
Single low-dose lipopolysaccharide preconditioning: neuroprotective against axonal injury and modulates glial cells

Abstract
Aim: Over 7 million traumatic brain injuries (TBI) are reported each year in the United States. However, treatments and neuroprotection following TBI are limited because secondary injury cascades are poorly understood. Lipopolysaccharide (LPS) administration before controlled cortical impact can contribute to neuroprotection. However, the underlying mechanisms and whether LPS preconditioning confers neuroprotection against closed-head injuries remains unclear.
Methods: The authors hypothesized that preconditioning with a low dose of LPS (0.2 mg/kg) would regulate glial reactivity and protect against diffuse axonal injury induced by weight drop. LPS was administered 7 days prior to TBI. LPS administration reduced locomotion, which recovered completely by time of injury.
Results: LPS preconditioning significantly reduced the post-injury gliosis response near the corpus callosum, possibly by downregulating the oncostatin M receptor. These novel findings demonstrate a protective role of LPS preconditioning against diffuse axonal injury. LPS preconditioning successfully prevented neurodegeneration near the corpus callosum, as measured by fluorojade B.
Conclusion: Further work is required to elucidate whether LPS preconditioning confers long-term protection against behavioral deficits and to elucidate the biochemical mechanisms responsible for LPS-induced neuroprotective effects.
Keywords
Introduction
Endotoxin preconditioning with lipopolysaccharide (LPS) was previously shown to be neuroprotective in models of ischemic stroke and traumatic brain injury (TBI).[1] The neuroprotective mechanisms of LPS remain unclear, particularly with regards to vascular-mediated effects. Prior studies have studies this using contusion-based models of neurotrauma. The contusion model irreversibly injures a focal brain region, creating a gross structural void associated with significant vascular injury.[2] The mechanisms of LPS preconditioning are poorly understood and have not been studied in a model of diffuse axonal injury. There is emerging evidence that pro-inflammatory regulation is neuroprotective in models of neural injury,[3] particularly the regulation of pro-inflammatory cytokines and microglial phenotype changes. Microglia is pleiotropic and transition between continuum states. These states are termed M1 or M2 depending on the inciting events.[4] Microglia may have protective and destructive roles, depending on the transition state.[5] The ratio of microglial states can predict the subacute outcome following injury. M1 microglia are pro-inflammatory and can cause long-term deficits.[6] Although LPS has been shown to influence astrocyte phenotypes, it is not clear whether LPS preconditioning modulates microglial phenotypes following diffuse axonal injury.[7]
In this study, we investigated the neuroprotective effect of LPS preconditioning in a closed-head model of diffuse axonal injury for the first time. This model of TBI has increased clinical relevance due to axonal shearing seen with diffuse neurotrauma.[8] We hypothesized that LPS preconditioning would reduce inflammation and neurodegeneration following diffuse axonal injury. In addition, we predicted that LPS preconditioning would regulate the oncostatin M receptor (OSMR) in astrocytes and activate the M1 microglia phenotype after TBI. TBI promotes pro-inflammatory cascades and increases the expression of glial fibrillary acidic protein (GFAP);[2,9] however, a link between astrocyte regulation and the microglia phenotype after TBI has not been investigated. In this study, we explored the effect of LPS preconditioning on astrocytes and microglia and addressed the relationship between cytokine receptor expression, astrocyte reactivity, and microglial phenotype. Improving our understanding of the protective effects of LPS preconditioning may accelerate the identification of novel therapeutic targets that reduce damage after TBI in individuals at risk of concussion.
Methods
Animals
All procedures involving live animals were approved by the Institutional Animal Care and Use Committee of West Virginia University and were performed according to the principles of the Guide for the Care and Use of Laboratory Animals, published by the Institute of Laboratory Resources, National Research Council (National Institutes of Health publication 85-23-2985). Thirty-two male Sprague Dawley rats (Hilltop) at 3-4 months of age were used in this study. Animals were given standard rat chow and water ad libitum.
LPS preconditioning
Rats were pretreated with a single intraperitoneal injection of either 0.2 mg/kg LPS (Sigma-Aldrich) or 0.9% saline (equal volume) 7 days prior to TBI.
Locomotor behavior
After LPS injection, the development of sickness behavior was monitored using activity chambers as described by Godbout et al.[10] Locomotor activity was assessed using an automated activity monitoring system (San Diego Instruments, San Diego, CA) that recorded beam breaks in the x, y, and z planes. Animals were acclimated to the room for 1 h prior to testing. Testing chambers consisted of square Plexiglass housing and 16 × 16 photobeam arrays to detect lateral movements. An 8 × 8 array, located above the 16 × 16 array, detected rearing-associated movements. Activity was quantified over 30 min and the sum of fine, ambulatory, and rearing beam breaks was calculated to give the total number of beam breaks. These recordings were completed at 2, 4, 24, and 48 h post-injection.
