
Immune-to-brain signaling and substrates of altered behavior during inflammation


Abstract
During the systemic inflammatory response to acute infection, and when in a safe environment, endothermic mammals typically display reduced activity and food intake, increased sleep, and the adoption of a curled-up position. These changes in behavior, in concert with fever, are adaptive in that they contribute to host survival. The present review addresses the immune-to-brain signaling pathways as well as possible neural substrates mediating reduced exploration and food intake during acute systemic inflammation. These involve rapid activation of peripheral nerves and glutamatergic brainstem circuits as well as slower IL-1β action in the brain activating limbic and possibly ventral hypothalamic structures. Although mostly adaptive acutely, behavioral changes during inflammation may also reflect brain dysfunction in severe sepsis-associated delirium or become maladaptive and result in depression due to medical conditions that involve long-term inflammatory episodes with pain or discomfort. The mechanisms underlying these conditions are presently ill-understood even though neuroinflammation and neurodegeneration occur during and subsequent to sepsis-associated brain dysfunction, respectively.
Keywords
Introduction
Fever and reduced activity and food intake as adaptive host responses to infection
The finding in the 1970s that peripheral administration of non-steroid anti-inflammatory drug-type antipyretics lowered survival of different species of animals after their inoculation with bacteria provided a conclusive piece evidence in favor of the idea that fever was beneficial for survival of infected organisms.[1-3] But fever is an energetically costly response often requiring an increase in metabolism of 30-50%.[4] Text books of human and veterinary medicine often have mentioned reduced activity and appetite along with the occurrence of fever during infectious disease.[4] From an energy balance point of view on endotherms, it makes sense to reduce energy expenditure in the form of physical activity, such as exploration of one’s environment, during fever. But, given the adaptive value as well as the high energy costs of increasing body temperature in response to infection, one may wonder how come then that the infected organism refrains from taking in more energy? In fact, reducing food intake upon infection may be an adaptive response as well since force-feeding mice during acute bacterial infection, it was indeed found to increase mortality.[5]
Sickness behavior as motivated behavior
Benjamin Hart[4] stated that in a “sickness behavior” perspective “the sleepy or depressed or inactive animal is less motivated to move about using energy that could fuel metabolic increases associated with fever”. To consider sickness a motivation, like fear, hunger, thirst and other motivational states, implies that its expression is flexible depending on other motivations. Thus, it is important to show that its occurrence does indeed depend on environmental conditions. Interestingly, rats that depend entirely on hoarded food for their consumption, when rendered sick by bacterial lipopolysaccharide (LPS) endotoxins, continued to hoard more food, even though they did not consume it, than did animals injected with LPS that had the possibility to hoard, but also received food in their cage.[6] Thus, the expression of sickness behavior depends on the external conditions and, in this case, likely on the motivation to hoard food. Based on these and other observations, sickness behavior is now considered as the expression of a motivational system that reorganizes the organism’s perception and action.
Sepsis-associated encephalopathy and delirium
Septic encephalopathy or brain dysfunction occurs in up to 70% of sepsis patients.[7] Encephalopathy was replaced by delirium due to a general medical condition in Diagnostic and Statistical Manual of Mental Disorders-IV and described as a disturbance in consciousness or perception or change in cognition characterized by reduced ability to focus or sustain attention and fluctuating changes in mental status, ranging from confusion to coma. However, functioning of the entire neuraxis and peripheral nerves can be disturbed during sepsis. Indeed, abnormal or slowed postural or protective reflexes have often been reported to occur during sepsis.[8-10] Therefore, and notwithstanding the fact that sickness behavior can clearly be adaptive in response to an acute infection, it should also be clear that during severe sepsis important cerebral dysfunction can occur.
Why immune-to-brain signaling?
Fever can be defined as “a state of elevated core temperature” that is “due to an elevation of the set-point of body temperature, according to which the higher temperature is actively established by the operation of thermo-effectors”.[11] Since the set-point of body temperature is regulated by the preoptic hypothalamus, this gave rise to the question how the immune system signals the brain to bring about fever when animals are infected with bacteria.
The view of sickness behavior as being due to a motivation also implies immune-to-brain signaling. Indeed, even though the brain circuits underlying every single postulated motivational system are not known in full detail, motivations are mediated by brain circuits comprising the hypothalamus and limbic system. Thus, the occurrence of sickness behavior in response to exposure of animals to bacteria also begged the question as to how such events are signaled to the brain. In what follows the actions of the pro-inflammatory cytokine interleukin-1 (IL-1) on peripheral nerves, brain circumventricular organs and the blood-brain barrier (BBB), IL-1 transport across the BBB and IL-1 synthesis in the brain will be discussed as immune-to-brain signaling pathways.
From immune-to-brain signaling to neuroinflammation?
Intereukin-1 as a mediator of immune-to-brain signaling that cannot passively cross the BBB
Once bacteria or their components have entered host tissues, they activate innate immune cells, including monocytes-macrophages and neutrophils, to generate an inflammatory response mediated by cytokines, such an interleukin-1β (IL-1β).[12,13] Peripheral injection of IL-1β mimics the symptoms of sickness and the signs of disease normally seen after infection.[14] Conversely, systemic administration of the naturally occurring IL-1 receptor antagonist (IL-1ra) alleviates or blocks systemic bacterial LPS-induced fever in rats.[15,16] In addition, peripheral IL-1ra also attenuates the reduction in locomotor activity and social interactions after systemic LPS injection.[17] Thus, IL-1 mediates, at least in part, fever and sickness behavior when these occur in response to the administration of bacterial LPS. However, the fact that IL-1 is a hydrophilic large peptide of 17 kDa means that it cannot passively cross the BBB separating the brain parenchyma from blood. Consequently, proposing and testing IL-1-mediated immune-to-brain signaling pathways became a topic of intense research activity from the 1990s onwards.
