
Endocannabinoid metabolism in neurodegenerative diseases


Endocannabinoids are endogenous lipid mediators contributing to a variety of physiological, pharmacological, and pathological processes primarily through acting on cannabinoid receptors (CB1R and CB2R), which are targets of Δ9-tetrahydrocannabinol (Δ9-THC), the major psychoactive ingredient in marijuana.[1] Although N-arachidonoyl ethanolamide is the first identified endocannabinoid, 2-arachidonoylglycerol (2-AG), the second identified endocannabinoid, is the most abundant ligand produced in our body and a full agonist for CB1R and CB2R.[2] It has been well recognized that 2-AG is a retrograde messenger modulating synaptic transmission and plasticity at both inhibitory GABAergic and excitatory glutamatergic synapses in the brain.[2-4] In particular, augmentation of 2-AG signaling by inhibition of its metabolism has been attracted attention recently due to its profound anti-inflammatory and neuroprotective properties.[4,5]
2-AG is synthesized largely from diacylglycerol by diacylglycerol lipase α and β and primarily hydrolyzed by the enzyme monoacylglycerol lipase (MAGL) to glycerol and arachidonic acid (AA), a precursor of prostaglandins and leukotrienes [Figure 1]. And 2-AG is also degraded by the enzymes α/β hydrolase domain-containing protein 6 and 12 and metabolized oxidatively by cyclooxygenase 2 (COX-2). Apparently, 2-AG is a very unstable bioactive lipid mediator, and it is easily and rapidly degraded by these enzymes upon its synthesis.[2] It has been estimated that 85% of 2-AG is metabolized by MAGL in the brain.[6] Arachidonic acid-delivered prostaglandins and leukotrienes are generally proinflammatory and neurotoxic, whereas 2-AG is capable of resolving neuroinflammation and protecting neurons from harmful insults.[4,5,7] This suggests that disruption of MAGL would be a promising strategy to enhance anti-inflammatory and neuroprotective 2-AG signaling, while reducing proinflammatory and neurotoxic eicosanoid (e.g. PGE2) levels [Figure 1].
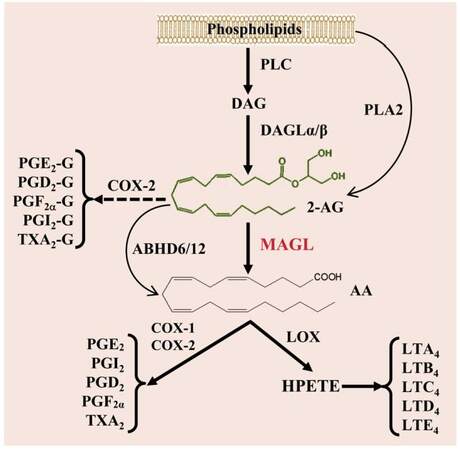
Figure 1. Pathways of 2-AG synthesis and metabolism. 2-AG is largely synthesized from DAG, which is formed from membrane phospholipids through PLC, by DAGLα/β. 2-AG in the brain is primarily hydrolyzed by MAGL to glycerol and AA. It is also hydrolyzed by ABHD6/12 to AA and metabolized oxidatively by COX-2 to form a new type of prostaglandin glycerol esters when expression and activity of COX-2 are excessively elevated during inflammation. AA is a precursor of prostaglandins through the enzymes COX-1/2 and HPETE through the enzyme arachidonate 5-LOX to form leukotrienes (LTA4 to E4). 2-AG: 2-arachidonoylglycerol; PLC: phospholipase C; DAG: diacylglycerol; DAGLα/β: diacylglycerol lipase α and β; PLA2: phospholipase A2; AA: arachidonic acid; COX-2: cyclooxygenase-2; MAGL: monoacylglycerol lipase; ABHD6/12: α/β hydrolase domaincontaining protein 6 and 12; HPETE: hydroperoxyeicosatetraenoic acid; LOX: lipoxygenase.