TBI induction
Animals were divided into four groups: sham surgery (n = 8), sham surgery with LPS pretreatment (n = 8), impact-acceleration injury following saline injection (n = 8), and impact-acceleration injury with LPS pretreatment (n = 8). Anesthesia was induced and maintained using isoflurane (4% induction, 2% maintenance). Body temperature was controlled with a homeothermic heating blanket equipped with a rectal probe. Rats received an impact-acceleration injury as described previously.[11-13] Briefly, a 10-mm diameter and 3-mm thick stainless steel disk was affixed to the skull with cyanoacrylate between bregma and lambda. The animal was placed in a prone position on a foam bed with a metal disk directly beneath a 2-m tall Plexiglass tube. A 450-g weight was dropped from the top of the tube, striking the metal disk. The disk was then removed while the rat was under anesthesia, the skull inspected, and the wound sutured. The animal was then returned to its cage, which was placed on a heating pad. Recovery from injury and anesthesia were monitored. No mortality was observed with the current injury parameters, and no gross lesions were apparent at the time of sacrifice indicating mild diffuse axonal injury.
Tissue preparation
Seven days after TBI, animals were anesthetized and perfused transcardially with physiological saline. Brains were removed and sectioned for histological analysis. The frontal cortex was selected for histological analysis. Tissue sections were placed in 4% paraformaldehyde for a minimum of 1 week. Following fixation, brains were processed using a Tissue-Tek VIP 5 Automatic Tissue Processor (Sakura Finatek, Torrence, CA). Processed tissues were paraffin-embedded with Tissue-Tek Tec 5 embedding system (Sakura Finatek, Torrence, CA) and sliced (6 µm) using a Leica RM2235 microtome (Leica Microscopes, Buffalo Grove, IL). Sections were mounted on glass slides and heat-fixed. Immediately prior to staining, tissues were de-paraffinized with xylene and alcohol washes.[14]
Fluoro-Jade B (FJB) staining
FJB staining was used to identify neural degeneration. For FJB labelling, slides were rehydrated through a series of alcohol and deionized (dH2O) water rinses then incubated in 0.06% potassium permanganate for 10 min. Then, slides were rinsed for 2 min in dH2O water and incubated with FJB in 0.1% acetic acid for 20 min. After staining, slides were washed three times in dH2O.
GFAP staining
Tissues were incubated in rabbit anti-cow GFAP antibody (Dako, Glostrup, Denmark) at a dilution of 1:500 in 4% horse serum in Dulbecco’s phosphate buffered (DPBS) overnight. Then, tissues were washed three times in DPBS and incubated in biotinylated anti-rabbit IgG antibody (Vector Laboratories, Burlingame, CA) diluted at 1:10,000 in 4% horse serum in DPBS for 4 h. Next, tissue was treated with avidin D-horeradish peroxidase (Vector Laboratories, Burlingame, CA) diluted at 1:1,000 in DPBS for 1 h. Sections were then stained in DAB chromogen solution (Vector Laboratories) for 5 min, then tissues were rinsed three times in DPBS and dried overnight.
M1 microglia staining
Tissues were incubated in mouse anti-rat CD68 antibody (AbD Serotec, Kidlington, UK) at a dilution of 1:100 in 4% horse serum in DPBS overnight. Sections were washed three times in DPBS and incubated in a biotinylated anti-mouse IgG secondary antibody (Vector Laboratories) diluted at 1:10,000 in 4% horse serum in DPBS for 4 h. Following secondary antibody incubation, tissues were incubated in alkaline phosphatase (Life Technology, Carlsbad, CA) diluted at 1:100 in Tris-bovine serum albumin for 1 h. Then, tissues were rinsed three times in DPBS and incubated in Fast Blue BB salt (Santa Cruz Biotechnology, Santa Cruz, CA) for 5 min. Tissues were washed in xylene, mounted using an antifade agent, and cover slipped. The slides were sealed with acrylic and stored in the dark in a laboratory refrigerator.
GFAP and OSMR staining
Tissues were labelled with rabbit against anti-cow GFAP (DAKO) antibody at a dilution of 1:500 in 5% horse serum in PBS overnight at 4°C. Next, sections were washed twice for 10 min each in PBS prior to application of Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen, Grand Island, NY) at a dilution of 1:100 in PBS for 3 h. Following secondary antibody incubation, slides were rinsed twice for 10 min each in PBS. Then, tissue was labelled with a goat anti-mouse OSMR antibody (LifeSpan Biosciences, Seattle, WA) at a dilution of 1:200 in 5% horse serum in PBS overnight at 4°C. Following incubation, slides were rinsed twice for 10 min each in PBS prior before applying biotinylated anti-goat IgG (Vector Laboratories) at a dilution of 1:10,000 in 5% horse serum in PBS for 2 h. Next, slides were rinsed twice for 10 min each in PBS and incubated in Streptavidin Alexa Fluor 546 (Life Technology) at a dilution of 1:100 in PBS for 1 h. Slides were rinsed in PBS for 10 min and then coverslipped with Vectashield Mounting Media containing DAPI (Vector Laboratories). Finally, slides were sealed with acrylic and stored in the dark in a laboratory refrigerator at 4°C. Images were acquired using a Zeiss Axio Imager 2 microscope and quantified using ImageJ with standard co-localization quantification techniques and the co-localization plugin established by Bolte et al.[15]
Histological quantification
Stereology and optical fractionation were used to quantify histological results as previously described.[16-18] Briefly, a region of interest encompassing the corpus callosum was drawn at low power using an Olympus AX70 microscope and StereoInvestigator software. The region encompassing the corpus callosum was chosen because it undergoes robust biochemical changes following impact-acceleration TBI.[19] The software selected random 75-µm counting frames with a depth of 6 µm, and the object of interest was marked by an investigator blinded to treatment. The region of interest volume was previously identified, and the number of cells marked by the observer was quantified.