Circulating IL-1 acting in brain circumventricular organs lacking a BBB
In the early 1980s, antipyretics were already known to inhibit the synthesis of prostaglandins, a family of small lipophilic mediators. IL-1 was subsequently found to induce the formation of prostaglandin E2 (PGE2) by stimulating the synthesis of the rate-limiting enzyme cyclooxygenase in monocytes, fibroblasts, muscular and nervous tissue.[18] Around the same time, lesions of the anteroventral wall of the third brain ventricle, which contains the organum vasculosum of the lamina terminalis (OVLT), a brain cirumventriular organ where the blood-brain barrier is absent, were shown to suppress the fever response to peripheral administration of bacterial LPS or IL-1.[19,20] In addition, local injection of PGE2 into the OVLT resulted in higher fever than its administration in the preoptic area.[21] This led to the first hypothesis of IL-1-mediated immune-to-brain signaling pathway according to which circulating IL-1 acts in the OVLT to induce the production of PGE, which, in turn, modulates thermosensitive neurons in the preoptic area [Figure 1A-1].[22]
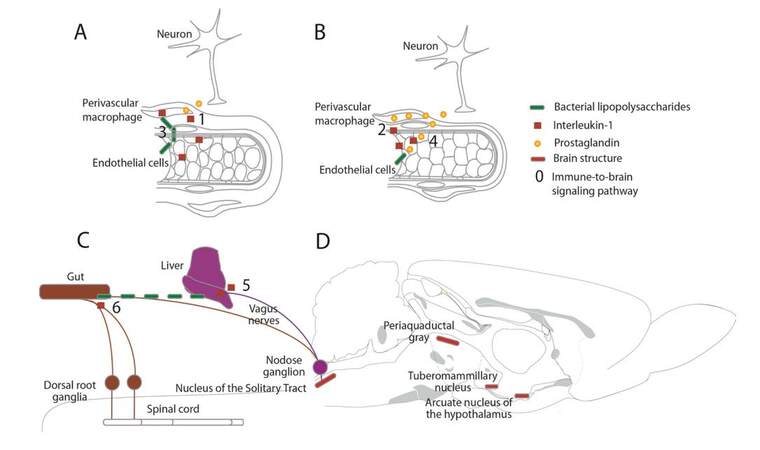
Figure 1. Immune-to-brain signaling pathways involving interleukin-1 in brain circumventricular organs (A), at the blood-brain barrier (B) and peripheral nerves (C) and substrates of altered behavior (D) during peripheral inflammation secondary to infection
Transport of circulating IL-1 across the BBB
A more general alternative hypothesis of immune-to-brain signaling was put forward after it was shown that intravenously administered radioactive recombinant IL-1α or β entered the brain by a saturable transport mechanism [Figure 1B-2].[23,24] Subsequent studies provided evidence indicating that transport over the brain endothelium making up the BBB contributed more to the presence of intravenously injected IL-1 in the brain than the contained leakage from a circumventricular organ.[25] At the same time evidence for the presence of IL-1 receptors in the brain accumulated.[26-34] Moreover, administration of IL-1ra into the lateral brain ventricle was found to attenuate the reduction in social exploration and food-motivated behavior, but not the fever response, after systemic IL-1β injection.[35,36] These findings thus clearly indicate that IL-1 can act in the brain to bring about changes in behavior after its peripheral injection.
Brain production of IL-1
As early as 1984, bioactive IL-1 had been detected in the brains of mice that were given systemic bacterial LPS endotoxin and showing signs of sickness behavior.[37] In 1992, the presence of IL-1β immunoreactive mononuclear cells around blood vessels of the central nervous system (CNS) was observed several hours after an intravenous injection of a high dose of bacterial LPS in rats.[38] When using lower doses of bacterial LPS injected either intravenously or intraperitoneally, cells in the circumventricular organs and choroid plexus were found to synthesize IL-1β [Figure 1A-3].[39-43] Soon after this, mRNA for the LPS-recognizing receptor Toll-like Receptor 4 was found to be expressed in these organs.[44] Interestingly, IL-1β was also found to be synthesized by cells with microglial morphology in brain parenchyma adjacent to circumventricular organs, including in the arcuate nucleus of the hypothalamus and in the nucleus of the solitary tract, beyond 4 h after peripheral LPS injection.[43] Bioactive IL-1 is found in plasma but not in the brain 2 h after systemic LPS administration, whereas at 6 h IL-1 is bioactive in the brain but not in plasma.[45] When IL-1ra was given into the lateral brain ventricle at the time that brain IL-1 was bioactive, it was found to attenuate reduced social exploration without affecting the fever response after peripheral LPS injection.[46] The subsequent finding that peripheral administration of a neutrophil-neutralizing antiserum attenuates brain IL-1β expression as well as the reduction in locomotor activity 24 h after systemic injection of LPS suggests that neutrophil infiltration into brain provides an important source of IL-1β at later time points.[47] Hence, brain IL-1 production and action sustains sickness behavior after systemic LPS administration, but not necessarily fever.
