
Autoimmune encephalopathies in children: diagnostic clues and therapeutic challenges

Abstract
Neuronal surface antibody syndromes (NSAS) encompass a variety of disorders associated with “neuronal surface antibodies”. These share clinical and neuroradiological features that pose challenges related to their recognition and treatment. Recent epidemiological studies show a clear predominance for the glutamate-N-methyl-D-aspartate receptor encephalitis in both adults and pediatric population. Despite this, the overall NSAS’s incidence remains underestimated, and diagnosis persists to be not always easy to achieve. Based on current literature data, in this paper the authors propose a diagnostic pathway to approach and treat pediatric NSAS. An autoimmune etiology can be suggested through the integration of clinical, immunological, electrophysiological and neuroradiological data. On that basis, a target treatment can be started, consisting of corticosteroids and intravenous immunoglobulin or plasma exchange as a first-line immunotherapy, followed by second-line drugs including rituximab, cyclophosphamide or mycophenolate mophetil, if the case. In children a prompt diagnosis and a targeted treatment may lead to a better clinical outcome. Nevertheless further studies are required to assess the need of more tailored treatments according to long-term outcome findings and prognostic factors in different NSAS.
Keywords
Introduction
Over the last decade there has been an increase in the identification of forms of encephalitis associated with “neuronal surface antibodies” (NSAbs). These have been labelled as “neuronal surface antibody syndromes” (NSAS).[1]
NSAS differ from encephalitis due to antibodies directed against intracellular neuronal antigens for a different etiopathogenetic mechanism, a weaker association with paraneoplastic syndromes, a better response to immunotherapy and a higher incidence in the pediatric population.[2-6] Pathogenesis predominantly involves humoral immune response, while cellular immune response activation may coexist in a variable proportion, according to the different forms. Target antigens include proteins with various roles in neuronal function, ranging from synaptic transmission and plasticity to ions channels’ clustering and modulation, and including also glutamic acid decarboxylase (GAD) enzyme when exposed on cellular surface during exocitosis.[1,7]
A number of studies reporting NSAS in infancy suggest that, so far, their incidence has been probably underestimated, due to the fact that they are still often unrecognized or identified at a later stage.[8,9]
In pediatric forms, as in adults one, females can be over-represented, and a history of other antibody-mediated condition is easily detectable.[10] Conversely, in children rather than adults, a paraneoplastic cause is less probable and the role of fever or intercurrent infections in supporting the autoimmune process is less clear.[1,10]
Glutamate-N-methyl-D-aspartate receptor encephalitis is the most frequent form of NSAS in children.[11-14] According to up-to-date researches it is also the most common pediatric form of encephalitis, with the only exception of acute demyelinating encephalomyelitis.[15,16] With reference to other pediatric NSAS, reports are mostly anecdotal, with the only exception of the forms associated with voltage-gated potassium channel complex (VGKC) antibodies.[17-20][Table 1].
Details of NSAS encephalitis in adults and children
| Target | Antigen | Adult forms | Pediatric forms | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patients | Clinical signs | Neuroimaging | EEG pattern | Tumor association (%) | Outcome | Incidence | Peculiar symptoms | ||
| Glutamate-N-methyl-D-aspartate receptor (NMDA-R) | |||||||||
| Glutamate- N-methyl- D-aspartate receptor subunits | GluNR1/GluNR2/ | F 80% Age:1-80 y (median 20 y) | Psychosis or epileptic seizures followed by insomnia, amnesia, MD, catatonia, AI, coma[11-13] | Normal (50%) Non specific pattern or DMN involvement (50%) | -GS and FIED -Extreme delta brush (25%) | Age dependent -OT (10-50% > 18 y)[11] -ADC (rare) | Monophasic (75%): excellent if early treated (good response to IM/tumor removal). Relapsing-remitting (25%): normal in between | 40% of the total[14] | Seizures > psychosis occurrence (age dependent) EEG: Rarely extreme delta brush.[11-13] |
| Voltage-gated potassium channel (VGK) | |||||||||
| VGKC-complex | VGKC undefined targets (intracellular epitope? interacting proteins?) | M 60% Age:1-86 (median 55) | Peripheral nerve hyperexcitability, Morvan’s syndrome, encephalopathy, behavioural changes, seizures, short term memory loss. Possible coexistence with LEMS.[1,17] | LE, possible atrophy at MRI follow-up | GS/FS (temporal) and bilateral (temporal) FIED | Age dependant Thymoma, SCLC/ADC/HM (rare) | Variable response to IM | F = M Age: 1-16 y (< 50 cases) | Broad clinical spectrum: fever followed by SE, encephalopathy, GDR, MD (Cho, My, Tr, MT), CA, AI, RS,TLE; RE.[18-20] |
| Leucine-rich glioma inactivated-1 component | LGi1 | M 65% Age: 30-80 y (median 60 y) | Prodromal faciobrachial seizures, short term memory loss, psycosis, hyponatremia (60%), tonic seizures/myoclonus.[21] | LE generally progressing to hippocampal/whole brain atrophy | -GS/FS (temporal) and bilateral (temporal) FIED | -Thymoma (< 10%) -SCLC/ADC/HM (rare) | Monophasic with complete recovery (rare) or relapsing desease course evolving to memory impairment, TLE, psychiatric disorders. | Not reported | Not reported |