Statistical analysis
Data were analyzed using GraphPad Prism version 4.0. One-way ANOVA with post-hoc Tukey’s test was used to compare histological findings across control and various experimental groups. Repeated measures two-way ANOVA was used to analyze the total activity data. An overlap coefficient of 0.6 or greater indicated strong co-localization, a coefficient between 0.4 and 0.6 indicated medium co-localization, and a coefficient < 0.4 indicated weak co-localization. A P value of < 0.05 was considered statistically significant for all studies.
Results
LPS induces transient acute sickness behavior
LPS injection induces systemic sickness in rodents as evidenced by an acute reduction in activity.[10,20]Figure 1 shows a significant reduction in total activity after LPS administration compared with saline-treated animals based on time (F3,66 = 8.14, P < 0.001), treatment (F1,66 = 18.67, P < 0.001), and the time treatment interaction (F3,66 = 22.92, P < 0.001) using two-way repeated measures ANOVA. Total activity was reduced by approximately 71% and 59% at 2 and 4 h post-injection, respectively. This was resolved within 24 h, indicating that sickness behavior was acute.
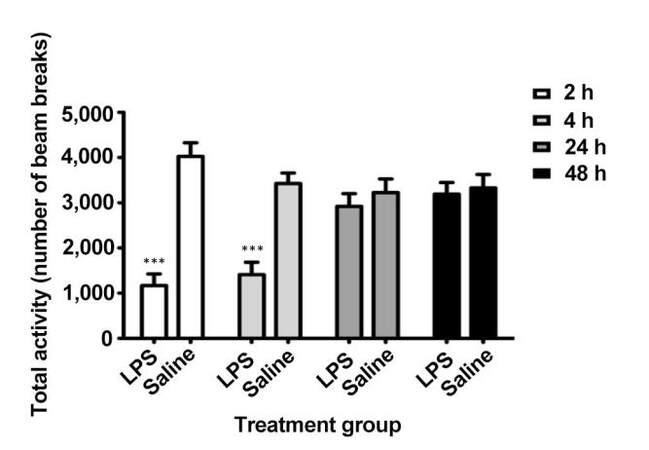
Figure 1. Total locomotor activity after LPS injection by number of beam breaks at 2, 4, 24, and 48 h after injection. Acute sickness was present at 2 and 4 h but resolved by 24 h. ***P < 0.001. LPS: lipopolysaccharide
LPS preconditioning reduces neuronal degeneration and glial activation following TBI
Previous studies have demonstrated a neuroprotective effect of LPS preconditioning following controlled cortical impact injury with large vascular insult.[2]Figure 2 shows a significant difference in cortical FJB expression between experimental groups (F3,32 = 59.79; P < 0.001). LPS preconditioning significantly reduced FJB levels following TBI according to one-way ANOVA with Tukey’s post-hoc test (q = 8.50, P < 0.001). No difference was observed between sham-injured and LPS-treated animals (q = 0.13, P > 0.05), indicating that peripheral administration of LPS did not induce neurodegeneration.
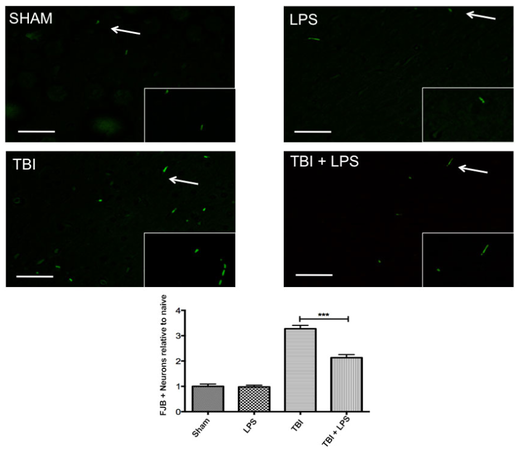
Figure 2. Neural degeneration increased following traumatic brain injury. Fluoro jade B significantly increased following traumatic brain injury but LPS preconditioning ameliorated this effect. Scale bar = 50 µm. ***P < 0.001. LPS: lipopolysaccharide; TBI: traumatic brain injuries; FJB: Fluoro-Jade B
GFAP expression in the cortex differed significantly between experimental groups (F3,32 = 57.92; P < 0.001) [Figure 3]. LPS preconditioning significantly reduced GFAP levels after TBI according to one-way ANOVA and post-hoc Tukey’s test (q = 8.70, P < 0.001). No difference was observed between sham-injured and LPS-treated animals (q = 2.89, P > 0.05), indicating that peripheral administration of LPS did not activate astrocytes.
