
Huge supratentorial cortical ependymoma in a young child: case report and literature review

Abstract
Supratentorial cortical ependymomas are uncommon in the pediatric population and extremely rare in very young children. Histologically, tumors of the anaplastic type are also less common in children. The authors report one case of anaplastic cortical ependymoma in a 16-month-old girl who presented with a 7-day history of left side weakness and rapid neurological deterioration. Brain imaging with computed tomography and magnetic resonance imaging scanning showed a huge right fronto-parietal cystic and solid lesion compressing the brain parenchyma. The young child was operated via a transparietal approach with gross total resection of the lesion. The tumor’s histology was anaplastic ependymoma. Intensive chemotherapy was given post operatively and the patient remained well without recurrence after 20 months of follow-up.
Keywords
Introduction
Ependymomas are rare neuroectodermal tumors arising from ependymal cells of the ventricular system, choroid plexus, filum terminale or the central canal of the spinal cord. They account for 1.2-7.8% of all intracranial neoplasms.[1-3] Pure supratentorial cortical ependymomas (CE) are uncommon. To our knowledge, 49 cases of CE have been reported, of which 16 involved pediatric patients, with only 4 occurring in very young children (less than 3 years).[1-5] Here we report the 5th case of CE in very young child, treated surgically and with chemotherapy with a good outcome at 20 months of follow-up.
Case report
A 16-month-old female, with normal development milestones and without other past medical history, was admitted to our department of neurosurgery after presenting with 7 days of left-sided weakness. At initial neurological examination, the young child was conscious, had a left hemiparesis mainly affecting the upper limb, a normal head circumference for age and no papilledema. Brain computed tomography (CT) scanning revealed a huge right fronto-parietal cystic-solid lesion mesearing 76 mm × 70 mm × 70 mm. It was slightly hyperdense in its solid component, with thin calcifications and was well-demarcated from brain parenchyma [Figure 1]. During hospitalization, the patient had a focal seizure followed by rapid deterioration in consciousness and right pupillary dilatation. The patient was transferred immediately to the operating theatre and underwent in emergency cyst puncture with solid component biopsy. The outcome was good, with improvement in her clinical state and resolution of the hemiparesis.
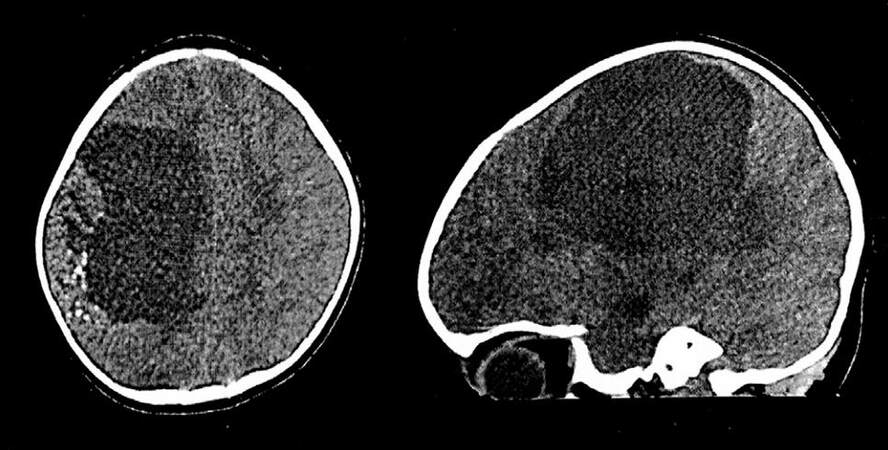
Figure 1. Preoperative brain computed tomography scan without contrast; axial view and sagittal reconstruction, showing a huge right fronto-parietal cyst and a slightly hyperdense cortical solid lesion with thin calcifications
The brain magnetic resonance imaging (MRI) performed after the first surgery revealed a large cortical fronto-parietal lesion, hypointense on T1, hyperintense on T2, markedly enhanced with contrast, with moderate surrounding edema [Figure 2]. The tumor was totally resected in an elective procedure using a right transparietal approach. The postoperative course was uneventful, without complications. Histological examination of the resected tumor showed perivascular pseudo rosettes of small round cells with mitosis (5 mitosis/10 fields) and necrosis. Immunohistochemical studies showed positivity to glial fibrillary acidic protein (GFAP), poly styrene 100, and keratin, but not to synaptophysin. Kalium iodidum (Ki)-67 was positive in 35%. These findings are in keeping with an anaplastic ependymoma, World Health Organization (WHO) classification grade III [Figure 3]. Cerebrospinal fluid studies were negative for malignant cells and no drop metastases were detected on neuroaxis MRI. Intensive chemotherapy with BBSFOPP protocol consisting of seven cycles of 3 courses alternating 2 drugs at each course (carboplatin/procarbazine, cisplatin/etoposide and vincristine/cyclophosphamide) was given post operatively for 16-month period. After 20 months of follow up, the patient remained neurologically normal without recurrence or metastasis on surveillance MRI [Figure 4].