Circulating IL-1 inducing prostaglandin synthesis at the BBB
In the early 1990s, a second form of the rate-limiting prostaglandin synthesizing enzyme cyclooxygenase (COX) was identified and found to be induced along brain blood vessels after peripheral administration of bacterial LPS or IL-1β.[48-50] Concurrently, it was shown that most IL-1 receptors in the rodent brain were expressed along blood vessels making up the BBB [Figure 1B-4].[31-34] This led to the hypothesis according to which circulating IL-1 acts on its signaling receptor expressed by brain endothelial cells to induce COX-2-mediated prostaglandin production, which, given their lipophilic profile, can diffuse across the BBB and activate prostaglandin receptors on neurons to bring about sickness symptoms.[48,49] Testing of this hypothesis using mice in which endothelial cells were deficient in COX-2 or PGE synthase showed that although the fever response to an intraperitoneal injection of IL-1β was abolished in these animals, the reduction in locomotor activity was not affected.[51,52] So, in accordance with a hypothesis put forward in 2002,[53] BBB prostaglandin synthesis underlies IL-1β-induced fever, but not necessarily sickness behavior.
IL-1 action on peripheral nerves
At least two of the classical symptoms of local inflammation, heat and pain, correspond to sensory modalities and thus involve neural activation. Interestingly, local IL-1β application under the skin of a rat paw was shown in 1994 to increase the sensitivity to mechanical and heat stimuli and to augment electric activity of sensory nerve fibers.[54] Based on these considerations, IL-1 was proposed to act on neural sensory afferents to signal the brain and bring about symptoms of sickness [Figure 1C-5]. In accordance with this hypothesis, subdiaphragmatic vagotomy was shown to attenuate the reduction in social exploration and food-motivated behavior, conditioned taste aversion, increased sleep and hyperalgesia as early as 2 h after intraperitoneal administration of IL-1β or bacterial LPS.[55-59] Reversible inactivation of the dorsal vagal complex, which contains the central terminals of vagal sensory fibers, by local anesthesia also restored social exploration after intraperitoneal LPS administration.[60] Moreover, the febrile responses to systemic administration of low doses of IL-1β or LPS were also attenuated by prior subdiaphragmatic vagotomy, whereas fevers after higher doses were unaffected by this procedure.[61-68] Furthermore, selective chemical lesions of C-fiber afferents after intraperitoneal injection of capsaicin in adult rodents were found to also attenuate the first phase of the fever response in response to systemic administration of LPS.[69] This suggests that LPS-induced rapid fever responses may involve vagally-mediated immune-to-brain signaling with later fever peaks or prolonged fever depending on prostaglandin synthesis at blood-brain interfaces.
Soon after the first vagotomy studies, intravenous IL-1β administration was found to increase afferent discharge activity of the hepatic and gastric branches of the vagus nerve in a prostaglandin-dependent way.[70-72] Subsequently, vagal paraganglia and the nodose ganglion containing the neuronal cell bodies of the vagus nerve were observed to bind IL-1ra and to express the signaling IL-1 receptor.[72,73] In addition, spinal sensory afferent cell bodies in dorsal root ganglia also express mRNA coding the signaling IL-1 receptor and their peripheral fibers respond to local administration of IL-1β by increasing their activity as well as their sensitivity to heat in vitro.[74,75] Interestingly, ganglia of both vagal and spinal sensory nerves express TLRs and some bacteria have been shown to directly activate sensory neurons.[76-78] Taken together, these findings suggest that low doses of IL-1β or bacterial fragments may act on sensory nerve fibers to signal the central nervous system to give rise to early fever, hyperalgesia and sickness behavior [Figure 1C-6].
Activation of neural substrates or initiation of neurodegeneration during systemic inflammation?
Neural substrates of acute sickness behavior
Possible neural substrates of bacterial LPS-induced hypophagia
The basomedial hypothalamus plays an important role in the long-term regulation of food intake. Interestingly, lesions of the arcuate nucleus of the hypothalamus [Figure 1D] exacerbated the anorectic effect of peripheral IL-1β administration.[79] However; antagonizing the action of α-melanocyte stimulating hormone, which is produced by neurons of the arcuate nucleus of the hypothalamus, on central melanocortin receptors has been found to alleviate hypophagia after the peripheral administration of either IL-1β or LPS from 8 h onwards.[80,81] These findings indicate that the overall role of the arcuate hypothalamus is to counter reduced food intake, even though activation of some of its composing neurons does seem to play a role in sustained inflammation-associated hypophagia.
The brainstem mediates short-term regulation of food intake and glutamatergic projections from the nucleus of the solitary tract [Figure 1D] to the parabrachial nuclei reduce food intake.[82] Interestingly, brainstem metabotropic glutamate receptor antagonism was found to attenuate hypophagia and to increase food intake during the first 6 h after peripheral LPS to a greater extent than in vehicle-treated animal.[83] In parallel, intra fourth ventricle administration of this metabotropic glutamate receptor antagonist also reduced expression of the cellular transcription activation marker c-Fos in the nucleus of the solitary tract and lateral parabrachial nuclei.[83] These findings suggest that brainstem glutamatergic circuits are part of the neuronal substrates that rapidly reduce food intake under inflammatory conditions.