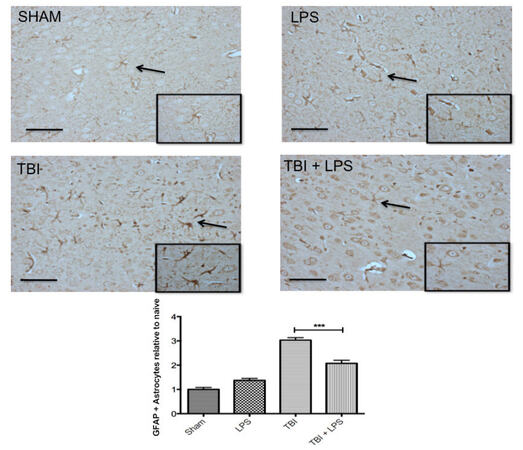
Figure 3. Astrocyte reactivity increased following TBI. GFAP increased significantly following TBI but LPS preconditioning ameliorated the effect. Scale bar = 50 µm. ***P < 0.001. LPS: lipopolysaccharide; TBI: traumatic brain injuries; GFAP: glial fibrillary acidic protein
LPS preconditioning reduces M1 microglia activation after TBI
To investigate the effect of LPS preconditioning on classically activated microglia, CD68 expression was quantified by stereology. Figure 4 shows a significant difference in CD68 expression between experimental groups (F3,32 = 28.22; P < 0.001). The presence of M1 microglia (CD68 expression) was significantly reduced after TBI following LPS preconditioning compared with no LPS pretreatment according to one-way ANOVA with post-hoc Tukey’s test (q = 9.77, P < 0.001). Importantly, no significant differences in CD68 expression were observed between injured animals with LPS preconditioning and sham-injured animals (q = 2.121, P > 0.05). These findings were consistent with IBA-1 staining for undifferentiated microglia, which showed a qualitative reduction in LPS pre-conditioned animals [Figure 5].
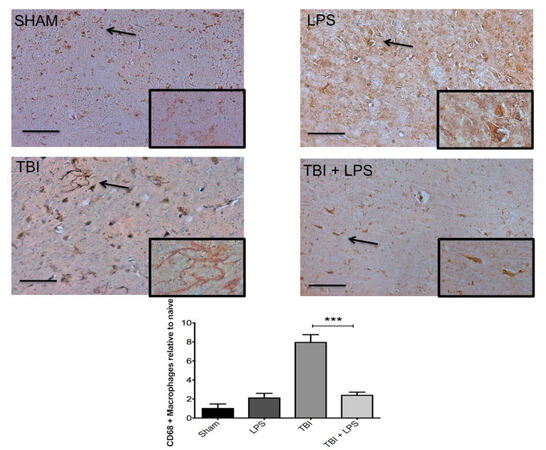
Figure 4. M1 microglia activation increased significantly following TBI. CD68 was significantly increased following TBI but LPS preconditioning ameliorated the effect. Scale bar = 50 µm. ***P < 0.001. LPS: lipopolysaccharide; TBI: traumatic brain injuries
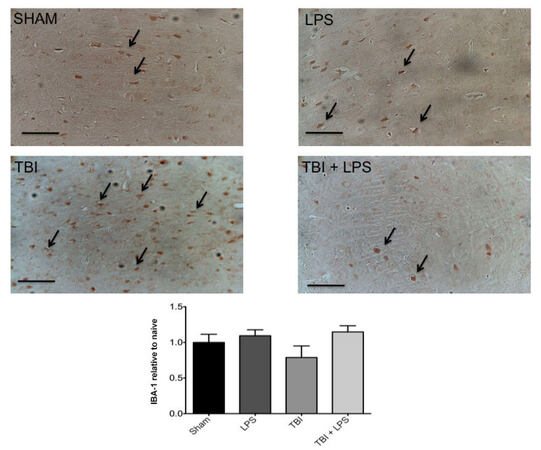
Figure 5. No significant differences were observed in IBA-1 microglia staining between groups. LPS: lipopolysaccharide; TBI: traumatic brain injuries
LPS preconditioning reduces OSMR expression in astrocytes after TBI
One of the primary mechanisms for regulating astrocyte activation is neuropoietic cytokine signaling through the gp130 receptor-signaling complex.[21] TBI associated with significant vascular injury upregulates members of the neuropoietic cytokine family, including OSMR.[22] We observed significant OSMR upregulation following diffuse axonal injury [Figure 6]. OSMR expression differed significantly between experimental groups (F3,32 = 11.80; P < 0.05). OSMR expression was reduced after TBI in LPS pre-conditioned animals compared with no LPS pretreatment according to one-way ANOVA with post-hoc Tukey’s test (q = 6.51, P < 0.05). No difference was observed between LPS-treated animals and sham-injured rats at this time point (q = 0.45, P > 0.05).