| contactin-associated protein-like 2 | CASPR2 | M 85% Age: 45-80 y (median 60 y) | Morvan’s syndrome; psychosis, seizures, memory impairment hyponatremia (60%).[22] | -Normal (especially in Morvan’s syndrome) -aspecific T2/FLAIR abnormalities -LE (25%) No progression to hippocampal atrophy/sclerosis | -GS/FS and GED/FIED | Thymoma (0-40%) SCLC/ADC/HM (rare) | Generally monophasic with good outcome (good response to IM). Relapses may occur. | M* Age: 2 y,6 y (2 cases) | GBS without encephalitis.[23]Complete recover |
| Alpha-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid- glutamate receptor (AMPA-R) | |||||||||
| GluA1/2 | F 90% Age: 40-80 y (median 60 y) | Seizures, short term memory loss, disorientation, psychosis. Frequent coexisting autoimmunities.[24] | LE | GS or FS and GED/FIED | Lung (70%), breast, thymoma (rare) | Monophasic (50%) good response to IM/tumor removal Relapsing remitting disease course (50%). | Not reported | Not described (GluA3). RE (occasional reports) doubt pathogenicity[25] | |
| Metabotropic gutamate receptors | |||||||||
| Type 1 | mGluR1 | F 100%* Age: 20-50 y* | Cerebellitis.[26] | -Normal (generally) -cerebellar T2/FLAIR abnormalities evolving in atrophy -LE (rare) | Normal | HL (70%) | Monophasic or chronic disease course (correlation with titer). Good response to IM | Not reported | Not reported |
| Type 5 | mGluR5 | F 50%* Age: 15-45 y* | Ophelia syndrome | LE | FS (bi-temporal) or FIED; FED or GED | HL (70%) | Monophasic or chronic disease course (correlation with titer). Generally good response to IM | M 15 y (1 case) | Confusion, anxiety, fear, extreme agitation, auditory/visual hallucination, GS. Complete spontaneous recovery.[27] |
| Gamma-aminobutyric acid receptors (GABAA-R and GABAB-R) | |||||||||
| GABAA-R associated protein (clustering and anchoring receptors) | GABARAP, α1/β3 | M 60% Age: 2-74 y (median 22 y)* | SE/RES, encephalopathy, RS, SPS, CA | LE | FS or GS + FED and FIED | None | Variable | Age: 2-16 y (7 cases ) | SE/RES, encephalopathy, Refractory seizures, SPS (rare).[28]Association with HL (1 case) |
| GABAB-R subunit 1/2 | GABAB1 GABAB2 | F 50% Age: 25-75 y (median 60 y) | Prominent seizures (FS, GS), short term memory loss, psychosis | LE (often asymmetric) | FS or GS + FED and FIED | SCLC/thymus/NET (50%) | Monophasic or chronic disease course: good response to IM. Occasional relapses. | Age: 3 y, 16 y (2 cases) | Lethargy, MD, opsoclonus, ataxia, seizures. Variable outcome MRI: diffuse changes in cortical and subcortical structures. [29-31] |
| Dopamine receptor | |||||||||
| Type 2 | D2-R | M 50% Age: 6-31 y (median 20 y) | SydCho, PANDAS, TS, encephalopathy, psychosis, sleep disorders | T2/FLAIR Basal ganglia abnormalities | Generally GS | Exceptional | Monophasic or relapsing remitting; good response to IM. | F = M Age: 2-17 y (median 7 y) | Comparable to adult forms Relapsing remitting course.[32] |
| Glycine receptor (Gly-R) | |||||||||
| Gly-R subunit α1 | Gly-Rα1 | M 80% Age: 30-60 y (median 50 y) | Prominent PERM and SPS; hyperekplexia, spinal My, CA, encephalitis, FS, brainstem disfunction (rare) | Normal (ca. 70%) LE or unspecific (rare) | Normal | Typically none; thymoma/lung (exceptional) | Monophasic or chronic disease course: good response to IM. Occasional relapses. | F 80% Age:14 M-5 y, (3 cases) | -PERM, hyperekplexia rigidity, My, no consciousness impairment; -explosive-onset epileptic encephalopathy; RFS, speech and behavioural disturbance. Variable outcome.[33,34] |
| Dipeptidyl-peptidase-like protein 6 | |||||||||
| Cell surface auxiliary subunit of the Kv4.2 potassium channel | DPPX | M 80% Age: 13-75 y (median 53 y) | Prodromal severe gastrointestinal dysfunction, encephalopathy, agitation, hallucinations, tremor, PERM, My, startle with muscle rigidity, sleep disturbances, AI seizures (rare) | -Normal -Aspecific T2/FLAIR abnormalities (rare) | Normal | None | Good response to IM but generally relapsing | One case reported (13 y) | Comparable to adult forms.[35] |
| Neuronal cell adhesion molecule IgLON5 | |||||||||
| Neuronal cell adhesion protein IgLON5 | IgLON5 | M 50% Age: 52-76 y (median 59 y)* | Prominent sleep dysfunction, abnormal sleep movements, OSAS, Cho, CA, progressive memory loss, mild gait imbalance.[36] | Normal | Normal | None | Chronic disease course refractory to IM | Not reported | Not reported |
| Glutamic-acid-decarboxylase | |||||||||
| Glutamic-acid-decarboxylase isoenzime65 | GAD65 | F 80% Age:15-80 y (median 60 y) | Neurocognitive disorders, LE, TLE with RE, SPS, PERM, CA; DM1 (if titer < 20 U/ml)-Frequent coexisting autoimmunities;[37]EMG abnormalities | -Normal -LE or mesio-temporal, brain stem, cerebellum and spinal cord T2/FLAIR abnormalities evolving into atrophy. | -Normal -GS or FS, FED (SE) and/or GED | Thymoma, lung, colon, pancreas, breast, thyroid, renal cell carcinoma (rare) | Chronic disease course: usually poor response to IM (more resistant than those with cell surface antibodies) | M = F Age: 5-16 y (median10 y) | Comparable to adult forms Variable response.[38] |
In this paper the authors propose a diagnostic pathway based both on literature and the experience that may help to obtain accurate identification of pediatric NSAS, with the aim to start an adequate and early treatment, and achieve a better clinical outcome.
