
A case of Hashimoto's encephalopathy presenting with seizures and cognitive impairment

Abstract
Hashimoto's encephalopathy (HE) is a rare disease with unknown pathogenesis. An epileptic seizure is reported in association with HE. Here, the author reported an 18-year-old girl with a history of hyperthyroidism for one year. She was admitted to the hospital due to status epilepticus. Serum thyroid function test showed that the concentration of anti-thyroid peroxidase antibodies and thyroglobulin antibody were significantly elevated. Brain magnetic resonance imaging showed that multiple abnormalities varied from bilateral frontal, parietal, occipital-temporal lobe to cerebellum hemisphere. The patient’s symptoms were significantly relieved after methylprednisolone therapy. At 3-month follow-up visits, she had been symptom free. HE is a diagnosis of exclusion and should be considered when evaluating a patient with cognitive dysfunction and high titers of anti-thyroid antibodies as it responds dramatically to steroids.
Keywords
Introduction
Hashimoto’s encephalopathy (HE), also termed as "steroid-responsive encephalopathy associated with autoimmune thyroiditis", is a rare immune-mediated encephalopathic event which affects children and adolescents. It is characterized by altered mental status, seizures, and cognitive dysfunction. It was reported seizures happened in 66% of HE, among which status epilepticus in 12%.[1] We reported a young female patient with generalized, tonic-clonic seizures and cognitive impairment changes who responded well to steroid treatment.
Case report
An 18-year-old girl who suffered from status epilepticus was admitted to the hospital. She had a history of hyperthyroidism for one year, and was treated with propylthiouracil (50 mg/day). There was no family history of psychiatric diseases, seizures, or other problems. Her initial vital signs were stable. On physical examination, she was conscious, cooperative, and oriented to person and time. However, she had short-term memory and computing power loss [the mini-mental state examination (MMSE) score was 22, which was lower than normal] as well as bilateral limbs dystaxia. The white blood cell count was 7.55 × 109/L, hemoglobin 156 g/L, and C-reactive protein 0.09 mg/dL. Routine biochemical analyses of liver, renal, and blood glucose were all within normal limits. The serum sodium was 131 mmol/L, potassium 3.3 mmol/L, and chlorine 91 mmol/L. The cerebrospinal fluid (CSF) analysis displayed normal pressure (145 mm H2O) and a normal cell count and protein and glucose levels. The serum and CSF of TORCH [toxoplasmosis, other (viruses), rubella cytomegalovirus, herpes (simplex viruses)] as well as the serum rapid plasma regain test, anti-human immunodeficiency virus antibody, epatitis B surface antigen and antineutrophil cytoplasmic antibody were negative. The plasma concentrations of sex hormone and cortisol were normal. Serum thyroid function test showed that triiodothyronine (T3) < 0.300 nmol/L(reference intervals 1.30-3.10 nmol/L), free triiodothyronine (FT3) < 0.400 pmol/L (reference intervals 3.10-6.80 pmol/L), upersensitive thyroidstimulating hormone (S-TSH) 0.01 mIU/L (reference intervals 0.27-4.20 mIU/L), anti-thyroid peroxidase antibodies (TPOAb) > 600.0 IU/mL (reference intervals 0.00-34.00 IU/L), thyroglobulin antibody (TGAb) 2,189.00 IU/mL (reference intervals 10.00-115.00 IU/mL). Electroencephalography showed a marked slowing of background rhythm as an indicator of encephalopathy but no activity corresponded with seizures. The color Doppler ultrasound examination of thyroid and abdomen was normal. Results of computed tomography scan of the head were unremarkable. Brain magnetic resonance images (MRI) showed that multiple abnormalities varied from bilateral frontal, parietal, occipital-temporal lobe to cerebellum hemisphere on axial T2-weighted images (T2WI) [Figure 1A]. The anticonvulsant, Tegretol (100 mg bid) was given to control the seizures. Given the indications for HE, the patient was firstly treated with intravenous injection of Dexamethasone 10 mg/day for 7 days, followed by 5 mg/day for 7 days. Finally, she was given with oral methylprednisolone 12 mg/day. Her symptoms improved siginificantly and neuropsychiatric symptoms fully resolved. Retest brain MRI showed that initial multiple abnormalities disappeared compared with initial brain MRI [Figure 1B]. Serum thyroid function test showed that T3 1.22 nmol/L, FT3 2.65 pmol/L, S-TSH 4.17 mIU/L, TPOAb 369.80 IU/mL, TGAb 1,102.00 IU/mL. On the 17th day, she was discharged on tapered doses of oral methylprednisolone. On 3-month follow-up visits as an outpatient, she had been symptom free without any seizure and MMSE score was 30.