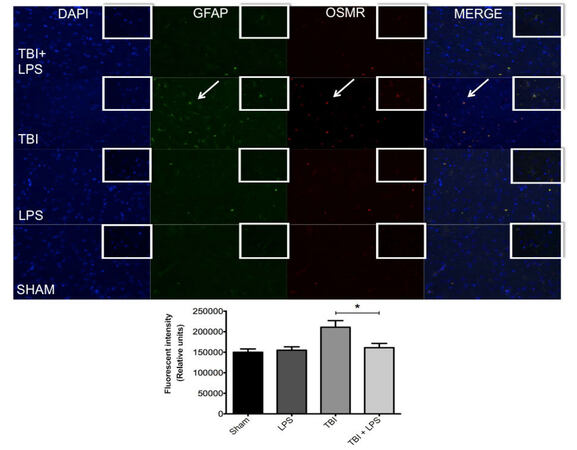
Figure 6. Colocalization of GFAP and OSMR with DAPI. GFAP (green) and OSMR (red) were significantly increased in the same cells (merged yellow) following TBI. LPS preconditioning prevented OSMR upregulation. *P < 0.05. LPS: lipopolysaccharide; TBI: traumatic brain injuries; GFAP: glial fibrillary acidic protein; OSMR: oncostatin M receptor
GFAP expression was quantified to measure astrocyte activation and was significantly different between experimental groups (F3,32 = 6.30; P < 0.05) [Figure 6]. LPS preconditioning significantly reduced GFAP expression after TBI as shown by one-way ANOVA and post-hoc Tukey’s test (q = 4.44; P < 0.05). Again, no difference was observed between LPS-treated and sham-injured rats at this time point (q = 0.45; P > 0.05). Importantly, there was a strong correlation between GFAP and OSMR expression evidenced by the yellow overlay (overlap coefficient r = 0.722), indicating a high degree of overlap within the same astrocyte. This was partially mitigated by LPS preconditioning (overlap coefficient r = 0.478).
Discussion
Diffuse axonal injury is induced consistently by the weight drop method without producing a grossly visible lesion.[23] We have previously confirmed that the injury parameters used in this study induce diffuse axonal injury by measuring β-amyloid precursor protein expression.[24] The weight drop model is ideal for testing neuroprotective strategies because it induces consistent axonal damage and a characteristic progression of traumatic axonal injury in rodents.[25] Axonal injury reduces cerebral blood flow following neurotrauma.[26] Therefore, it is potentially worthwhile to investigate compounds that contribute to vascular preconditioning. Vascular preconditioning by heat activation reduces TBI severity and the extent of axonal damage by selectively activating hypoxia-inducible factor 1α.[27] Low-dose LPS pretreatment has also been used for successful vascular preconditioning in penetrating models of TBI.[28] One proposed mechanism for LPS is a reduction of inflammatory mediators before injury.[29] Inflammatory mediators can activate gliosis.[30]
In this study, we show for the first time that low-dose LPS preconditioning is protective in a closed-head model of diffuse axonal injury. LPS preconditioning has previously been shown to be protective in penetrating models, but there was significant vascular disruption in these models and they were generally more severe than our model of diffuse axonal injury. The findings of the present study are significant in that they demonstrate that LPS preconditioning regulates microglia and OSMR in a model of diffuse axonal injury. Furthermore, these protective effects are sustained at one week post-injury. Protection was established in a mild injury model with no mortality or gross pathological changes, indicating that LPS pretreatment may also protect against mild neurotrauma.
Longhi et al.[2] showed that LPS preconditioning alters IL-6 and OSM expression following TBI. In this study, we demonstrated that LPS may exert a neuroprotective effect against diffuse axonal injury through modulation of neurodegeneration and the gliosis response. This supports the notion that LPS induces neuroprotective effects originally proposed by Longhi et al.[2] We observed a transient acute sickness induced by LPS pretreatment. However, LPS preconditioning had the following effects, including: reduced FJB, OSMR, GFAP, and CD68 expression. Decreased FJB staining was indicative of reduced neurodegeneration. This has been demonstrated in vitro by Zhu et al.,[31] and our findings have now confirmed this in vivo. Increased FJB staining has been associated with motor deficits following TBI.[32] In a future study, we plan to look at the role of LPS preconditioning in preventing motor deficits following TBI.
Reactive astrocytes can inhibit axonal regrowth after an axon is severed.[33] OSM activity increases in demyelinated areas, leading to an upregulation of OSMR. This upregulation indicates a loss of detrimental connectivity,[22] while decreased OSMR indicates preservation of myelin integrity and axonal tracts. Our findings show that LPS preconditioning significantly reduces OSMR levels [Figure 7]. Interestingly, GFAP expression was also downregulated, which may reflect an interaction between LPS and toll like receptor 4 (TLR4). Low-dose LPS administration can stimulate TLR4, which activates signaling cascades to suppress innate immunity and astrocyte activation. These cell-signaling events permit axonal regeneration without the threat of glial scar formation.[34] As long as myelin integrity is maintained, neurons can re-innervate most of their lost connections. The clinical use of LPS preconditioning is obviously limited because injury cannot be predicted, but targeting TLR4 pharmacologically might represent a reasonable strategy. Further work is needed to determine the exact interactions between low-dose LPS administration and TLR4.