Diagnostic clues
When a healthy child presents with unexpected symptoms such as seizures, sudden behavioral changes and movement disorders, causes like infections and traumas must be ruled out, together with toxic, metabolic and neoplastic factors. Another issue to be excluded is a previous central nervous system disease history. Once left aside all this, an autoimmune etiology should be always taken into account.
Longitudinal clinical, neurophysiological and neuroradiological findings facilitate the diagnostic pathway, and often provide information suggestive of specific NSAS variants [Figure 1].
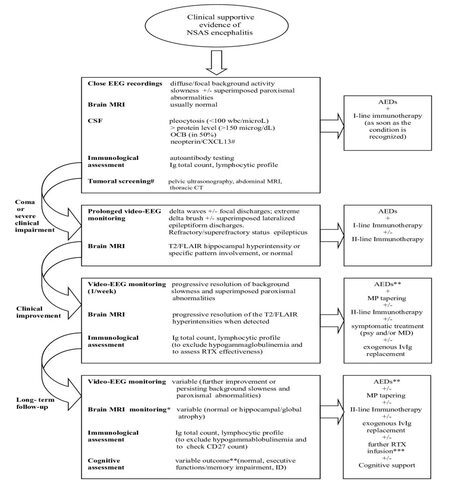
Figure 1. Diagnostic investigations and treatment of encephalitis related to neuronal surface antibody syndrome (NSAS) in children according to clinical steps. IvMP + IvIg +/-PE: intravenous methylprednisolone + intravenous immunoglobulin + plasma exchange (I-line immunotherapy). EGG: electroencephalogram; MRI: magnetic resonance imaging; CSF: cerebrospinal fluid; OCB: oligoclonal band; CXCL13: chemokine (C-X-C motif) ligand 13; CT: computed tomography; II-line immunotherapy: RTX (rituximab), Cyc (cyclophosphamide) or MMF(mycophenolate mophetil); AEDs: anti epileptic drugs; #: in anti NMDA-R encephalitis; *: timing according to patient an specific disorder; **: withdrawing according to patients/specific disorder and EEG findings; ***: according to immunological assessment at follow-up; psy: psychosis; MD: movement disorders; ID: intellectual disability; MP: methylprednisolone
Moreover, abnormalities at electroencephalogram (EEG) and magnetic resonance imaging (MRI) may be roughly specific for the different phases of the disease, allowing monitoring its progression.
As reported in adults, the different forms of pediatric NSAS share an overlap of clinical signs and symptoms. Typically, onset is acute/subacute (< 12 weeks) and occurs simultaneously or after a stressful event, such as infections, fever or cephalalgia. Behavioral and sleep disorders, confusion and short term memory impairment are often present, but epileptic seizures are generally the first obvious sign in children, consisting in focal seizures arising from bi-temporal lobes or, less frequently, generalized ones.[8-10] In this phase EEG recordings usually shows a diffuse background activity slowness, sometimes associated with superimposed paroxysmal abnormalities,[9,39] and MRI findings are usually normal or non specific.
Usually, a more acute stage follows, characterized by reduced consciousness, refractory/super-refractory epilepticus status and a progressive sinking into a coma lasting days to weeks.[40,41] Ventilator and/or other vital parameters support are often required,[13] as well as a heavy sedation that might easily hide eventual associated symptoms.[28]
In this phase, interictal EEG recordings may show a diffuse high delta waves activity with focal discharges, refractory multifocal status epilepticus[38,39] or less frequently, extreme delta-brush with superimposed periodic lateralized epileptiform discharges.[39] A close EEG monitoring is mandatory, inclusive of at least one 24 h video-EEG recording (duration to increase according to the clinical picture). On MRI, in this stage common findings are a T2/FLAIR hyperintensity in the medial temporal lobes involving the hippocampus, and/or the prefrontal areas and the cingulate gyrus. Only few exceptions are detectable, for example a frequent persistent negative findings in dipeptidyl-peptidase-like protein-6 encephalitis, an extensive multifocal or widespread diffuse abnormalities in NMDAR and GABAA-R encephalitis, and a basal ganglia involvement in D2-R encephalitis.[42,43]
A slowly progressive improvement of the interictal activity usually follows, sometimes preceded by a previous sleep background activity reorganization (personal observation), which generally gives way to a drug-resistant epilepsy although, in a minority of cases, a complete recovery is achieved.[44]
According to Suleiman classification, in order to help in the diagnosis 5 different autoimmune epilepsy categories are identifiable, on the basis both of the data obtained and of the immunotherapy response. Treatment may lead to remission, but the response depends on the specific form and possible association with malignancy.[7,45] In some forms, such as NMDA-R encephalitis, a complete recovery occurs in about 80% with a very low mortality rate.[13,14,46-48] When a complete remission does not occur, sequelae ranging from drug-resistant epilepsy associated with cognitive decline, to milder cognitive impairment are easily detectable.[1,12,13,47]
Following the treatment phase, EEG interictal activities slowly improve and longitudinal MRI studies can reveal a trend toward complete resolution, especially in some specific NSAS and when a rapid immunotherapy is administered. A more severe course towards a global atrophy predominantly affecting hippocampus, frontal and parietal regions usually characterizes the remaining cases.[43] An EEG monitoring including awakeness and sleep recordings (e.g. 40 min cad.) as well as a neuroradiological surveillance are thereby suggested.