Potential neural substrates of bacterial LPS-induced reduced exploration
Interestingly, all intervention strategies restoring social exploration after intraperitoneal LPS injection also reduce induction of the cellular transcription activation marker c-Fos in the central nucleus of the amygdala (CEA) and the oval bed nucleus of the stria terminalis (ovBNST).[46,60,66] The amygdala and the bed nucleus of the stria terminalis project to the ventrolateral periaqueductal gray (vlPAG) [Figure 1D] in the pons,[84] the stimulation of which induces immobility and reduced social interactions.[85] Thus, c-Fos expressing neurons in the CEA and ovBNST may inhibit GABAergic neurons projection to the vlPAG resulting in immobility and reduced social interactions.[53] In addition, reduced exploration of different environments and devices has been shown to be associated with c-Fos expression in the ventral tuberomammillary nucleus [Figure 1D] after peripheral bacterial LPS injection.[86,87] Reduced activation of the ventral tuberomammillary nucleus may therefore be part of the neural substrates underlying reduced environmental exploration during sickness.
The realization that endogenous IL-1β can act in the brain to bring about sickness behavior raised the question as to where in the brain it binds to the signaling IL-1 receptor to reduce social and environmental exploration. Although the hippocampus is one of the most prominent sites of neuronal IL-1 receptor expression (see transport of circulating IL-1 across the BBB), no published study to date seems to have critically addressed the involvement of hippocampal IL-1 receptors in mediating sickness behavior. It is important to point out that this is not because such approaches are not available. Indeed, several groups have employed hippocampal overexpression of the IL-1ra. These studies addressed the role of hippocampal IL-1 in mediating responses to psychological stressors, such as electrical shocks and chronic isolation, and not those occurring upon exposure to infectious microorganisms or their components.[88,89] However, the findings of Chaskiel et al.[83] show that selective lesioning of IL-1 receptor-expressing cells in the hippocampus does not alter the reduction in social exploration after intracerebroventricular administration of IL-1β in mice. Thus, IL-1 receptors in the hippocampus do not seem to mediate the component of sickness behavior that involves reduced exploration.
Severe sepsis may lead to neurodegeneration
Magnetic resonance imaging of septic patients with brain dysfunction has indicated the presence of vasospasms in the medial cerebral arteries and ischemic strokes in brain gray matter as well as white matter edema.[90-92] (see for review[93]) Post mortem examination of brains of patients who died from sepsis revealed intracerebral hemorrhage, necrotic vessels with infiltrating leukocytes, increased perivascular spaces, microglial activation, cerebral IL-1β and TNF-α expression, neuronal apoptosis as well as perivascular dissociation of myelinated fibers and demyelination.[91,94,95] Clinical research thus clearly indicates the occurrence of neuroinflammation that may, in turn, lead to neurodegeneration.
Recently, several groups have employed cecal ligature and puncture (CLP) in rodents to study CNS dysfunction associated with sepsis. In these models, food intake and social interactions were found to be reduced during the first days, while activity and body temperature were altered and some conditioning learning tasks impaired for several weeks after sepsis induction.[96-99] Increased cerebral pro-inflammatory cytokine expression, impaired BBB function, cortical perivascular edema, glial cell activation, brain leukocyte adhesion and infiltration as well as neuronal death and degeneration in cortical and subcortical areas have all been observed from the first day of CLP onwards.[96-105] Thus, relevant animal models of sepsis have been shown to result both in transient sickness behavior and in long-term learning deficits that are accompanied by neuroinflammation and neurodegeneration.
Conclusion
During the systemic inflammatory response to acute infection, and when in a safe environment, endothermic mammals typically display reduced activity and food intake, increased sleep, and the adoption of a curled-up position. These changes in behavior, in concert with fever, are adaptive in that they contribute to host survival. Although the precise neurobiological substrates still need to be worked out, they are brought about by immune-to-brain signaling pathways that involve rapid activation of peripheral nerves and glutamatergic brainstem circuits as well as slower IL-1β action in the brain activating limbic and possibly ventral hypothalamic structures. Notwithstanding the fact that they are mostly adaptive acutely, behavioral changes during inflammation may also reflect brain dysfunction in severe sepsis-associated delirium or become maladaptive and result in depression due to medical conditions that involve long-term inflammatory episodes with pain or discomfort. The mechanisms underlying these conditions are presently ill-understood even though neuroinflammation and neurodegeneration occur during and subsequent to sepsis-associated brain dysfunction, respectively.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Patient consent
No patient involved.
Ethics approval
This article does not contain any studies with human participants or animals.
REFERENCES
1. Bernheim HA, Kluger MJ. Fever: effect of drug-induced antipyresis on survival. Science 1976;193:237-9.
2. Kluger MJ, Vaughn LK. Fever and survival in rabbits infected with Pasteurella multocida. J Physiol 1978;282:243-51.
5. Murray MJ, Murray AB. Anorexia of infection as a mechanism of host defense. Am J Clin Nutr 1979;32:593-6.
6. Aubert A, Kelley KW, Dantzer R. Differential effect of lipopolysaccharide on food hoarding behavior and food consumption in rats. Brain Behav Immun 1997;11:229-38.
7. Sprung CL, Peduzzi PN, Shatney CH, Schein RM, Wilson MF, Sheagren JN, Hinshaw LB. Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med 1990;18:801-6.
8. Wilson JX, Young GB. Progress in clinical neurosciences: sepsis-associated encephalopathy: evolving concepts. Can J Neurol Sci 2003;30:98-105.
9. Ebersoldt M, Sharshar T, Annane D. Sepsis-associated delirium. Intensive Care Med 2007;33:941-50.
11. Commission IT. Glossary of terms for thermal physiology. Second edition. Revised by The Commission for Thermal Physiology of the International Union of Physiological Sciences. Pflugers Arch 1987;410:567-87.
12. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008;8:958-69.
14. Kent S, Bluthé RM, Kelley KW, Dantzer R. Sickness behavior as a new target for drug development. Trends Pharmacol Sci 1992;13:24-8.