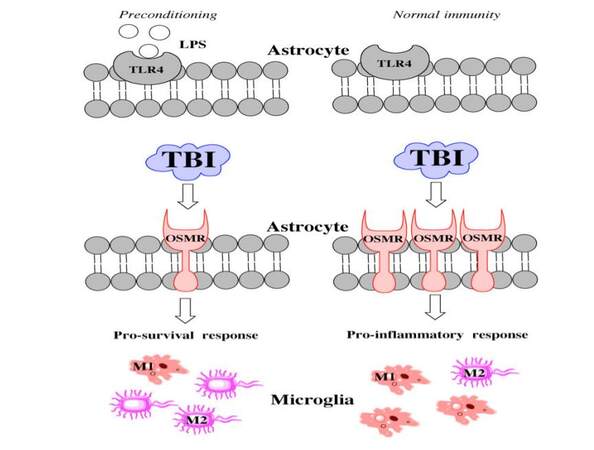
Figure 7. Hypothetical schematic showing the mechanism of LPS action. LPS: lipopolysaccharide; TBI: traumatic brain injuries; OSMR: oncostatin M receptor; TLR4: toll like receptor 4
In addition to effects on astrocytosis, LPS preconditioning has also been associated with microglial changes. LPS can promote the infiltration of macrophages into the brain, which helps resolve the microglia response after diffuse traumatic axonal injury.[35] Microglia is broadly grouped into pro-inflammatory M1 microglia and pro-survival anti-inflammatory M2 microglia.[36] The balance of M1 to M2 microglia is tightly controlled following injury.[37] M1 microglia can exacerbate axonal injury, thereby limiting functional recovery. We showed that LPS preconditioning selectively inhibited the M1 response. LPS caused an acute increase in pro-inflammatory markers, which may signal peripheral macrophages to cross brain vasculature by chemotaxis. Peripheral macrophages alter the inflammatory mileu of the brain while attenuating microglial activity.[38] In contrast, M2 microglia are largely unaffected due to the earlier peak of activation after injury, which we have shown using non-differentiated IBA-1 imaging.[39] The brain establishes functional recovery by shifting the balance away from M1 microglia.
LPS has been associated with inflammation. Suppressing inflammation limits the effect of LPS on human physiology.[40] LPS increases tissue necrosis factor α, interleukin 6, and interleukin 1 expression [Figure 6].[41] LPS preconditioning reduces neuronal loss and microglia activation in other injury models, such as global hypoxia.[42] In the present study, LPS preconditioning also significantly reduced neuronal degeneration following diffuse axonal injury. Chronic LPS-mediated inflammation is detrimental to the brain, but acute LPS preconditioning provides “inflamaprepping” that primes the nervous system for response to injury.[43] Although inflamaprepping is not a universal to all TBIs, it offers a potential therapeutic advantage for a certain subset of individuals, namely soldiers and athletes. Soldiers and athletes are at increased risk of concussion and subconcussive injury. Prevent injury and enhancing recovery in these individuals is receiving increasing attention. Inflamaprepping with a systemically injected agent such as low-dose LPS is clinically feasible and may limit gliosis and subsequent glial scar formation by preparing the brain for trauma. Ultimately this would facilitate rapid axonal regeneration guided by preserved myelin tracts. In the current study, we have shown that inflamaprepping inhibits gliosis, downregulates the OSMR receptor, and shifts the microglia phenotype balance away from the pro-inflammatory M1 state, thereby decreasing neurodegeneration and promoting neuroprotection. The benefits of this neuroprotection on preventing behavioral decline will be investigated in a future study.
There are some limitations to the present study. Firstly, we did not assess post-injury behavior. Based on our histopathologic findings, we expect that LPS preconditioning prevents behavioral deficits following TBI, but needs to be verified in a future study. Secondly, we did not or examine glial marker expression later than 7 days after LPS treatment. This data would have indicated the current state of gliosis and inflammation at the time of injury.
In conclusion, we have shown for the first time that low-dose LPS preconditioning has protective effects in a diffuse axonal injury model. LPS preconditioning prevented both astrocyte and microglia activation through downregulation of the OSMR receptor. This protective effect was verified by reduced FJB staining, indicating decreased degeneration. Preconditioning and inflamaprepping may be viable targets for TBI treatment and may prevent long-term behavioral sequelae in patients. Future work will examine the long-term functional changes that lead to neurodegenerative disease progression and tauopathies. We will elucidate whether LPS preconditioning reduces tau hyperphosphorylation and improves behavior following repetitive injury. Mediating the gliosis response with LPS preconditioning may decrease neurodegeneration and slow the development of tauopathy.
Financial support and sponsorship
Aric Logsdon and Brandon Lucke-Wold received an American Foundation of Pharmaceutical Education Pre-doctoral Fellowship. Brandon Lucke-Wold also received a Neurosurgery Research and Education Foundation Medical Student Summer Research Fellowship, a Sigma Xi Grants in Aid of Research, American Association of Pharmaceutical Scientists Pre-doctoral fellowship, and American Medical Association Foundation Seed Grant.
Conflicts of interest
There are no conflicts of interest.
Patient consent
No patients were involved.
Ethical approval
Ethical approval was obtained prior to the commencement of the study.
REFERENCES
1. Walshe CM, Laffey JG, Kevin L, O'Toole D. Sepsis protects the myocardium and other organs from subsequent ischaemic/reperfusion injury via a MAPK-dependent mechanism. Intensive Care Med Exp 2015;3:35.