Ideally, CSF and autoimmune responses should be investigated as soon as there is suspicion of autoimmune encephalitis.
CSF findings
As in adult, CSF usually shows a mild to moderate lymphocytic pleocytosis (< 100 white blood cells/μL), increased protein concentration (< 150 mg/dL), normal glucose level and frequently elevated IgG index; oligoclonal bands result detectable in about 50% of cases.
In children, elevated CSF neopterin can be used as an additional marker of CNS inflammation.[10,49]
A recent study underlies the role of the CXCL13 chemokine as a potential CSF biomarker of clinical outcome in anti-NMDAR encephalitis, its prolonged or secondary elevation suggesting a limited response to immunotherapy, an higher risk of relapses, and thereby the need to a more aggressive therapeutic approach.[50]
Immunological findings
As soon as an autoimmune condition is suspected (at prodromal or early acute stage) and before immunotherapy is started, a serum and/or CSF sample must be taken for autoantibody testing, and a Ig total level as well as a study of lymphocytic profile should be performed, in order to get a value to compare later on with [Figure 2].
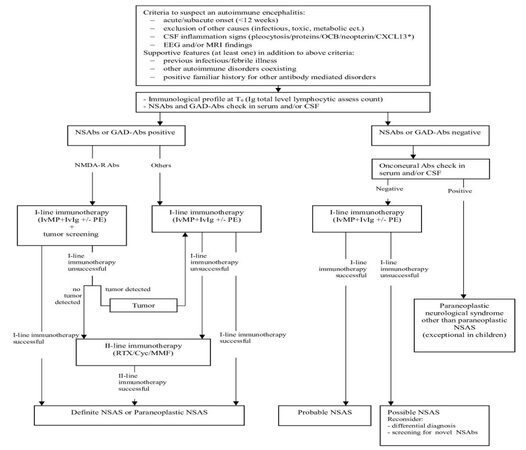
Figure 2. Flowchart for approaching the recognition of encephalitis related to neuronal surface antibody syndrome (NSAS) in children [modified from Zuliani et al.[1] 2012 and Suleiman et al.[8] 2013]. NSAbs: neuronal surface antibodies; CSF: cerebrospinal fluid; CXCL13: chemokine (C-X-C motif) ligand 13; MRI: magnetic resonance imaging; *: in NMDA-R encephalitis; GAD-Abs: antibodies to glutamic acid decarboxylase; *: GAD positivity is defined as > 1,000 u/mL.[8] OCB: oligoclonal bands; IvMP: intravenous methylprednisolone; IvIg: intravenous immunoglobulin; PE: plasma exchange; RTX: Rituximab; Cyc: cyclophosphamide; MMF: mycophenolate mophetil
If NSAbs are detected, the diagnosis is usually easily achieved. On a practical level, the expanding NSAbs spectrum could make it difficult to choose which antibody to check first. In order to direct the diagnostic pathway, indirect immunohistochemistry on rat brain tissue or immunocytochemistry in primary rat neuronal culture can highlight staining patterns evocative for surface (e.g. neuropil) or intracellular antigens.
This preliminary screening must be confirmed by more specific tecniques, such as cell-based assay, ELISA or radioimmunoassay.[51] A blended approach reduces the false positive rate,[52] providing an efficient diagnostic tool for pediatric NSAS.
A paraneoplastic cause is much less probable in children, so that testing for onconeural antibodies (Hu, Ma2, CV2/CRMP5, Ri, amphiphysin) can be not strictly necessary, at first instance at least. Nevertheless, if a paraneoplastic clinical picture shows-up, a commercial immunoblotting assay specifically designed can be properly used to manage a differential diagnosis.
Tumor surveillance
Because of the low prevalence of malignancy in pediatric NSAS, many authors consider tumor surveillance not strictly necessary in the first instance, with the only exception related to NMDA-R encephalitis. Nevertheless it may become increasingly relevant in patients who are older at the time of clinical onset.[13]
Treatment
Due to antibodies pathogenicity, treatment is focused on reducing the serum antibodies titer. There is no consensus on the immunotherapy approach to carry out, but it has become increasingly clear that starting treatment as early as possible is crucial to achieve a better clinical outcome.[12,48]
At seizure onset, antiepileptic therapy, with few exceptions, usually results ineffective.[9]
During the acute phase, as the diagnostic work out may take time, starting immunotherapy empirically is highly recommended. Although some patients undergo a complete recovery spontaneously, this is not frequent and it is not possible to identify the patients with a favorable outcome. Taking time before treatment waiting for immunological results or tumoral screening in anti-NMDAR cases is not recommended, not only because of the severity of the clinical findings but also because patients not promptly treated may be at higher risk for relapse.[12,53]
To date no consensus has been achieved on the treatment scheme to be used, and the available protocols are heterogeneous.
The first-line therapy usually includes a short course of high-dose steroids (methylprednisolone MP; 30 mg/kg/day i.v. per 3-5 days) followed by or combined with intravenous immunoglobulin (IvIg) administration (0.4 g/kg/day per 5 days).