15. Smith BK, Kluger MJ. Human IL-1 receptor antagonist partially suppresses LPS fever but not plasma levels of IL-6 in Fischer rats. Am J Physiol 1992;263:R653-5.
16. Luheshi G, Miller AJ, Brouwer S, Dascombe MJ, Rothwell NJ, Hopkins SJ. Interleukin-1 receptor antagonist inhibits endotoxin fever and systemic interleukin-6 induction in the rat. Am J Physiol 1996;270:E91-5.
17. Bluthé RM, Dantzer R, Kelley KW. Effects of interleukin-1 receptor antagonist on the behavioral effects of lipopolysaccharide in rat. Brain Res 1992;573:318-20.
18. Bernheim HA. Is prostaglandin E2 involved in the pathogenesis of fever? Effects of interleukin-1 on the release of prostaglandins. Yale J Biol Med 1986;59:151-8.
19. Blatteis CM, Bealer SL, Hunter WS, Llanos-Q J, Ahokas RA, Mashburn TA Jr. Suppression of fever after lesions of the anteroventral third ventricle in guinea pigs. Brain Res Bull 1983;11:519-26.
20. Stitt JT. Evidence for the involvement of the organum vasculosum laminae terminalis in the febrile response of rabbits and rats. J Physiol 1985;368:501-11.
21. Stitt JT. Prostaglandin E as the neural mediator of the febrile response. Yale J Biol Med 1986;59:137-49.
22. Stitt JT. Passage of immunomodulators across the blood-brain barrier. Yale J Biol Med 1990;63:121-31.
23. Banks WA, Kastin AJ, Durham DA. Bidirectional transport of interleukin-1 alpha across the blood-brain barrier. Brain Res Bull 1989;23:433-7.
24. Banks WA, Ortiz L, Plotkin SR, Kastin AJ. Human interleukin (IL) 1 alpha, murine IL-1 alpha and murine IL-1 beta are transported from blood to brain in the mouse by a shared saturable mechanism. J Pharmacol Exp Ther 1991;259:988-96.
25. Maness LM, Kastin AJ, Banks WA. Relative contributions of a CVO and the microvascular bed to delivery of blood-borne IL-1alpha to the brain. Am J Physiol 1998;275:E207-12.
26. Farrar WL, Kilian PL, Ruff MR, Hill JM, Pert CB. Visualization and characterization of interleukin 1 receptors in brain. J Immunol 1987;139:459-63.
27. Katsuura G, Gottschall PE, Arimura A. Identification of a high-affinity receptor for interleukin-1 beta in rat brain. Biochem Biophys Res Commun 1988;156:61-7.
28. Takao T, Tracey DE, Mitchell WM, De Souza EB. Interleukin-1 receptors in mouse brain: characterization and neuronal localization. Endocrinology 1990;127:3070-8.
29. Cunningham ET Jr, Wada E, Carter DB, Tracey DE, Battey JF, De Souza EB. Localization of interleukin-1 receptor messenger RNA in murine hippocampus. Endocrinology 1991;128:2666-8.
30. Deyerle KL, Sims JE, Dower SK, Bothwell MA. Pattern of IL-1 receptor gene expression suggests role in noninflammatory processes. J Immunol 1992;149:1657-65.
31. Cunningham ETJ, Wada E, Carter DB, Tracey DE, Battey JF, De Souza EB. In situ histochemical localization of type I interleukin-1 receptor messenger RNA in the central nervous system, pituitary, and adrenal gland of the mouse. J Neurosci 1992;12:1101-14.
32. Yabuuchi K, Minami M, Katsumata S, Satoh M. Localization of type I interleukin-1 receptor mRNA in the rat brain. Brain Res Mol Brain Res 1994;27:27-36.
33. Wong ML, Licinio J. Localization of interleukin 1 type I receptor mRNA in rat brain. Neuroimmunomodulation 1994;1:110-5.
34. Ericsson A, Liu C, Hart RP, Sawchenko PE. Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neurol 1995;361:681-98.
35. Coceani F, Lees J, Redford J, Bishai I. Interleukin-1 receptor antagonist: effectiveness against interleukin-1 fever. Can J Physiol Pharmacol 1992;70:1590-6.
36. Kent S, Bluthe RM, Dantzer R, Hardwick AJ, Kelley KW, Rothwell NJ, Vannice JL. Different receptor mechanisms mediate the pyrogenic and behavioral effects of interleukin 1. Proc Natl Acad Sci U S A 1992;89:9117-20.
37. Fontana A, Weber E, Dayer JM. Synthesis of interleukin 1/endogenous pyrogen in the brain of endotoxin-treated mice: a step in fever induction? J Immunol 1984;133:1696-8.
38. van Dam AM, Brouns M, Louisse S, Berkenbosch F. Appearance of interleukin-1 in macrophages and in ramified microglia in the brain of endotoxin-treated rats: a pathway for the induction of non-specific symptoms of sickness? Brain Res 1992;588:291-6.
39. Nakamori T, Morimoto A, Yamaguchi K, Watanabe T, Long NC, Murakami N. Organum vasculosum laminae terminalis (OVLT) is a brain site to produce interleukin-1 beta during fever. Brain Res 1993;618:155-9.
40. Nakamori T, Morimoto A, Yamaguchi K, Watanabe T, Murakami N. Interleukin-1 beta production in the rabbit brain during endotoxin-induced fever. J Physiol(Lond) 1994;476:177-86.