2. Longhi L, Gesuete R, Perego C, Ortolano F, Sacchi N, Villa P, Stocchetti N, De Simoni MG. Long-lasting protection in brain trauma by endotoxin preconditioning. J Cereb Blood Flow Metab 2011;31:1919-29.
3. Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat 2015;11:97-106.
4. Schaafsma W, Zhang X, van Zomeren KC, Jacobs S, Georgieva PB, Wolf SA, Kettenmann H, Janova H, Saiepour N, Hanisch UK, Meerlo P, van den Elsen PJ, Brouwer N, Boddeke HW, Eggen BJ. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav Immun 2015;48:205-21.
5. Hayakawa K, Okazaki R, Morioka K, Nakamura K, Tanaka S, Ogata T. Lipopolysaccharide preconditioning facilitates M2 activation of resident microglia after spinal cord injury. J Neurosci Res 2014;92:1647-58.
6. Kumar A, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. Microglial/macrophage polarization dynamics following traumatic brain injury. J Neurotrauma 2016;33:1732-50.
7. Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci 2012;32:6391-410.
8. Johnson VE, Meaney DF, Cullen DK, Smith DH. Animal models of traumatic brain injury. Handb Clin Neurol 2015;127:115-28.
9. Oliva AA Jr, Kang Y, Sanchez-Molano J, Furones C, Atkins CM. STAT3 signaling after traumatic brain injury. J Neurochem 2012;120:710-20.
10. Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J 2005;19:1329-31.
11. Mills JD, Bailes JE, Sedney CL, Hutchins H, Sears B. Omega-3 fatty acid supplementation and reduction of traumatic axonal injury in a rodent head injury model. J Neurosurg 2011;114:77-84.
12. Mills JD, Bailes JE, Turner RC, Dodson SC, Sakai J, Maroon JC. Anabolic steroids and head injury. Neurosurgery 2012;70:205-9; discussion 209-10.
13. Mills JD, Hadley K, Bailes JE. Dietary supplementation with the omega-3 fatty acid docosahexaenoic acid in traumatic brain injury. Neurosurgery 2011;68:474-81; discussion 481.
14. Turner RC, Naser ZJ, Bailes JE, Smith DW, Fisher JA, Rosen CL. Effect of slosh mitigation on histologic markers of traumatic brain injury: laboratory investigation. J Neurosurg 2012;117:1110-8.
15. Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 2006;224:213-32.
16. Lyeth BG, Jenkins LW, Hamm RJ, Dixon CE, Phillips LL, Clifton GL, Young HF, Hayes RL. Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res 1990;526:249-58.
17. Maughan PH, Scholten KJ, Schmidt RH. Recovery of water maze performance in aged versus young rats after brain injury with the impact acceleration model. J Neurotrauma 2000;17:1141-53.
18. Meyer DL, Davies DR, Barr JL, Manzerra P, Forster GL. Mild traumatic brain injury in the rat alters neuronal number in the limbic system and increases conditioned fear and anxiety-like behaviors. Exp Neurol 2012;235:574-87.
19. Adelson PD, Robichaud P, Hamilton RL, Kochanek PM. A model of diffuse traumatic brain injury in the immature rat. J Neurosurg 1996;85:877-84.
20. Huang Y, Henry CJ, Dantzer R, Johnson RW, Godbout JP. Exaggerated sickness behavior and brain proinflammatory cytokine expression in aged mice in response to intracerebroventricular lipopolysaccharide. Neurobiol Aging 2008;29:1744-53.
21. Hsu MP, Frausto R, Rose-John S, Campbell IL. Analysis of IL-6/gp130 family receptor expression reveals that in contrast to astroglia, microglia lack the oncostatin M receptor and functional responses to oncostatin M. Glia 2015;63:132-41.
22. Glezer I, Rivest S. Oncostatin M is a novel glucocorticoid-dependent neuroinflammatory factor that enhances oligodendrocyte precursor cell activity in demyelinated sites. Brain Behav Immun 2010;24:695-704.
23. Viano DC, Hamberger A, Bolouri H, Saljo A. Evaluation of three animal models for concussion and serious brain injury. Ann Biomed Eng 2012;40:213-26.
24. Smith DW, Bailes JE, Fisher JA, Robles J, Turner RC, Mills JD. Internal jugular vein compression mitigates traumatic axonal injury in a rat model by reducing the intracranial slosh effect. Neurosurgery 2012;70:740-6.
25. Ohta M, Higashi Y, Yawata T, Kitahara M, Nobumoto A, Ishida E, Tsuda M, Fujimoto Y, Shimizu K. Attenuation of axonal injury and oxidative stress by edaravone protects against cognitive impairments after traumatic brain injury. Brain Res 2013;1490:184-92.
26. Okamoto T, Hashimoto K, Aoki S, Ohashi M. Cerebral blood flow in patients with diffuse axonal injury -- examination of the easy Z-score imaging system utility. Eur J Neurol 2007;14:540-7.
