimmune activation and potentiates human immunodeficiency virus-1 neurotoxicity. PLoS One 2010;5:e12856.
38. Grover VP, Pavese N, Koh SB, Wylezinska M, Saxby BK, Gerhard A, Forton DM, Brooks DJ, Thomas HC, Taylor-Robinson SD. Cerebral microglial activation in patients with hepatitis C: in vivo evidence of neuroinflammation. J Viral Hepat 2012;19:e89-96.
39. Bokemeyer M, Ding XQ, Goldbecker A, Raab P, Heeren M, Arvanitis D, Tillmann HL, Lanfermann H, Weissenborn K. Evidence for neuroinflammation and neuroprotection in HCV infection-associated encephalopathy. Gut 2011;60:370-7.
40. Cauli O, Rodrigo R, Piedrafita B, Boix J, Felipo V. Inflammation and hepatic encephalopathy: ibuprofen restores learning ability in rats with portacaval shunts. Hepatology 2007;46:514-9.
41. Butterworth RF, Norenberg MD, Felipo V, Ferenci P, Albrecht J, Blei AT, Members of the ISHEN Commission on Experimental Models of HE. Experimental models of hepatic encephalopathy: ISHEN guidelines. Liver Int 2009;29:783-8.
42. Erceg S, Monfort P, Hernandez-Viadel M, Rodrigo R, Montoliu C, Felipo V. Oral administration of sildenafil restores learning ability in rats with hyperammonemia and with portacaval shunts. Hepatology 2005;41:299-306.
43. Cauli O, Rodrigo R, Piedrafita B, Llansola M, Mansouri MT, Felipo V. Neuroinflammation contributes to hypokinesia in rats with hepatic encephalopathy: ibuprofen restores its motor activity. J Neurosci Res 2009;87:1369-74.
44. Cauli O, Llansola M, Erceg S, Felipo V. Hypolocomotion in rats with chronic liver failure is due to increased glutamate and activation of metabotropic glutamate receptors in substantia nigra. J Hepatol 2006;45:654-61.
45. Rodrigo R, Cauli O, Gomez-Pinedo U, Agusti A, Hernandez-Rabaza V, Garcia-Verdugo JM, Felipo V. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology 2010;139:675-84.
46. Hermenegildo C, Montoliu C, Llansola M, Munoz MD, Gaztelu JM, Minana MD, Felipo V. Chronic hyperammonemia impairs the glutamate-nitric oxide-cyclic GMP pathway in cerebellar neurons in culture and in the rat in vivo. Eur J Neurosci 1998;10:3201-9.
47. Aguilar MA, Minarro J, Felipo V. Chronicmod erate hyperammonemia impairs active and passive avoidance behavior and conditional discrimination learning in rats. Exp Neurol 2000;161:704-13.
48. Cauli O, Rodrigo R, Llansola M, Montoliu C, Monfort P, Piedrafita B, El Mlili N, Boix J, Agusti A, Felipo V. Glutamatergic and gabaergic neurotransmission and neuronal circuits in hepatic encephalopathy. Metab Brain Dis 2009;24:69-80.
49. Monfort P, Cauli O, Montoliu C, Rodrigo R, Llansola M, Piedrafita B, El Mlili N, Boix J, Agusti A, Felipo V. Mechanisms of cognitive alterations in hyperammonemia and hepatic encephalopathy: therapeutical implications. Neurochem Int 2009;55:106-12.
50. Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, Nedergaard M. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med 2013;19:1643-8.
51. Ackerman Z, Cominelli F, Reynolds TB. Effect of misoprostol on ibuprofen-induced renal dysfunction in patients with decompensated cirrhosis: results of a double-blind placebo-controlled parallel group study. Am J Gastroenterol 2002;97:2033-9.
52. Hawkey CJ. Non -steroidal anti-inflammator y drug gastropathy: causes and treatment. Scand J Gastroenterol Suppl 1996;220:124-7.
53. Agusti A, Cauli O, Rodrigo R, Llansola M, Hernandez-Rabaza V, Felipo V. p38 MAP kinase is a therapeutic target for hepatic encephalopathy in rats with portacaval shunts. Gut 2011;60:1572-9.
54. Strassburger M, Braun H, Reymann KG. Anti-inflammatory treatment with the p38 mitogen-activated protein kinase inhibitor SB239063 is neuroprotective, decreases the number of activatedmicroglia and facilitates neurogenesis in oxygen-glucose-deprived hippocampal slice cultures. Eur J Pharmacol 2008;592:55-61.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Montoliu C, Llansola M, Felipo V. Neuroinflammation and neurological alterations in chronic liver diseases. Neurosciences 2015;2:138-44. http://dx.doi.org/10.4103/2347-8659.160845
AMA Style
Montoliu C, Llansola M, Felipo V. Neuroinflammation and neurological alterations in chronic liver diseases. Neuroimmunology and Neuroinflammation. 2015; 2: 138-44. http://dx.doi.org/10.4103/2347-8659.160845
Chicago/Turabian Style
Montoliu, Carmina, Marta Llansola, Vicente Felipo. 2015. "Neuroinflammation and neurological alterations in chronic liver diseases" Neuroimmunology and Neuroinflammation. 2: 138-44. http://dx.doi.org/10.4103/2347-8659.160845
ACS Style
Montoliu, C.; Llansola M.; Felipo V. Neuroinflammation and neurological alterations in chronic liver diseases. Neurosciences. 2015, 2, 138-44. http://dx.doi.org/10.4103/2347-8659.160845
About This Article
Copyright

Data & Comments
Data


 Cite This Article 22 clicks
Cite This Article 22 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.