41. Van Dam AM, Bauer J, Tilders FJ, Berkenbosch F. Endotoxin-induced appearance of immunoreactive interleukin-1 beta in ramified microglia in rat brain: a light and electron microscopic study. Neuroscience 1995;65:815-26.
42. Quan N, Whiteside M, Herkenham M. Time course and localization patterns of interleukin-1beta messenger RNA expression in brain and pituitary after peripheral administration of lipopolysaccharide. Neuroscience 1998;83:281-93.
43. Konsman JP, Kelley K, Dantzer R. Temporal and spatial relationships between lipopolysaccharide-induced expression of Fos, interleukin-1beta and inducible nitric oxide synthase in rat brain. Neuroscience 1999;89:535-48.
44. Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. Faseb J 2001;15:155-63.
45. Quan N, Sundar SK, Weiss JM. Induction of interleukin-1 in various brain regions after peripheral and central injections of lipopolysaccharide. J Neuroimmunol 1994;49:125-34.
46. Konsman JP, Veeneman J, Combe C, Poole S, Luheshi GN, Dantzer R. Central nervous action of interleukin-1 mediates activation of limbic structures and behavioural depression in response to peripheral administration of bacterial lipopolysaccharide. Eur J Neurosci 2008;28:2499-510.
47. Aguilar-Valles A, Kim J, Jung S, Woodside B, Luheshi GN. Role of brain transmigrating neutrophils in depression-like behavior during systemic infection. Mol Psychiatry 2014;19:599-606.
48. Cao C, Matsumura K, Yamagata K, Watanabe Y. Induction by lipopolysaccharide of cyclooxygenase-2 mRNA in rat brain; its possible role in the febrile response. Brain Res 1995;697:187-96.
49. Cao C, Matsumura K, Yamagata K, Watanabe Y. Endothelial cells of the rat brain vasculature express cyclooxygenase-2 mRNA in response to systemic interleukin-1 beta: a possible site of prostaglandin synthesis responsible for fever. Brain Res 1996;733:263-72.
50. Elmquist JK, Breder CD, Sherin JE, Scammell TE, Hickey WF, Dewitt D, Saper CB. Intravenous lipopolysaccharide induces cyclooxygenase 2-like immunoreactivity in rat brain perivascular microglia and meningeal macrophages. J Comp Neurol 1997;381:119-29.
51. Ching S, Zhang H, Belevych N, He L, Lai W, Pu XA, Jaeger LB, Chen Q, Quan N. Endothelial-specific knockdown of interleukin-1 (IL-1) type 1 receptor differentially alters CNS responses to IL-1 depending on its route of administration. J Neurosci 2007;27:10476-86.
52. Wilhelms DB, Kirilov M, Mirrasekhian E, Eskilsson A, Kugelberg UO, Klar C, Ridder DA, Herschman HR, Schwaninger M, Blomqvist A, Engblom D. Deletion of prostaglandin E2 synthesizing enzymes in brain endothelial cells attenuates inflammatory fever. J Neurosci 2014;34:11684-90.
53. Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci 2002;25:154-9.
54. Fukuoka H, Kawatani M, Hisamitsu T, Takeshige C. Cutaneous hyperalgesia induced by peripheral injection of interleukin-1 beta in the rat. Brain Res 1994;657:133-40.
55. Bluthé RM, Walter V, Parnet P, Layé S, Lestage J, Verrier D, Poole S, Stenning BE, Kelley KW, Dantzer R. Lipopolysaccharide induces sickness behaviour in rats by a vagal mediated mechanism. C R Acad Sci III 1994;317:499-503.
56. Watkins LR, Wiertelak EP, Goehler LE, Mooney-Heiberger K, Martinez J, Furness L, Smith KP, Maier SF. Neurocircuitry of illness-induced hyperalgesia. Brain Res 1994;639:283-99.
57. Bret-Dibat JL, Bluthé RM, Kent S, Kelley KW, Dantzer R. Lipopolysaccharide and interleukin-1 depress food-motivated behavior in mice by a vagal-mediated mechanism. Brain Behav Immun 1995;9:242-6.
58. Goehler LE, Busch CR, Tartaglia N, Relton J, Sisk D, Maier SF, Watkins LR. Blockade of cytokine induced conditioned taste aversion by subdiaphragmatic vagotomy: further evidence for vagal mediation of immune-brain communication. Neurosci Lett 1995;185:163-6.
59. Zielinski MR, Dunbrasky DL, Taishi P, Souza G, Krueger JM. Vagotomy attenuates brain cytokines and sleep induced by peripherally administered tumor necrosis factor-alpha and lipopolysaccharide in mice. Sleep 2013;36:1227-38, 38A.
60. Marvel FA, Chen CC, Badr N, Gaykema RP, Goehler LE. Reversible inactivation of the dorsal vagal complex blocks lipopolysaccharide-induced social withdrawal and c-Fos expression in central autonomic nuclei. Brain Behav Immun 2004;18:123-34.
61. Watkins LR, Goehler LE, Relton JK, Tartaglia N, Silbert L, Martin D, Maier SF. Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: evidence for vagal mediation of immune-brain communication. Neurosci Lett 1995;183:27-31.
62. Sehic E, Blatteis CM. Blockade of lipopolysaccharide-induced fever by subdiaphragmatic vagotomy in guinea pigs. Brain Res 1996;726:160-6.
63. Hansen MK, Krueger JM. Subdiaphragmatic vagotomy blocks the sleep- and fever-promoting effects of interleukin-1beta. Am J Physiol 1997;273:R1246-53.