Steroids are then tapered using 1-2 mg/kg/day orally, on average for another 12 weeks, adjusting the dose according to patient tolerability or possible side effects. If no benefit is noticed during steroid treatment, plasma exchange (PE), 3-5 cycles, should be considered.
In case the first-line treatment is unsatisfactory, a second-line immunotherapy should be started. It usually consists of rituximab 375 mg/mq per week every other week for 4 weeks,[54,55] cyclophosphamide (Cyc) 750 mg/m2, 3 times or mycophenolate mophetil 600 mg/m2, alone or in combination.[1,13]
The immunotherapy’s effectiveness can be checked with HIC on frozen rat brain tissue to assess the lack of immunostaining.
In the meanwhile, antiepileptic treatment is usually continued, even though its real impact in modifying the epileptic course remains uncertain as long as the immune mechanism starts to decrease itself. The decision whether to withdraw antiepileptic drugs or not should be made according to the patient, the specific disorder and EEG findings in the follow-up.
Psychiatric symptoms and involuntary movements, when present, can be treated symptomatically, and medications with a broad effect on multiple symptoms are usually recommended. Long acting benzodiazepines, sedatives such as clonidine, and anticonvulsivant drugs may be helpful in improving abnormal movements and mood instability. The management of psychiatric symptoms is more challenging: sedative and sleep medications other than benzodiazepines seem to be the most effective, while antipsychotic drugs are less efficacious and often associated with adverse events.[56]
Finally, during the remission and stabilization phases a gammaglobuline total check should be repeated in order to detect rituximab induced hypogammaglobinemia, that can eventually be treated with a replacement of an extra dose of exogenous IvIg.[10,57]
Regarding relapse risk prevention, no data are available so far on the preventive value of chronic long term IvIg administration, but encouraging results come from the chance of monitoring the CD19+ and CD27+ lymphocytes value every 2 months, re-administering rituximab in case of their further increase.[58]
Discussion
The early recognition of an immune mechanism underlying a neurologic disorder provides a chance to start early treatment and to achieve a better outcome.
Guidelines for NSAS in children have been recently developed,[10] extrapolated from a previous study by Zuliani[1] referred to adults, and mainly differing from it since focused on the higher epilepsy occurrence among pediatric symptoms.[10,59] As in Zuliani’s, the role given to immunotherapy response becomes a retrospective feature that helps with the classification itself. This points out that, whenever a specific antibody is detected, the diagnosis of NSAS is easily achieved; conversely, the hypothesis lies in a shady area. In this paper, based on a review of the literature and the experience, the authors provide a simplified pathway that may facilitate the identification and the early treatment of these forms. Concerning the diagnostic algorithm many questions remain unanswered.
A field that requires further work is the differential diagnosis among the individual forms of NSAS but this was beyond the aim of the paper. The spectrum of signs and symptoms is wide and it is often difficult to achieve a specific diagnosis on clinical ground only because of the overlapping of clinical signs. The recognition of some highly characteristic clinical features is sometimes possible and further work using an integrated approach combining EEG, neuroimaging and early identification of the underlying immunological mechanism is highly recommendable, as it can lead to an early appropriate treatment and to the possibility of a perceivable clinical improvement.
Few reports are available to date about the predictive value of electrophysiological findings in NSAS.[60] Identifying specific EEG pathological patterns may help not only in distinguishing the different forms, but also in recognizing them at an initial stage, with the aim of administering an early treatment. Moreover MRI-patterns, despite providing a supportive feature in the diagnostic flow-chart orientation, are often quite a specific,[43] and thereby usually insufficient, if alone, to get specific NSAS distinguishable from each other, especially at clinical onset.
Correlations between electrophysiological and neuroradiological data are then mandatory, but remain partly unexplored. Modern neuroradiological techniques are now increasingly available and could be useful to better understand pathophysiological mechanism and disclose predictive outcome data in different NSAS.[43]
Moreover, no consensus is obtained to date about the sensibility and specificity of serum vs. CSF testing. Although a study performed by Gresa-Arribas et al.[61] in 2014 demonstrated the higher CSF testing reliability in NMDA-R encephalitis, consensual data lack on the others NSAS. Moreover, the same work stressed out a positive correlation between the antibody titer and the risk of relapse, but no predictive threshold value has been established to help in deciding to resort to a retreatment or a chronic immunotherapy.
Finally, concerning the therapeutic approach, strategies tailored to the individual syndrome should be outlined, considering a less aggressive approach for those with a usually better outcome, such as NMDA-R encephalitis.[46,47] This must be done by taking into account the possible side effects of immunotherapic drugs and a “risks vs. benefits” assessment per single patient.[62]
Conclusion
In children NSAS clinical picture is heterogeneous, often overlapping and still poorly outlined. Anyhow, the recognition of some characteristic clinical features are sometimes possible, and can help the diagnostic approach with the aim to start a proper and early treatment. Nevertheless, further studies on larger prospective pediatric cohorts and randomized treatment trials are required in order to assess the need to tailor more or less aggressive treatments according to long term outcome findings and prognostic factors in different NSAS.
Acknowledgments
We thank Dr. Angela Vincent and Dr. Raffaele Iorio for performing diagnostic laboratory workup.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012;83:638-45.
3. Melzer N, Meuth SG, Wiendl H. Paraneoplastic and non-paraneoplastic autoimmunity to neurons in the central nervous system. J Neurol 2013;260:1215-33.
4. Lancaster E, Dalmau J. Neuronal autoantigens-pathogenesis, associated disorders and antibody testing. Nat Rev Neurol 2012;8:380-90.
