
Progressive muscle cramps with pain as atypical initial presentations of amyotrophic lateral sclerosis: a case report


Abstract
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease and is a progressive and devastating neurodegenerative disease that affects both lower and upper motor neurons. Muscle cramps, which are characterized by a sudden, painful, involuntary contraction of muscles, are not rare in ALS patients. However, muscle cramps do not normally present early in ALS and therefore not used for the initial diagnosis of ALS. In this paper the authors present a case of ALS with initial manifestation of progressive painful muscle cramps in the absence of muscle weakness. This case might help people to recognize atypical foremost presentations of ALS and therefore formulate effective therapies.
Keywords
Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive, fatal neurodegenerative disorder.[1,2] The worldwide incidence of ALS is around 1.7-2.3% per 100,000 population per year and the average age of onset is 61.8 and 49.8 for Western countries and China individually. The occurrence rate of men is higher than women.[3-6] The pathogenesis of motor neuron disease (MND) remains largely unclear, genetic factors and various exogenous risk factors are suggested to be associated with MND, including rural residency, alcohol consumption, smoking, toxic substance, glutamate metabolism disorder and aberrant autoimmunity.[4-6]
Typical clinical presentations of ALS include asymmetric weakness of limbs (60-80%), bulbar symptoms (20%), respiratory muscle weakness (1-3%), generalized weakness in limbs and bulbar muscles (1-9%), axial onset with muscle weakness, muscle atrophy and fasciculations.[7,8] Muscle cramping with pain presents in 7-12% of ALS patients.[9] Muscle cramps, are not rare in ALS patients, but rarely act as initial symptom without muscle weakness of the ALS patients. Some studies reported that muscle cramps could appear during the early phase or prodromal phase of ALS, and muscle cramps could help in the early diagnosis of ALS.[10,11] In this paper we present a rare case with initial manifestation of progressive painful muscle cramps in the absence of obvious muscle weakness.
Case report
A 56-year-old Chinese woman presented with progressive limb muscle cramps with pain and walk difficulty for the last 7 months. She had no special medical history before. At the beginning, the muscle cramps started in her left lower limb, 4 months after the onset, muscle cramps were aggravated, and slowly progressed to her right lower limb and left upper limb in turn. Muscle cramps could be induced by touching her skin of limbs and chest. Muscle cramps frequently broke out during sleeping, and could be alleviated slightly by standing. Stiffness and weakness developed in the lower limbs for 4 months after symptom onset. Fasciculations, combined with tight feeling of the hips, was seen in both lower extremities and left upper limb. Then she had difficulty in walking, button-up her clothes and bradykinesia. There was no numbness and muscle atrophy of her tongue, face, hands and limbs. She had no complaints of dysphagia, dysarthria or dyspnea and history of alcohol drinking or cigarette smoking, no family history of neurological diseases.
Physical examination revealed a normal body figure, well-oriented in place, time and person, with no unconsciousness. Cranial nerve examination revealed no abnormalities. Examination of limbs revealed spastic rigidity in both lower limbs, with 4/5 muscle power in both lower limbs, 4/5 muscle power in left upper limb, and 4/5 to 5/5 muscle power in right upper limb. There was no atrophy of the limbs, trunk and lingual muscles. Sensations and cerebellar examinations were unremarkable. Deep tendon reflexes in the 4 limbs were asymmetrical that left limbs were hyperactive and right limbs brisk. Hoffman’s reflexes and patellar clonus were negative. Left ankle clonus was positive. Babinski’s signs were presented in both sides. For laboratory examinations, blood cell count, urine analyses, liver function, renal function, muscle enzymes (creatine kinase, creatine kinase-MB, aspartate aminotransferase, lactate dehydrogenase, hydroxybutyrate dehydrogenase), serum vitamin B12, autoantibodies and tumor biomarkers were normal. Intracranial pressure and cerebral spinal fluid analysis were normal. Brain and spine magnetic resonance imaging (MRI) suggest Arnold-Chiari malformation type I [Figure 1A], which was not explainable for the symptoms and abnormalities of physical examinations. Spine MRI revealed slight prominence of intervertebral disks and no abnormality was observed in the whole spinal cord [Figure 1A]. Electromyography (EMG) showed neurogenic changes in the left biceps brachii, the left thenar muscles and bilateral gastrocnemius [Figure 1B-D]. Evoked potentials examination showed prolonged latency period in the left side of the visual evoked potential and event-related potential-P300. Motor evoked potential, somatosensoroy evoked potential and brainstem auditory evoked potential, nerve conduction, ambulatory electroencephalogram and electrocardiogram were normal.