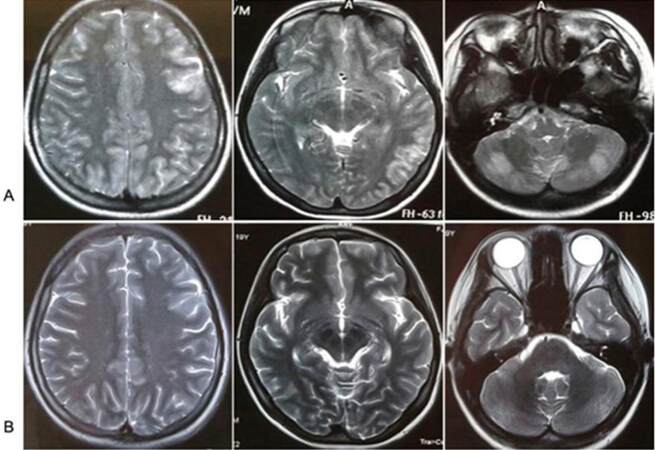
Figure 1. Brain magnetic resonance images (MRI) showed that multiple abnormalities varied from bilateral frontal, parietal, occipital-temporal lobe to cerebellum hemisphere on axial T2-weighted images (T2WI). Multiple abnormalities disappeared after steroids therapy. (A) MRI of the brain obtained before steroids therapy; (B) MRI of the brain obtained after steroids therapy
Discussion
As a rare steroid responsive neuropsychiatric syndrome, HE is associated with the serologic evidence of anti-thyroid antibodies when other causes of encephalopathy are excluded. The clinical manifestations of HE include cognitive impairment, various types of epileptic seizures, dystaxia and tremor, sleep disturbance and headache.[2] In this case, this female patient suffered from four generalized, tonic-clonic seizures and mild cognitive impairment as well as limbs dystaxia. Recently, HE has received extensive attention due to its treatability and unclear pathogenesis. In China, Hashimoto’s encephalopathy is still not fully recognized because of its complex clinical manifestions and absence of specific biomarkers. The previous research demonstrated that cognitive impairment (84.6%) and psychiatric symptoms (38.5%) were the most frequent symptoms, however, seizures (30.8%) and myoclonus (7.7%) were relatively infrequent in thirteen consecutive patients with HE.[3] Therefore, presenting symptoms of HE may be quite variable.
The diagnosis of HE should be considered in patients presenting with the characteristic neuropsychiatric manifestations excluding other causes of encephalopathy. Generally, high levels of anti-thyroid antibodies in serum or CSF are important and helpful in the diagnosis of HE. They have no alteration in the CSF and/or imaging tests compatible with infectious, vascular, or neoplastic etiology, and response well to immunosuppressive therapy.[4] Non-specific electroencephalogram abnormalities are presented in the vast majority of patients, and brain MRI may display abnormalities in 49%, such as cerebral atrophy, focal cortical abnormality, diffuse subcortical abnormality and non-specific subcortical focal white matter abnormality.[5] In this female patient, the analysis of CSF as well as serum inflammation biomarkers was normal, indicating the exclusion of intracranial infection. Although the elevated CSF protein is common in HE, this change depends on the severity of the illness. Her brain MRI showed that multiple abnormalities varied from bilateral frontal, parietal, occipital-temporal lobe to cerebellum hemisphere. Neuroimaging results have no reliable diagnostic value in HE.
In the context of the typical clinical picture, high titres of antithyroid antibodies, in particular TPOAb, are diagnostic.[6] Recently, Blanchin et al.[7] reported that TPOAb from Hashimoto’s encephalopathy patients could bind cerebellar astrocytes in HE patients but not in Hashimoto thyroiditis patients. This may support the role of TPOAb in the pathogenesis of Hashimoto’s encephalopathy. TPOAb is present in 95-100% and TGAb in 73% of patients with HE. Elevated serum level of TPOAb may be related with vasculitic type Hashimoto’s encephalopathy and elevated serum levels of TPOAb and TGAb may be with diffuse progressive type of HE. However, the elevated titres of these antibodies can be tested in the healthy population. Therefore, the role of those antibodies and their pathophysiology are unknown. In addition, corticosteroid treatment is successful in most cases, which can further support the diagnosis of HE. Furthermore, other common causes of encephalopathy should be ruled out, such as intracranial infection, metabolic disease, electrolyte imbalance, poisoning or toxins, neoplasm, and the central nervous system involvement of vasculitic syndromes. With the aid of a detailed medical history and related auxiliary examination (such as CSF and MRI), it is easy to rule out these diseases.