5. Alavi S. Paraneoplastic neurologic syndromes in children: a review article. Iran J Child Neurol 2013;7:6-14.
6. Hacohen Y1, Wright S, Waters P, Agrawal S, Carr L, Cross H, De Sousa C, Devile C, Fallon P, Gupta R, Hedderly T, Hughes E, Kerr T, Lascelles K, Lin JP, Philip S, Pohl K, Prabahkar P, Smith M, Williams R, Clarke A, Hemingway C, Wassmer E, Vincent A, Lim MJ. Paediatric autoimmune encephalopathies: clinical features, laboratory investigations and outcomes in patients with or without antibodies to known central nervous system autoantigens. J Neurol Neurosurg Psychiatry 2013;84:748-55.
7. Dalmau J, Bataller L. Limbic encephalitis: the new cell membrane antigens and a proposal of clinical-immunological classification with therapeutic implications. Neurologia 2007;22:526-37. (in Spanish)
8. Suleiman J, Wright S, Gill D, Brilot F, Waters P, Peacock K, Procopis P, Nibber A, Vincent A, Dale RC, Lang B. Autoantibodies to neuronal antigens in children with new-onset seizures classified according to the revised ILAE organization of seizures and epilepsies. Epilepsia 2013;54:2091-100.
9. Suleiman J, Dale RC. The recognition and treatment of autoimmune epilepsy in children. Dev Med Child Neurol 2015;57:431-40.
10. Suleiman J, Brilot F, Lang B, Vincent A, Dale RC. Autoimmune epilepsy in children: case series and proposed guidelines for identification. Epilepsia 2013;54:1036-45.
11. Dalmau J, Tüzün E, Wu HY, Masjuan J, Rossi JE, Voloschin A, Baehring JM, Shimazaki H, Koide R, King D, Mason W, Sansing LH, Dichter MA, Rosenfeld MR, Lynch DR. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61:25-36.
12. Wright S, Hacohen Y, Jacobson L, Agrawal S, Gupta R, Philip S, Smith M, Lim M, Wassmer E, Vincent A. N-methyl-D-aspartate receptor antibody-mediated neurological disease: results of a UK-based surveillance study in children. Arch Dis Child 2015;100:521-6.
13. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011;10:63-74.
14. Florance NR, Davis RL, Lam C, Szperka C, Zhou L, Ahmad S, Campen CJ, Moss H, Peter N, Gleichman AJ, Glaser CA, Lynch DR, Rosenfeld MR, Dalmau J. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol 2009;66:11-8.
15. Granerod J, Ambrose HE, Davies NW, Clewley JP, Walsh AL, Morgan D, Cunningham R, Zuckerman M, Mutton KJ, Solomon T, Ward KN, Lunn MP, Irani SR, Vincent A, Brown DW, Crowcroft NS; UK Health Protection Agency (HPA) Aetiology of Encephalitis Study Group. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 2010;10:835-44.
16. Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis 2012;54:899-904.
17. Paterson RW1, Zandi MS, Armstrong R, Vincent A, Schott JM. Clinical relevance of positive voltage-gated potassium channel (VGKC)-complex antibodies: experience from a tertiary referral centre. J Neurol Neurosurg Psychiatry 2014;85:625-30.
18. Dhamija R, Renaud DL, Pittock SJ, McKeon A, Lachance DH, Nickels KC, Wirrell EC, Kuntz NL, King MD, Lennon VA. Neuronal voltage-gated potassium channel complex autoimmunity in children. Pediatr Neurol 2011;44:275-81.
19. Lin JJ, Lin KL, Hsia SH, Wang HS, Chiu CH; CHEESE Study Group. VGKC complex antibodies in pediatric severe acute encephalitis: a study and literature review. Brain Dev 2013;35:630-5.
20. Hacohen Y, Singh R, Rossi M, Lang B, Hemingway C, Lim M, Vincent A. Clinical relevance of voltage-gated potassium channel-complex antibodies in children. Neurology 2015;85:967-75.
21. Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR, Schott JM, Armstrong RJ, S Zagami A, Bleasel A, Somerville ER, Smith SM, Vincent A. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892-900.
22. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, Peles E, Buckley C, Lang B, Vincent A. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734-48.
23. Rosch RE, Bamford A, Hacohen Y, Wraige E, Vincent A, Mewasingh L, Lim M. Guillain-Barré syndrome associated with CASPR2 antibodies: two paediatric cases. J Peripher Nerv Syst 2014;19:246-9.
24. Höftberger R1, Van Sonderen A, Leypoldt F, Houghton D, Geschwind M, Gelfand J, Paredes M, Sabater L, Saiz A, Titulaer MJ, Graus F, Dalmau J. Encephalitis and AMPA receptor antibodies: Novel findings in a case series of 22 patients. Neurology 2015;84:2403-12.
25. Nibber A, Clover L, Pettingill P, Waters P, Elger CE, Bien CG, Vincent A, Lang B. Antibodies to AMPA receptors in Rasmussen's encephalitis. Eur J Paediatr Neurol 2016;20:222-7.
26. Marignier R, Chenevier F, Rogemond V, Sillevis Smitt P, Renoux C, Cavillon G, Androdias G, Vukusic S, Graus F, Honnorat J, Confavreux C. Metabotropic glutamate receptor type 1 autoantibody-associated cerebellitis: a primary autoimmune disease? Arch Neurol 2010;67:627-30.