Alzheimer’s disease (AD) is the most common cause of dementia in elderly. While the etiology of AD is multifactorial, accumulating evidence implicates traumatic brain injury (TBI) as an epigenetic risk factor in AD development and dementia. Chronic traumatic encephalopathy (CTE) is the most recently defined TBI-caused neurodegenerative disease, and neuropathology and neurocognitive deficits in CTE are similar to those in AD.[4,8-10] The significant similarities and overlap in the spectrum of changes in neuropathology, neurobiology, synaptic and neurocognitive deficits between CTE and AD suggest that CTE, in essence, is a TBI-triggered AD-like neurodegenerative disease. Repetitive or multiple brain injuries may lead to AD-like neuropathology, impairments in synaptic and cognitive functions, and dementia.[8-10] However, there are currently no effective therapies to prevent and treat AD and TBI-caused AD-like neurodegenerative disease or to halt progression of diseases. Earlier studies show that 2-AG protects neurons against brain trauma in a mouse model of closed head injury.[7] Recent studies provide evidence that inactivation of MAGL reduces neuroinflammation, Aβ accumulation and deposition, tau phosphorylation, and neurodegeneration and improves synaptic and neurocognitive functions in several animal models of neurodegenerative diseases, including AD, Parkinson’s disease (PD), and TBI.[8,11-14] This suggests that manipulations of 2-AG metabolism may provide novel pharmacotherapies for these intractable neurodegenerative diseases.[4,8,11,12]
Although disruption of MAGL ameliorates neuropathology and prevent synaptic and cognitive declines in animal models of neurodegenerative diseases,[4,8,11-14] the signaling pathways that mediate these beneficial effects produced by MAGL inhibition are still unclear. MAGL loss-of-function enhances anti-inflammatory and neuroprotective 2-AG signaling and decreases proinflammatory and neurotoxic prostaglandins and leukotrienes [Figure 1]. It is possible that the beneficial effects produced by MAGL inhibition are mediated either by enhanced 2-AG signaling through CB1R or CB2R-depdendent mechanism or by reduced eicosanoid levels through cannabinoid receptor-independent mechanisms.[4,8,11-14] It has been proposed that peroxisome proliferator-activated receptor γ (PPARγ), a nuclear receptor that displays significant anti-inflammatory properties, is a target of endocannabinoids.[15,16] Recent studies demonstrated that suppression of neuroinflammation and alleviation of AD neuropathology by 2-AG or inactivation of MAGL are mediated via PPARγ,[5,11,16] suggesting that PPARγ is likely a downstream signaling molecule of 2-AG. Despite the fact that the mechanisms by which inhibition of 2-AG metabolism alleviates neuroinflammation and neuropathology and prevents deterioration in synaptic and cognitive functions in animal models of AD, PD, and TBI remain to be elucidated, MAGL is a promising therapeutic target for neurodegenerative diseases.
Financial support and sponsorship
This article is financed by National Institutes of Health grants (No. NS076815).
Conflicts of interest
There are no conflicts of interest.
Patient consent
There is no patient involved.
Ethics approval
This article does not contain any studies with human participants or animals.
REFERENCES
1. Gaoni Y, Mechoulam R. Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc 1964;86:1646-7.
2. Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 2009;89:309-80.
3. Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron 2012;76:70-81.
4. Xu JY, Chen C. Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist 2015;21:152-68.
5. Zhang J, Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons by limiting COX-2 elevation. J Biol Chem 2008;283:22601-11.
6. Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 2007;14:1347-56.
7. Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 2001;413:527-31.
8. Zhang J, Teng Z, Song Y, Hu M, Chen C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J Cereb Blood Flow Metab 2015;35:443-53.
9. Gupta R, Sen N. Traumatic brain injury: a risk factor for neurodegenerative diseases. Rev Neurosci 2016;27:93-100.
10. Djordjevic J, Sabbir MG, Albensi BC. Traumatic brain injury as a risk factor for Alzheimer's disease: is inflammatory signaling a key player? Curr Alzheimer Res 2016;13:730-8.
11. Zhang J, Hu M, Teng Z, Tang YP, Chen C. Synaptic and cognitive improvements by inhibition of 2-AG metabolism are through upregulation of microRNA-188-3p in a mouse model of Alzheimer's disease. J Neurosci 2014;34:14919-33.
12. Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang YP, Teng Z, Chen C. Monoacylglycerol lipase is a therapeutic target for Alzheimer's disease. Cell Rep 2012;2:1329-39.
13. Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 2011;334:809-13.
14. Piro JR, Benjamin DI, Duerr JM, Pi Y, Gonzales C, Wood KM, Schwartz JW, Nomura DK, Samad TA. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer's disease. Cell Rep 2012;1:617-23.
15. O'Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol 2007;152:576-82.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Chen C. Endocannabinoid metabolism in neurodegenerative diseases. Neurosciences 2016;3:268-70. http://dx.doi.org/10.20517/2347-8659.2016.46
AMA Style
Chen C. Endocannabinoid metabolism in neurodegenerative diseases. Neuroimmunology and Neuroinflammation. 2016; 3: 268-70. http://dx.doi.org/10.20517/2347-8659.2016.46
Chicago/Turabian Style
Chen, Chu. 2016. "Endocannabinoid metabolism in neurodegenerative diseases" Neuroimmunology and Neuroinflammation. 3: 268-70. http://dx.doi.org/10.20517/2347-8659.2016.46
ACS Style
Chen, C. Endocannabinoid metabolism in neurodegenerative diseases. Neurosciences. 2016, 3, 268-70. http://dx.doi.org/10.20517/2347-8659.2016.46
About This Article
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 2 clicks
Cite This Article 2 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.