27. Umschweif G, Alexandrovich AG, Trembovler V, Horowitz M, Shohami E. Hypoxia-inducible factor 1 is essential for spontaneous recovery from traumatic brain injury and is a key mediator of heat acclimation induced neuroprotection. J Cereb Blood Flow Metab 2013;33:524-31.
28. Lin HY, Wu CL, Huang CC. The Akt-endothelial nitric oxide synthase pathway in lipopolysaccharide preconditioning-induced hypoxic-ischemic tolerance in the neonatal rat brain. Stroke 2010;41:1543-51.
29. Merry HE, Wolf PS, Fitzsullivan E, Keech JC, Mulligan MS. Lipopolysaccharide pre-conditioning is protective in lung ischemia-reperfusion injury. J Heart Lung Transplant 2010;29:471-8.
30. Tarassishin L, Suh HS, Lee SC. LPS and IL-1 differentially activate mouse and human astrocytes: Role of CD14. Glia 2014;62:999-1013.
31. Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X, Chen F, Wang CS, Feng H, Lin JK. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. J Neuroinflammation 2014;11:59.
32. Lopez NE, Lindsay G, Karina LR, Mary HA, Putnam J, Eliceiri B, Coimbra R, Bansal V. Ghrelin decreases motor deficits after traumatic brain injury. J Surg Res 2014;187:230-6.
33. Cua RC, Lau LW, Keough MB, Midha R, Apte SS, Yong VW. Overcoming neurite-inhibitory chondroitin sulfate proteoglycans in the astrocyte matrix. Glia 2013;61:972-84.
34. Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J Pain 2014;15:712-25.
35. Raposo C, Schwartz M. Glial scar and immune cell involvement in tissue remodeling and repair following acute CNS injuries. Glia 2014;62:1895-904.
36. Crain JM, Nikodemova M, Watters JJ. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J Neurosci Res 2013;91:1143-51.
37. Stirling DP, Cummins K, Mishra M, Teo W, Yong VW, Stys P. Toll-like receptor 2-mediated alternative activation of microglia is protective after spinal cord injury. Brain 2014;137:707-23.
38. Morganti JM, Jopson TD, Liu S, Gupta N, Rosi S. Cranial Irradiation Alters the Brain's Microenvironment and Permits CCR2+ Macrophage Infiltration. PLoS One 2014;9:e93650.
39. Turtzo LC, Lescher J, Janes L, Dean DD, Budde MD, Frank JA. Macrophagic and microglial responses after focal traumatic brain injury in the female rat. J Neuroinflammation 2014;11:82.
40. Kaushik DK, Thounaojam MC, Mitra A, Basu A. Vespa tropica venom suppresses lipopolysaccharide-mediated secretion of pro-inflammatory cyto-chemokines by abrogating nuclear factor-kappa B activation in microglia. Inflamm Res 2014;63:657-65.
41. Song X, Zhang W, Wang T, Jiang H, Zhang Z, Fu Y, Yang Z, Cao Y, Zhang N. Geniposide plays an anti-inflammatory role via regulating TLR4 and downstream signaling pathways in lipopolysaccharide-induced mastitis in mice. Inflammation 2014;37:1588-98.
42. Lange S, Rocha-Ferreira E, Thei L, Mawjee P, Bennett K, Thompson PR, Subramanian V, Nicholas AP, Peebles D, Hristova M, Raivich G. Peptidylarginine deiminases: novel drug targets for prevention of neuronal damage following hypoxic ischemic insult (HI) in neonates. J Neurochem 2014;130:555-62.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Turner RC, Naser ZJ, Lucke-Wold BP, Logsdon AF, Vangilder RL, Matsumoto RR, Huber JD, Rosen CL. Single low-dose lipopolysaccharide preconditioning: neuroprotective against axonal injury and modulates glial cells. Neurosciences 2017;4:6-15. http://dx.doi.org/10.20517/2347-8659.2016.40
AMA Style
Turner RC, Naser ZJ, Lucke-Wold BP, Logsdon AF, Vangilder RL, Matsumoto RR, Huber JD, Rosen CL. Single low-dose lipopolysaccharide preconditioning: neuroprotective against axonal injury and modulates glial cells. Neuroimmunology and Neuroinflammation. 2017; 4: 6-15. http://dx.doi.org/10.20517/2347-8659.2016.40
Chicago/Turabian Style
Turner, Ryan C., Zachary J. Naser, Brandon P. Lucke-Wold, Aric F. Logsdon, Reyna L. Vangilder, Rae R. Matsumoto, Jason D. Huber, Charles L. Rosen. 2017. "Single low-dose lipopolysaccharide preconditioning: neuroprotective against axonal injury and modulates glial cells" Neuroimmunology and Neuroinflammation. 4: 6-15. http://dx.doi.org/10.20517/2347-8659.2016.40
ACS Style
Turner, RC.; Naser ZJ.; Lucke-Wold BP.; Logsdon AF.; Vangilder RL.; Matsumoto RR.; Huber JD.; Rosen CL. Single low-dose lipopolysaccharide preconditioning: neuroprotective against axonal injury and modulates glial cells. Neurosciences. 2017, 4, 6-15. http://dx.doi.org/10.20517/2347-8659.2016.40
About This Article
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.