27. Lancaster E, Martinez-Hernandez E, Titulaer MJ, Boulos M, Weaver S, Antoine JC, Liebers E, Kornblum C, Bien CG, Honnorat J, Wong S, Xu J, Contractor A, Balice-Gordon R, Dalmau J. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology 2011;77:1698-701.
28. Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R, McCracken L, Martinez-Hernandez E, Mason WP, Kruer MC, Ritacco DG, Grisold W, Meaney BF, Alcalá C, Sillevis-Smitt P, Titulaer MJ, Balice-Gordon R, Graus F, Dalmau J. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014;13:276-86.
29. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, Friedman D, Skeen MB, Grisold W, Kimura A, Ohta K, Iizuka T, Guzman M, Graus F, Moss SJ, Balice-Gordon R, Dalmau J. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010;9:67-76.
30. Höftberger R1, Titulaer MJ, Sabater L, Dome B, Rózsás A, Hegedus B, Hoda MA, Laszlo V, Ankersmit HJ, Harms L, Boyero S, de Felipe A, Saiz A, Dalmau J, Graus F. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology 2013;81:1500-6.
31. Kruer MC, Hoeftberger R, Lim KY, Coryell JC, Svoboda MD, Woltjer RL, Dalmau J. Aggressive course in encephalitis with opsoclonus, ataxia, chorea, and seizures: the first pediatric case of γ-aminobutyric acid type B receptor autoimmunity. JAMA Neurol 2014;71:620-3.
32. Dale RC, Merheb V, Pillai S, Wang D, Cantrill L, Murphy TK, Ben-Pazi H, Varadkar S, Aumann TD, Horne MK, Church AJ, Fath T, Brilot F. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain 2012;135:3453-68.
33. Joana Damásio J, Leite MI, Coutinho E, Waters P, Woodhall, M, Santos MA, Carrilho I, Vincent A. Progressive Encephalomyelitis With Rigidity and Myoclonus: the first pediatric case with glycine receptor antibodies. JAMA Neurol 2013;70:498-501.
34. Wuerfel E, Bien CG, Vincent A, Woodhall M, Brockmann K. Glycine receptor antibodies in a boy with focal epilepsy and episodic behavioral disorder. J Neurol Sci 2014;343:180-2.
35. Tobin WO, Lennon VA, Komorowski L, Probst C, Clardy SL, Aksamit AJ, Appendino JP, Lucchinetti CF, Matsumoto JY, Pittock SJ, Sandroni P, Tippmann-Peikert M, Wirrell EC, McKeon A. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 2014;83:1797-803.
36. Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J, Torres-Vega E, Contreras A, Giometto B, Compta Y, Embid C, Vilaseca I, Iranzo A, Santamaría J, Dalmau J, Graus F. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol 2014;13:575-86.
37. Gresa-Arribas N, Ari-o H, Martínez-Hernández E, Petit-Pedrol M, Sabater L, Saiz A, Dalmau J, Graus F. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS One 2015;10:e0121364.
38. Mishra N, Rodan LH, Nita DA, Gresa-Arribas N, Kobayashi J, Benseler SM. Anti-glutamic acid decarboxylase antibody associated limbic encephalitis in a child: expanding the spectrum of pediatric inflammatory brain diseases. J Child Neuro 2014;29:677-83.
39. Armangue T, Titulaer MJ, Malaga I, Malaga I, Bataller L, Gabilondo I, Graus F, Dalmau J Pediatric; Spanish Anti-N-methyl-D-Aspartate Receptor (NMDAR) Encephalitis Work Group. Pediatric anti-N-methyl-D-aspartate receptor encephalitis—clinical analysis and novel findings in a series of 20 patients. J Pediatr 2013;162:850-6.e2.
41. Dalmau J. Status epilepticus due to paraneoplastic and nonparaneoplastic encephalitides. Epilepsia 2009;50 Suppl 12:58-60.
42. Albert DV, Pluto CP, Weber A, Vidaurre J, Barbar-Smiley F, Abdul Aziz R, Driest K, Bout-Tabaku S, Ruess L, Rusin JA, Morgan-Followell B. Utility of neurodiagnostic studies in the diagnosis of autoimmune encephalitis in children. Pediatr Neurol 2016;55:37-45.
43. Heine J, Prüss H, Bartsch T, Ploner CJ, Paul F, Finke C. Imaging of autoimmune encephalitis-Relevance for clinical practice and hippocampal function. Neuroscience 2015;309:68-83.
44. Pillai SC, Mohammad SS, Hacohen Y, Tantsis E, Prelog K, Barnes EH, Gill D, Lim MJ, Brilot F, Vincent A, Dale RC. Postencephalitic epilepsy and drug-resistant epilepsy after infectious and antibody-associated encephalitis in childhood: Clinical and etiologic risk factors. Epilepsia 2016;57:e7-11.
45. Quek AM, Britton JW, McKeon A, So E, Lennon VA, Shin C, Klein C, Watson RE Jr, Kotsenas AL, Lagerlund TD, Cascino GD, Worrell GA, Wirrell EC, Nickels KC, Aksamit AJ, Noe KH, Pittock SJ. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol 2012;69:582-93.
46. Zekeridou A, Karantoni E, Viaccoz A, Ducray F, Gitiaux C, Villega F, Deiva K, Rogemond V, Mathias E, Picard G, Tardieu M, Antoine JC, Delattre JY, Honnorat J. Treatment and outcome of children and adolescents with N-methyl-D-aspartate receptor encephalitis. J Neurol 2015;262:1859-66.