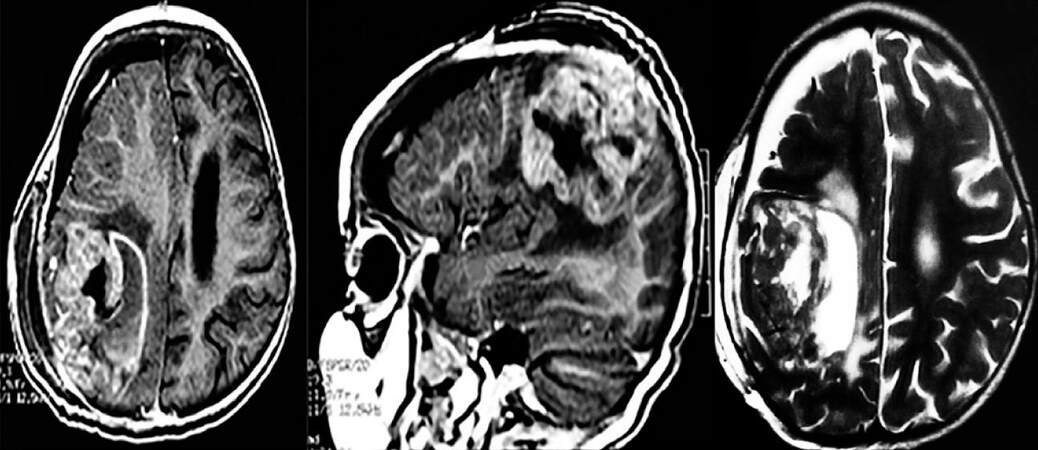
Figure 2. Post puncture magnetic resonance imaging (axial and sagittal T1 post gadolinium and axial T2 weighted images) showing a huge right parietal tumor, enhancing with contrast and with moderate peritumoral edema
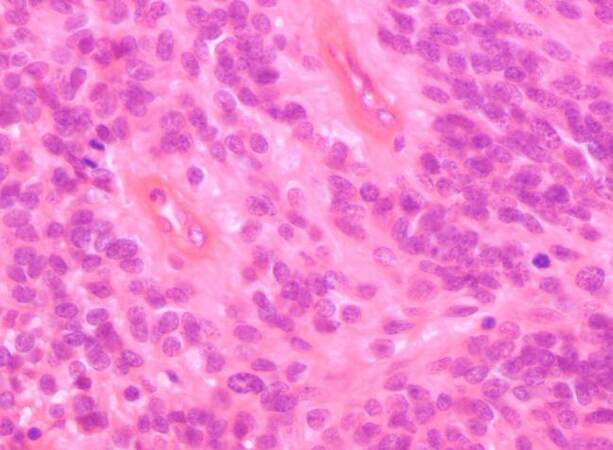
Figure 3. Photomicrograph of the tumor specimen showing marked hypercellularity, nuclear atypia and brisk mitotic activity. (HE, X40)
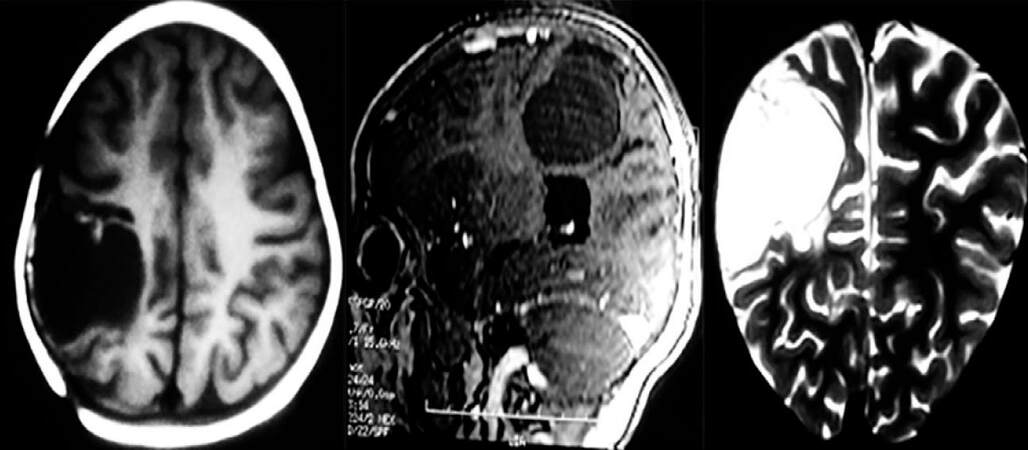
Figure 4. Follow-up magnetic resonance imaging at 20 months (axial T1, sagittal T1 with gadolinium and coronal T2 weighted images) showing complete removal of the lesion without recurrence
Discussion
Ependymomas account for 2-9% of all neuroepithelial tumors, and are graded as grade II (low grade), and grade III (anaplastic) according to 2007 WHO classification. They involve frequently the spinal cord and ventricular system, especially the fourth ventricle, and they commonly occur in children and young adults.[1]
Supratentorial CE is an uncommon ependymoma located in the superficial cortex and more often found in adults than children. There are only 49 cases reported in the literature.[1-5]
Ependymomas are known to arise from ependymal cells. The pathogenesis of the extraventricular tumor type remains uncertain. Pure cortical ependymomas may arise from embryonic remnants of ependymal tissue encapsulated in the developing cerebral hemispheres.[2]
CEs are rare in pediatric patients and exceedingly uncommon in very young children.
Among the 49 cases reported in the literature, 16 involved children and only 4 occurred in patients under the age of 3 years.[1,2,4,6]