64. Romanovsky AA, Simons CT, Székely M, Kulchitsky VA. The vagus nerve in the thermoregulatory response to systemic inflammation. Am J Physiol 1997;273:R407-13.
65. Opp MR, Toth LA. Somnogenic and pyrogenic effects of interleukin-1beta and lipopolysaccharide in intact and vagotomized rats. Life Sci 1998;62:923-36.
66. Konsman JP, Luheshi GN, Bluthe RM, Dantzer R. The vagus nerve mediates behavioural depression, but not fever, in response to peripheral immune signals; a functional anatomical analysis. Eur J Neurosci 2000;12:4434-46.
67. Luheshi GN, Bluthe RM, Rushforth D, Mulcahy N, Konsman JP, Goldbach M, Dantzer R. Vagotomy attenuates the behavioural but not the pyrogenic effects of interleukin-1 in rats. Auton Neurosci 2000;85:127-32.
68. Hansen MK, O Connor KA, Goehler LE, Watkins LR, Maier SF. The contribution of the vagus nerve in interleukin-1beta-induced fever is dependent on dose. Am J Physiol 2001;280:R929-34.
69. Dogan MD, Patel S, Rudaya AY, Steiner AA, Szekely M, Romanovsky AA. Lipopolysaccharide fever is initiated via a capsaicin-sensitive mechanism independent of the subtype-1 vanilloid receptor. Br J Pharmacol 2004;143:1023-32.
70. Niijima A. The afferent discharges from sensors for interleukin 1 beta in the hepatoportal system in the anesthetized rat. J Auton Nerv Syst 1996;61:287-91.
71. Kurosawa M, Uvnäs-Moberg K, Miyasaka K, Lundeberg T. Interleukin-1 increases activity of the gastric vagal afferent nerve partly via stimulation of type A CCK receptor in anesthetized rats. J Auton Nerv Syst 1997;62:72-8.
72. Ek M, Kurosawa M, Lundeberg T, Ericsson A. Activation of vagal afferents after intravenous injection of interleukin-1beta: role of endogenous prostaglandins. J Neurosci 1998;18:9471-9.
73. Goehler LE, Relton JK, Dripps D, Kiechle R, Tartaglia N, Maier SF, Watkins LR. Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Res Bull 1997;43:357-64.
74. Obreja O, Rathee PK, Lips KS, Distler C, Kress M. IL-1 beta potentiates heat-activated currents in rat sensory neurons: involvement of IL-1RI, tyrosine kinase, and protein kinase C. Faseb J 2002;16:1497-503.
75. Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, Samad TA. Nociceptors are interleukin-1beta sensors. J Neurosci 2008;28:14062-73.
76. Hosoi T, Okuma Y, Matsuda T, Nomura Y. Novel pathway for LPS-induced afferent vagus nerve activation: possible role of nodose ganglion. Auton Neurosci 2005;120:104-7.
77. Barajon I, Serrao G, Arnaboldi F, Opizzi E, Ripamonti G, Balsari A, Rumio C. Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J Histochem Cytochem 2009;57:1013-23.
78. Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, Wainger B, Strominger A, Muralidharan S, Horswill AR, Bubeck Wardenburg J, Hwang SW, Carroll MC, Woolf CJ. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013;501:52-7.
79. Reyes TM, Sawchenko PE. Involvement of the arcuate nucleus of the hypothalamus in interleukin-1-induced anorexia. J Neurosci 2002;22:5091-9.
80. Huang QH, Hruby VJ, Tatro JB. Role of central melanocortins in endotoxin-induced anorexia. Am J Physiol 1999;276:R864-71.
81. Whitaker KW, Reyes TM. Central blockade of melanocortin receptors attenuates the metabolic and locomotor responses to peripheral interleukin-1beta administration. Neuropharmacology 2008;54:509-20.
82. Wu Q, Clark MS, Palmiter RD. Deciphering a neuronal circuit that mediates appetite. Nature 2012;483:594-7.
83. Chaskiel L, Paul F, Gerstberger R, Hubschle T, Konsman JP. Brainstem metabotropic glutamate receptors reduce food intake and activate dorsal pontine and medullar structures after peripheral bacterial lipopolysaccharide administration. Neuropharmacology 2016;107:146-59.
84. Gray TS, Magnuson DJ. Peptide immunoreactive neurons in the amygdala and the bed nucleus of the stria terminalis project to the midbrain central gray in the rat. Peptides 1992;13:451-60.
85. Depaulis A, Keay KA, Bandler R. Quiescence and hyporeactivity evoked by activation of cell bodies in the ventrolateral midbrain periaqueductal gray of the rat. Exp Brain Res 1994;99:75-83.
86. Gaykema RP, Park SM, McKibbin CR, Goehler LE. Lipopolysaccharide suppresses activation of the tuberomammillary histaminergic system concomitant with behavior: a novel target of immune-sensory pathways. Neuroscience 2008;152:273-87.
87. Gaykema RP, Goehler LE. Ascending caudal medullary catecholamine pathways drive sickness-induced deficits in exploratory behavior: brain substrates for fatigue? Brain Behav Immun 2011;25:443-60.
88. Depino AM, Alonso M, Ferrari C, del Rey A, Anthony D, Besedovsky H, Medina JH, Pitossi F. Learning modulation by endogenous hippocampal IL-1: blockade of endogenous IL-1 facilitates memory formation. Hippocampus 2004;14:526-35.