47. Finke C, Kopp UA, Prüss H, Dalmau J, Wandinger KP, Ploner CJ. Cognitive deficits following anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry 2012;83:195-8.
48. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, Honig LS, Benseler SM, Kawachi I, Martinez-Hernandez E, Aguilar E, Gresa-Arribas N, Ryan-Florance N, Torrents A, Saiz A, Rosenfeld MR, Balice-Gordon R, Graus F, Dalmau J. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157-65.
49. Dale RC, Brilot F, Fagan E, Earl J. Cerebrospinal fluid neopterin in paediatric neurology: a marker of active central nervous system inflammation. Dev Med Child Neurol 2009;51:317-23.
50. Leypoldt F, Hoftberger R, Titulaer MJ, Armangue T, Gresa-Arribas N, Jahn H, Rostásy K, Schlumberger W, Meyer T, Wandinger KP, Rosenfeld MR, Graus F, Dalmau J. Investigations on CXCL13 in anti-N-methyl-d-aspartate receptor encephalitis: a potential biomarker of treatment response. JAMA Neurol 2015;72:180-6.
51. Nakahara E, Sakuma H, Kimura-Kuroda J, Shimizu T, Okumura A, Hayashi M. A diagnostic approach for identifying anti-neuronal antibodies in children with suspected autoimmune encephalitis. J Neuroimmunol 2015;285:150-5.
52. Armangue T, Santamaria J, Dalmau J. When a serum test overrides the clinical assessment. Neurology 2015;84:1379-81.
53. Gabilondo I, Saiz A, Galán L, González V, Jadraque R, Sabater L, Sans A, Sempere A, Vela A, Villalobos F, Vi-als M, Villoslada P, Graus F. Analysis of relapses in anti-NMDAR encephalitis. Neurology 2011;77:996-9.
54. Le Moigno L, Ternant D, Paintaud G, Thibault G, Cloarec S, Tardieu M, Lagrue E, Castelnau P. N-methyl-D-aspartate receptor antibody encephalitis: value of immunomodulatory therapy. Arch Pediatr 2014;21:620-3.
55. Dale RC, Pillai S, Brilot F. Cerebrospinal fluid CD19(+) B-cell expansion in N-methyl-D-aspartate receptor encephalitis. Dev Med Child Neurol 2013;55:191-3.
56. Mohammad SS, Jones H, Hong M, Nosadini M, Sharpe C, Pillai SC, Brilot F, Dale RC. Symptomatic treatment of children with anti-NMDAR encephalitis. Dev Med Child Neurol 2016;58:376-84.
57. Makatsori M, Kiani-Alikhan S, Manson AL, Verma N, Leandro M, Gurugama NP, Longhurst HJ, Grigoriadou S, Buckland M, Kanfer E, Hanson S, Ibrahim MA, Grimbacher B, Chee R, Seneviratne SL. Hypogammaglobulinaemia after rituximab treatment-incidence and outcomes. QJM 2014;107:821-8.
58. Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol 2013;70:1110-7.
59. Haberlandt E, Bast T, Ebner A, Holthausen H, Kluger G, Kravljanac R, Kröll-Seger J, Kurlemann G, Makowski C, Rostasy K, Tuschen-Hofstätter E, Weber G, Vincent A, Bien CG. Limbic encephalitis in children and adolescents. Arch Dis Child 2011;96:186-91.
60. Baysal-Kirac L, Tuzun E, Altindag E, Ekizoglu E, Kinay D, Bilgic B, Tekturk P, Baykan B. Are there any specific eeg findings in autoimmune epilepsies? Clin EEG Neurosci 2016;47:224-34.
61. Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, Gleichman AJ, Balice-Gordon R, Rosenfeld MR, Lynch D, Graus F, Dalmau J. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2014;13:167-77.
62. Mehrpooya M, Vaseghi G, Eshraghi A, Eslami N. Delayed Myocardial Infarction Associated With Rituximab Infusion: a case report and literature review. Am J Ther 2016;23:e283-7.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Olivieri G, Contaldo I, Ferrantini G, Musto E, Scalise R, Stefanini MC, Battaglia D, Mercuri E. Autoimmune encephalopathies in children: diagnostic clues and therapeutic challenges. Neurosciences 2016;3:147-55. http://dx.doi.org/10.20517/2347-8659.2016.09
AMA Style
Olivieri G, Contaldo I, Ferrantini G, Musto E, Scalise R, Stefanini MC, Battaglia D, Mercuri E. Autoimmune encephalopathies in children: diagnostic clues and therapeutic challenges. Neuroimmunology and Neuroinflammation. 2016; 3: 147-55. http://dx.doi.org/10.20517/2347-8659.2016.09
Chicago/Turabian Style
Olivieri, Giorgia, Ilaria Contaldo, Gloria Ferrantini, Elisa Musto, Roberta Scalise, Maria Chiara Stefanini, Domenica Battaglia, Eugenio Mercuri. 2016. "Autoimmune encephalopathies in children: diagnostic clues and therapeutic challenges" Neuroimmunology and Neuroinflammation. 3: 147-55. http://dx.doi.org/10.20517/2347-8659.2016.09
ACS Style
Olivieri, G.; Contaldo I.; Ferrantini G.; Musto E.; Scalise R.; Stefanini MC.; Battaglia D.; Mercuri E. Autoimmune encephalopathies in children: diagnostic clues and therapeutic challenges. Neurosciences. 2016, 3, 147-55. http://dx.doi.org/10.20517/2347-8659.2016.09
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 12
likes
Like This Article 12
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.