Our patient is the 5th case reported of CE in those under 3 years of age.
The age of these 5 young children ranged from 12-24 months, with a mean age of 19.4 months. There was male preponderance with male to female ratio of 3:2 [Table 1].
The summary of previous reported young children supratentorial cortical ependymoma with our case.
| References | Age (months)/gender | Location | Clinical presentation | Neuroimaging finding | WHO grade | Treatment | Follow-up |
|---|---|---|---|---|---|---|---|
| Lehman et al.[4] | 12/F | Rt frontal | Seizures | Solid lesion not enhanced | III [clear cells] | GTR | No recurrence in 48 months |
| Lee et al.[2] | 21/M | Rt fronto-parietal | Seizures | Diffuse enhancing mass, some focal calcifications | II | GTR | No recurrence in 12 months |
| Liu et al.[1] | 24/M | Lt frontal | Right side weakness | Large mass, irregular peripheral enhancement no edema | III | GTR | Recur at 3 years after surgery alive at 4 years |
| Kambe et al.[6] | 24/M | Rt parietal | Seizures | Solid mass, calcification homogeneous enhancement | II [tanycytic] | GTR | No recurrence in 20 months |
| Our case | 16/F | Rt fronto-parietal | Left side weakness, seizures, ICH signs | Large solid cystic lesion heterogeneous enhancement | III | GTR | No recurrence in 20 months |
The typical presentation of CE was with seizures and focal neurological deficits, and rarely with signs of raised intracranial pressure.[1,2,7]
Our patient presented with left hemiparesis, a seizure, and rapid deterioration with signs of intracranial hypertension, whereas the others 4 young children reported, presented either with seizures or with hemiparesis [Table 1].