89. Ben Menachem-Zidon O, Goshen I, Kreisel T, Ben Menahem Y, Reinhartz E, Ben Hur T, Yirmiya R. Intrahippocampal transplantation of transgenic neural precursor cells overexpressing interleukin-1 receptor antagonist blocks chronic isolation-induced impairment in memory and neurogenesis. Neuropsychopharmacology 2008;33:2251-62.
90. Bartynski WS, Boardman JF, Zeigler ZR, Shadduck RK, Lister J. Posterior reversible encephalopathy syndrome in infection, sepsis, and shock. Am J Neuroradiol 2006;27:2179-90.
91. Sharshar T, Carlier R, Bernard F, Guidoux C, Brouland JP, Nardi O, de la Grandmaison GL, Aboab J, Gray F, Menon D, Annane D. Brain lesions in septic shock: a magnetic resonance imaging study. Intensive Care Med 2007;33:798-806.
92. Polito A, Eischwald F, Maho AL, Polito A, Azabou E, Annane D, Chretien F, Stevens RD, Carlier R, Sharshar T. Pattern of brain injury in the acute setting of human septic shock. Crit Care 2013;17:R204.
93. Stubbs DJ, Yamamoto AK, Menon DK. Imaging in sepsis-associated encephalopathy--insights and opportunities. Nat Rev Neurol 2013;9:551-61.
94. Graham DI, Behan PO, More IA. Brain damage complicating septic shock: acute haemorrhagic leucoencephalitis as a complication of the generalised Shwartzman reaction. J Neurol Neurosurg Psychiatry 1979;42:19-28.
95. Sharshar T, Gray F, Poron F, Raphael JC, Gajdos P, Annane D. Multifocal necrotizing leukoencephalopathy in septic shock. Crit Care Med 2002;30:2371-5.
96. Granger JI, Ratti PL, Datta SC, Raymond RM, Opp MR. Sepsis-induced morbidity in mice: effects on body temperature, body weight, cage activity, social behavior and cytokines in brain. Psychoneuroendocrinology 2013;38:1047-57.
97. Michels M, Vieira AS, Vuolo F, Zapelini HG, Mendonca B, Mina F, Dominguini D, Steckert A, Schuck PF, Quevedo J, Petronilho F, Dal-Pizzol F. The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav Immun 2015;43:54-9.
98. Ji MH, Qiu LL, Tang H, Ju LS, Sun XR, Zhang H, Jia M, Zuo ZY, Shen JC, Yang JJ. Sepsis-induced selective parvalbumin interneuron phenotype loss and cognitive impairments may be mediated by NADPH oxidase 2 activation in mice. J Neuroinflammation 2015;12:182.
99. Singer BH, Newstead MW, Zeng X, Cooke CL, Thompson RC, Singer K, Ghantasala R, Parent JM, Murphy GG, Iwashyna TJ, Standiford TJ. Cecal Ligation and Puncture Results in Long-Term Central Nervous System Myeloid Inflammation. PLoS One 2016;11:e0149136.
100. Brooks HF, Osabutey CK, Moss RF, Andrews PL, Davies DC. Caecal ligation and puncture in the rat mimics the pathophysiological changes in human sepsis and causes multi-organ dysfunction. Metab Brain Dis 2007;22:353-73.
101. Brooks HF, Moss RF, Davies NA, Jalan R, Davies DC. Caecal ligation and puncture induced sepsis in the rat results in increased brain water content and perimicrovessel oedema. Metab Brain Dis 2014;29:837-43.
102. Hofer S, Bopp C, Hoerner C, Plaschke K, Faden RM, Martin E, Bardenheuer HJ, Weigand MA. Injury of the blood brain barrier and up-regulation of icam-1 in polymicrobial sepsis. J Surg Res 2008;146:276-81.
103. Kafa IM, Uysal M, Bakirci S, Ayberk Kurt M. Sepsis induces apoptotic cell death in different regions of the brain in a rat model of sepsis. Acta Neurobiol Exp (Wars) 2010;70:246-60.
104. Yokoo H, Chiba S, Tomita K, Takashina M, Sagara H, Yagisita S, Takano Y, Hattori Y. Neurodegenerative evidence in mice brains with cecal ligation and puncture-induced sepsis: preventive effect of the free radical scavenger edaravone. PLoS One 2012;7:e51539.
105. Hara N, Chijiiwa M, Yara M, Ishida Y, Ogiwara Y, Inazu M, Kuroda M, Karlsson M, Sjovall F, Elmer E, Uchino H. Metabolomic analyses of brain tissue in sepsis induced by cecal ligation reveal specific redox alterations--protective effects of the oxygen radical scavenger edaravone. Shock 2015;44:578-84.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Konsman JP. Immune-to-brain signaling and substrates of altered behavior during inflammation. Neurosciences 2016;3:207-14. http://dx.doi.org/10.20517/2347-8659.2016.19
AMA Style
Konsman JP. Immune-to-brain signaling and substrates of altered behavior during inflammation. Neuroimmunology and Neuroinflammation. 2016; 3: 207-14. http://dx.doi.org/10.20517/2347-8659.2016.19
Chicago/Turabian Style
Konsman, Jan Pieter. 2016. "Immune-to-brain signaling and substrates of altered behavior during inflammation" Neuroimmunology and Neuroinflammation. 3: 207-14. http://dx.doi.org/10.20517/2347-8659.2016.19
ACS Style
Konsman, JP. Immune-to-brain signaling and substrates of altered behavior during inflammation. Neurosciences. 2016, 3, 207-14. http://dx.doi.org/10.20517/2347-8659.2016.19
About This Article
Copyright

Data & Comments
Data


 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.