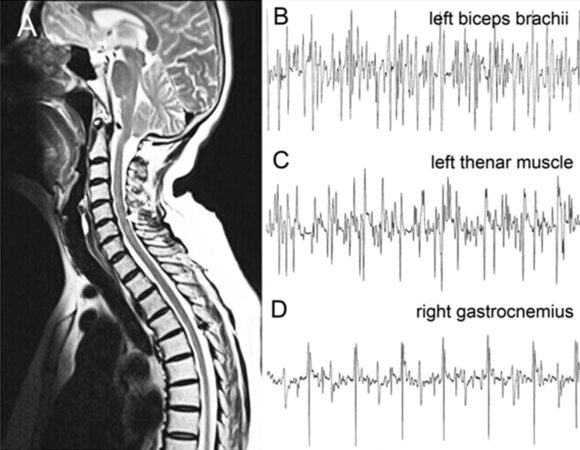
Figure 1. Brain and spine image and electromyography of the ALS case. (A) Brain MRI suggests Arnold-Chiari malformation type I (arrow). Spine MRI reveals slight prominence of intervertebral disks at C4/5, C6/7 and L4/5-L5/S1, and no abnormality was observed in the whole spinal cord. (B-D) Electromyography showed denervation potentials in the left biceps brachii (B), the left thenar muscle (C) and right gastrocnemius (D). ALS: amyotrophic lateral sclerosis; MRI: magnetic resonance imaging
According to the revised 2,000 criteria,[12] the patient was diagnosed with clinically probable ALS, which was confirmed by other hospitals in Beijing. The oral administration of diazepam and baclofen was initiated; muscle cramps were relieved a bit. The patient did periodic follow-ups in our outpatient clinics, but her symptoms did not improve with the use of the muscle-relaxants prescribed.
Discussion
ALS has many varied manifestations as first symptoms, which are vital to help for early diagnosis, and understand the natural course the disease progression.[13] Atypical presentations include cramps and fasciculations in the absence of muscle weakness,[14] frontal-temporal dementia,[15] weight loss[16] and cardiovascular consequences.[17] Clinical characteristics of our case include the progression of muscular painful cramps with upper neurons signs, limb weakness with lower motor neuron signs and electrodiagnostic (EMG/nerve conduction velocity) evidence of denervation. Besides MNDs, other etiology of muscle cramps includes metabolic disorders, electrolyte disturbances, medications, etc.[14] Our patient had no special medical history and laboratory examinations which support the other disease. As to pathophysiology, the muscle cramps may arise from spontaneous discharges of the motor nerves, this may origin at neuronal level and then transferred through the nerve trunks to the muscular fibers or it may begin at nerve trunks level by activation of abnormal cholinergic receptors that function as trigger points.[18] The neurodegeration in ALS might be caused by a complicated interaction of glutamate excitotoxicity, generation of free radicals, superoxide dismutase 1 enzymes, resulting in impaired axonal structure or transport defects.[2,14] A "dying-forward" process is proposed to explain these clinical characteristics.[19]
Hitherto, there is no disease-modifying therapeutics for ALS. The main managements for patients with ALS are the symptomatic treatments, including muscle relaxants, anticonvulsants for cramps, fasciculations and spasticity, physiotherapy for weakness or disability, and ventilator support for dyspnea,[2,14] These treatments can alleviate symptoms, improve the quality of life and increase the life expectancy.
In conclusion, our case presents a rare initial manifestation of progressive painful muscle cramps in the absence of muscle weakness. It helps to improve our early recognition of the atypical initial presentations of ALS, and formulate effective symptomatic therapies to improve the life quality and survival of the patients.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ, Millul A, Benn E, Beghi E, EURALS. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry 2010;81:385-90.
2. Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 2015;6:171.
3. Su XW, Broach JR, Connor JR, Gerhard GS, Simmons Z. Genetic heterogeneity of amyotrophic lateral sclerosis: implications for clinical practice and research. Muscle Nerve 2014;49:786-803.
4. Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, White LA. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 2013;41:118-30.
5. Chen L, Zhang B, Chen R, Tang L, Liu R, Yang Y, Yang Y, Liu X, Ye S, Zhan S, Fan D. Natural history and clinical features of sporadic amyotrophic lateral sclerosis in China. J Neurol Neurosurg Psychiatry 2015;86:1075-81.
6. Huynh W, Kiernan MC. A unique account of ALS in China: exploring ethnic heterogeneity. J Neurol Neurosurg Psychiatry 2015;86:1051-2.
7. Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013;14:248-64.
8. Shoesmith CL, Findlater K, Rowe A, Strong MJ. Prognosis of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry 2007;78:629-31.
9. Nalini A, Thennarasu K, Gourie-Devi M, Shenoy S, Kulshreshtha D. Clinical characteristics and survival pattern of 1,153 patients with amyotrophic lateral sclerosis: experience over 30 years from India. J Neurol Sci 2008;272:60-70.
10. Caress JB, Ciarlone SL, Sullivan EA, Griffin LP, Cartwright MS. Natural history of muscle cramps in amyotrophic lateral sclerosis. Muscle Nerve 2015;53:513-7.
11. Ioannides ZA, Henderson RD, Robertson T, Davis M, McCombe PA. When does ALS start? A novel SOD-1 p.Gly142Arg mutation causing motor neurone disease with prominent premorbid cramps and spasms. J Neurol Neurosurg Psychiatry 2015; doi: 10.1136/jnnp-2015-311582.
12. Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293-9.
13. Ringel SP, Murphy JR, Alderson MK, Bryan W, England JD, Miller RG, Petajan JH, Smith SA, Roelofs RI, Ziter F. The natural history of amyotrophic lateral sclerosis. Neurology 1993;43:1316-22.
14. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet 2011;377:942-55.
15. Ferguson TA, Elman LB. Clinical presentation and diagnosis of amyotrophic lateral sclerosis. NeuroRehabilitation 2007;22:409-16.
16. Moirangthem V, Ouseph MM. Atypical presentations of amyotrophic lateral sclerosis: a case report. J Neuropsychiatry Clin Neurosci 2011;23:362-4.
17. Shemisa K, Kaelber D, Parikh SA, Mackall JA. Autonomic etiology of heart block in amyotrophic lateral sclerosis: a case report. J Med Case Rep 2014;8:224.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Liu CH, Zhu C, Zeng F, Yang H, Wang YJ. Progressive muscle cramps with pain as atypical initial presentations of amyotrophic lateral sclerosis: a case report. Neurosciences 2016;3:170-2. http://dx.doi.org/10.20517/2347-8659.2015.49
AMA Style
Liu CH, Zhu C, Zeng F, Yang H, Wang YJ. Progressive muscle cramps with pain as atypical initial presentations of amyotrophic lateral sclerosis: a case report. Neuroimmunology and Neuroinflammation. 2016; 3: 170-2. http://dx.doi.org/10.20517/2347-8659.2015.49
Chicago/Turabian Style
Liu, Cheng-Hui, Chi Zhu, Fan Zeng, Heng Yang, Yan-Jiang Wang. 2016. "Progressive muscle cramps with pain as atypical initial presentations of amyotrophic lateral sclerosis: a case report" Neuroimmunology and Neuroinflammation. 3: 170-2. http://dx.doi.org/10.20517/2347-8659.2015.49
ACS Style
Liu, C.H.; Zhu C.; Zeng F.; Yang H.; Wang Y.J. Progressive muscle cramps with pain as atypical initial presentations of amyotrophic lateral sclerosis: a case report. Neurosciences. 2016, 3, 170-2. http://dx.doi.org/10.20517/2347-8659.2015.49
About This Article
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 39
likes
Like This Article 39
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.