Intracranial ependymomas appear on brain CT scans as isodense or mildly hyperdense soft tissue lesions with frequent large cyst, calcifications in 50%, hemorrhage in approximately 10% and, often, heterogeneous enhancement.[8]
MRI is the brain imaging modality of choice for ependymoma. CE generally appears as large, well-demarcated lesions, T1 hypointense and T2 hyperintense, showing cyst formation. After gadolinium administration, T1 weighted images usually show heterogeneous enhancement of the solid component.[8] Frontal and parietal region are the most common locations.[8]
These radiologic findings are, however, non-specific and glioblastomas, pleomorphic xanthoastrocytomas, oligodendrogliomas, primitive neuroectodermal tumors, astroblastomas and angiocentric gliomas should be considered in the differential diagnosis of a large supratentorial cortical lesion.[1,8-11]
Histologically, the classic cellular features of ependymomas include round to oval nuclei with evenly dispersed stippled chromatin, perivascular pseudo rosettes or true ependymal rosettes. Unusual morphological features like clear cells, spindle cells and giant cells can also be seen.[1,4]
Anaplastic ependymomas are characterized by increased cellularity, cytological atypia, increased mitotic activity, microvascular proliferation and/or pseudopalissading tumor necrosis. There is normally a clear interface between tumor and adjacent brain tissue and relative uniformity of tumor cell nuclei.[1,4] However, some authors have described ependymomas, that infiltrate at their peripheries.[4]
Immunohistochemical studies revealed frequent immunoreactivity for GFAP, S100, a perinuclear dot-like or ring-like positivity for epithelial membrane antigen and/or membranous with or without perinuclear dot-like staining pattern for cluster of differentiation 99.[1,4]
Immunohistochemical cell proliferation markers, that have some specificity for ependymomas are available. MIB-1 L1 and Ki-67 are associated with high-grade ependymomas, as in our case. Recently, further markers have been found, notably topo-II-α and p53 and murine double min 2 protein expression which are correlated with high-grade tumors and a poor prognosis.[12]
Although nearly 70% of all ependymomas diagnosed in the pediatric population are histologically benign, CEs are more frequently anaplastic in young children (3/5) [Table 1].
Given the superficial location, radical resection is the treatment of choice for CE.
There has been much debate about adjuvant radiotherapy and chemotherapy.
Radiotherapy was thought to increase the length of survival and reduce or postpone tumor recurrence. It is used in patients with anaplastic ependymomas in cases of partial resection of either benign or malignant tumor.[13,14] Whole brain irradiation with additional local fractions is recommended by many authors in malignant ependymoma.[14]
In very young children, delayed radiotherapy and a period of close observation after gross total resection may be chosen for some cases to defer the side-effects of radiotherapy.[4,6,13,15]
Chemotherapy has mainly been studied in children with incomplete tumor resections and multiple agents have been given in an effort to delay or avoid irradiation. These studies suggest that chemotherapy offers limited benefits.[15]
The 4 patients previously reported had gross total resection without adjuvant therapy, whereas our patient had intensive postoperative chemotherapy.
Recurrences of CE are less frequently seen than for the intraventricular type. Among the 5 young children reported, recurrence only occurred in one and at 3 years [Table 1].
CE appear to have a relatively favorable prognosis compared with other supratentorial ependymomas.[7,13] All the young children reported, including our case, are alive after a mean follow-up of 29.6 months.
We believe that successful gross total resection of CE is the best prognostic factor for long-term survival with both WHO grade II and grade III lesions.
In conclusion, supratentorial CE is a very rare tumor type that should be considered in the differential diagnosis of a large cortical lesion in very young children.
Gross total resection is the treatment of choice, which can afford good prognosis with long-term survival.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Liu Z, Li J, Liu Z, Wang Q, Famer P, Mehta A, Chalif D, Wang Y, Li JY. Supratentorial cortical ependymoma: case series and review of the literature. Neuropathology 2014;34:243-52.