Regarding treatment for HE, the patient’s symptom improved significantly and rapidly after initiation of corticosteroid treatment, and eventually achieved a long-term stable remission. Clinical improvement with corticosteroid therapy is usually observed in the first 4-6 weeks. This positive response has been considered to be part of the definition of HE, but does not occur in all patients.[8] Other therapies such as plasmapheresis and immunosuppressant medications have been successfully used in patients non-responsive to corticosteroids. It was reported that only a few HE patients have been treated with Intravenous immunoglobulins.[9] Moreover, since the antithyroid antibodies could not been used as relapse markers of encephalopathys, the question of the continuation of the immunomodulatory of immunosuppressive drugs remains an open debate.[10]
In conclusion, HE is a rare disease associated with encephalopathy and autoimmune thyroiditis. Our research suggests that a high degree of suspicion is necessary to diagnose HE, especially in those patients with high levels of antithyroid antibodies and presented with unexplained encephalopathy, such as seizures and cognitive dysfunction, as in our case. Corticosteroid treatment is successful for HE in most cases, however, clinicians should be aware that relapses can occur early or even late after tapering of steroid use; therefore, a long follow-up period should be recommended.
Financial support and sponsorship
This study was supported by the grants from the Fujian Province Natural Science Fund (2013J01275, 2014J01401).
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Chong JY, Rowland LP, Utiger RD. Hashimoto encephalopathy: syndrome or myth? Arch Neurol 2003;60:164-71.
2. Castillo P, Woodruff B, Caselli R, Vernino S, Lucchinetti C, Swanson J, Noseworthy J, Aksamit A, Carter J, Sirven J, Hunder G, Fatourechi V, Mokri B, Drubach D, Pittock S, Lennon V, Boeve B. Steroid-responsive encephalopathy associated with autoimmune thyroiditis. Arch Neurol 2006;63:197-202.
3. Wang J, Zhang J, Xu L, Shi Y, Wu X, Guo Q. Cognitive impairments in Hashimoto's encephalopathy: a case-control study. PLos One 2013;8:e55758.
4. Philip R, Saran S, Gutch M, Gupta K. An unusual presentation of Hashimoto's encephalopathy. Indian J Endocrinol Metab 2014;18:113-5.
5. Kothbauer-Margreiter I, Sturzenegger M, Komor J, Baumgartner R, Hess CW. Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment. J Neurol 1996;243:585-93.
6. Mocellin R, Walterfang M, Velakoulis D. Hashimoto's encephalopathy: epidemiology, pathogenesis and management. CNS Drugs 2007;21:799-811.
7. Blanchin S, Coffin C, Viader F, Ruf J, Carayon P, Potier F, Portier E, Comby E, Allouche S, Ollivier Y, Reznik Y, Ballet JJ. Anti-thyroperoxidase antibodies from patients with Hashimoto's encephalopathy bind to cerebellar astrocytes. J Neuroimmunol 2007;192:13-20.
8. Nakagawa H, Yoneda M, Fujii A, Kinomoto K, Kuriyama M. Hashimoto encephalopathy presenting with progressive cerebellar ataxia. BMJ Case Rep 2009;2009.
9. Jacob S, Rajabally YA. Current proposed mechanisms of action of intravenous immunoglobulins in inflammatory neuropathies. Curr Neuropharmacol 2009;7:337-42.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Chen XY, Wang YZ, Lei HX, Zhang X. A case of Hashimoto's encephalopathy presenting with seizures and cognitive impairment. Neurosciences 2016;3:117-9. http://dx.doi.org/10.20517/2347-8659.2015.57
AMA Style
Chen XY, Wang YZ, Lei HX, Zhang X. A case of Hashimoto's encephalopathy presenting with seizures and cognitive impairment. Neuroimmunology and Neuroinflammation. 2016; 3: 117-9. http://dx.doi.org/10.20517/2347-8659.2015.57
Chicago/Turabian Style
Chen, Xing-Yong, Yin-Zhou Wang, Hui-Xin Lei, Xu Zhang. 2016. "A case of Hashimoto's encephalopathy presenting with seizures and cognitive impairment" Neuroimmunology and Neuroinflammation. 3: 117-9. http://dx.doi.org/10.20517/2347-8659.2015.57
ACS Style
Chen, X.Y.; Wang Y.Z.; Lei H.X.; Zhang X. A case of Hashimoto's encephalopathy presenting with seizures and cognitive impairment. Neurosciences. 2016, 3, 117-9. http://dx.doi.org/10.20517/2347-8659.2015.57
About This Article
Copyright

Data & Comments
Data


 Cite This Article 0 clicks
Cite This Article 0 clicks

 Like This Article 36
likes
Like This Article 36
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.