2. Lee SK, Lim DJ, Kim SD. Supratentorial cortical ependymoma in a 21-month-old boy. J Korean Neurosurg Soc 2011;50:244-7.
3. Yurt A, Selcuki M, Ertürk AR, Kuplioglu A. Large supratentorial cortical ependymoma in a child. Clin Med Res 2010;1:25-7.
4. Lehman NL. Patterns of brain infiltration and secondary structure formation in supratentorial ependymal tumors. J Neuropathol Exp Neurol 2008;67:900-10.
5. Roncaroli F, Consales A, Fioravanti A, Cenacchi G. Supratentorial cortical ependymoma: report of three cases. Neurosurgery 2005;57:E192.
6. Kambe A, Kurosaki M, Watanabe T, Nakazato Y. Pediatric supratentorial cortical tanycytic ependymoma associated with absence seizures. Clin Neuropathol 2014;33:308-10.
7. Van Gompel JJ, Koeller KK, Meyer FB, Marsh WR, Burger PC, Roncaroli F, Worrell GA, Giannini C. Cortical ependymoma: an unusual epileptogenic lesion. J Neurosurg 2011;114:1187-94.
8. Yuh EI, Barkovich AJ, Gupta N. Imaging of ependymomas: MRI and CT. Childs Nerv Syst 2009;25:1203-13.
9. Ng DW, King NK, Foo AS, Sitoh YY, Lee HY, Ng WH. Anaplastic supratentorial cortical ependymoma presenting as a butterfly lesion. Surg Neurol Int 2012;3:107.
10. Tailor J, Jaunmuktane Z, Brandner S, Sethi H. Supratentorial ependymoma presenting as a cortical cyst with a mural nodule in an adult. J Surg Case Rep 2015; doi: 10.1093/jscr/rju124.
11. Mohaghegh MR, Chitsaz A, Okhovat AA, Pour EB. Supratentorial cortical ependymoma: an unusual presentation of a rare tumor. Adv Biomed Res 2015;4:72.
12. Sharma MC, Ghara N, Jain D, Sarkar C, Singh M, Mehta VS. A study of proliferative markers and tumor suppressor gene proteins in different grades of ependymomas. Neuropathology 2009;29:148-55.
13. Cage TA, Clark AJ, Aranda D, Gupta N, Sun PP, Parsa AT, Auguste KI. A systematic review of treatment outcomes in pediatric patients with intracranial ependymomas. J Neurosurg Pediatr 2013;11:673-81.
14. Pang KY, Wai-Shing Ho W, Wong CK. Supratentorial extraventricular ependymoma: Literature review and case report. Ann Coll Surg HK 2002;6:22-4.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Darmoul M, Kilani M, Ben Nsir A, Hattab MN. Huge supratentorial cortical ependymoma in a young child: case report and literature review. Neurosciences 2016;3:141-4. http://dx.doi.org/10.20517/2347-8659.2015.53
AMA Style
Darmoul M, Kilani M, Ben Nsir A, Hattab MN. Huge supratentorial cortical ependymoma in a young child: case report and literature review. Neuroimmunology and Neuroinflammation. 2016; 3: 141-4. http://dx.doi.org/10.20517/2347-8659.2015.53
Chicago/Turabian Style
Darmoul, Mehdi, Mohamed Kilani, Atef Ben Nsir, Mohamed Nejib Hattab. 2016. "Huge supratentorial cortical ependymoma in a young child: case report and literature review" Neuroimmunology and Neuroinflammation. 3: 141-4. http://dx.doi.org/10.20517/2347-8659.2015.53
ACS Style
Darmoul, M.; Kilani M.; Ben Nsir A.; Hattab MN. Huge supratentorial cortical ependymoma in a young child: case report and literature review. Neurosciences. 2016, 3, 141-4. http://dx.doi.org/10.20517/2347-8659.2015.53
About This Article
Copyright

Data & Comments
Data


 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 29
likes
Like This Article 29
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.