
When friend turns foe: central and peripheral neuroinflammation in central nervous system injury


Abstract
Injury to the central nervous system (CNS) is common, and though it has been well studied, many aspects of traumatic brain injury (TBI) and stroke are poorly understood. TBI and stroke are two pathologic events that can cause severe, immediate impact to the neurostructure and function of the CNS, which has been recognized recently to be exacerbated by the body’s own immune response. Although the brain damage induced by the initial trauma is most likely unsalvageable, the secondary immunologic deterioration of neural tissue gives ample opportunity for therapeutic strategists seeking to mitigate TBI’s secondary detrimental effects. The purpose of this paper is to highlight the cell death mechanisms associated with CNS injury with special emphasis on inflammation. The authors discuss sources of inflammation, and introduce the role of the spleen in the systemic response to inflammation after CNS injury.
Keywords
Introduction
Prevalence and incidence of stroke and TBI
Presently, there are over 500 defined neurological disorders caused by trauma, infections, degeneration, autoimmune disorders, structural defects, tumors, or strokes of the brain and spinal cord.[1] Of these disorders, which are characterized by progressive neuronal degeneration and adverse physical and cognitive impairments, stroke and traumatic brain injury (TBI) both have profound impacts on the American population and worldwide. According to the American Stroke Association, stroke is the 5th leading cause of death in the United States, killing 130,000 every year, and is the leading cause of preventable long-term disability. Healthcare services and medications for stroke alone cost a total of over $34 billion per year nationwide.[2] Stroke patients commonly suffer paralysis and other motor dysfunctions, which require extensive therapy.[3] Similarly, TBI accounted for approximately 2.4 million emergency department visits, hospitalizations, or deaths in 2010, and an estimated 5.3 million people are currently living with TBI-related disabilities in the United States.[4] Common features of TBI include bruising, torn tissues, bleeding, and physical damage to the brain resulting in long-term complications or death. Among other effects, characteristic symptoms of mild TBI include fatigue or lethargy, a change in sleep patterns, behavioral or mood changes, and trouble with memory, concentration, attention, or thinking, which are clear clinical manifestations of a neurological disorder.[4]
CNS inflammation in stroke and TBI
The CNS was previously believed to be an immune-privileged site, but a large body of research from the past few decades reveals a complex interplay between glial cells and systemic leukocytes in neuroinflammation. Neuroinflammation is a pathological hallmark of many neurological disorders. Following onset of both stroke and TBI, an acute inflammatory response is mounted to counter initial mechanical damage to brain tissue. Resident microglia become activated and carry out neuroprotective roles via secretion of pro-inflammatory cytokines.[5,6] However, infiltration of peripheral leukocytes through the compromised blood-brain barrier (BBB) coupled with chronically activated microglia propagates chronic inflammation and maintains a toxic environment that cyclically contributes to secondary axonal death.[7] Thus, sequestration of the neuroinflammatory response has been the target of recent therapeutic investigation to attenuate neurological damage. Our laboratory’s long-understanding neuroinflammation in preclinical models of stroke and TBI is the main theme of this review paper.
Therapeutic effects of acute neuroinflammation
It is important to note that CNS inflammation is not entirely detrimental. Acute central inflammation following stroke or TBI is deemed to be "neuroprotective".[3,4] Activated microglia and CNS-specific T cells, for example, help maintain neurogenesis and spatial learning abilities in the adult brain.[8] Ziv et al.[8] described how a protective immune response that intends to eliminate danger and minimize tissue (neuronal) loss must be “regulated and shaped by a well-balanced innate-adaptive dialogue” between microglia and systemic T-lymphocytes.
Acute inflammation in stroke
In the acute inflammatory stage associated with stroke, potent pro-inflammatory cytokines TNF-a, interleukin 6 (IL-6), and IL-1β are upregulated in the cerebrospinal fluid (CSF) and blood in humans.[9] There is evidence that microglia 1 (M1) activated microglia locally produce TNF-a and IL-1, while IL-6 is also produced by neurons.[9] Additionally, Beschorner et al.[10] demonstrated abundant expression of cluster of differentiation 14 (CD14) by ischemia-activated microglia. Since CD14 is key pattern recognition receptor of the innate immune system also found on peripheral monocytes involved in cellular activation, this implicates resident microglial contribution to acute ischemic inflammation.[10] Investigations therefore aim to shift classically activated M1 microglia to the alternatively activated M2 phenotype, which secretes anti-inflammatory cytokines and neurotrophic factors that may contribute to neuroregeneration.[6]
Acute inflammation in TBI
The TBI brain experiences a short, endogenous pro-cell-survival stage in acute neuroinflammation, though this is not sufficient for long-term neuroprotection against chronic neuroinflammation.[11] Primary damage caused by TBI is mechanical, including neuronal injury and disruption of the BBB.[11] Following are two stages of immune response similar to that of stroke: an acute "neuroprotective" stage and a chronic "neurodegenerative" stage.[6] Microglial cells become activated into their pro-inflammatory states,[6] while some afford neuroprotective/regenerative capabilities to combat such damage, but only acutely.[6] Giunta et al.[5] showed that microglia promoted widespread cellular proliferation and focal neurogenesis in the dentate gyrus of the hippocampus. However, this protection appears to be insufficient as activated microglia secreting pro-inflammatory cytokines prove to have a more powerful role in acute inflammation and beyond; chronically activated microglia were found in TBI patients up to two decades after the initial traumatic insult.[5,11] Neuroinflammation in the TBI brain appears to be more widespread. In TBI mouse model, there was significant upregulation of activated microglial cells in both gray and white matter not only at the TBI impacted cortical site but also at proximal adjacent ipsilateral areas and distal areas.[12]
Detrimental effects of chronic neuroinflammation
Chronic neuroinflammation is mediated by both central and peripheral sources.[6,13] If this delicate innate-adaptive, central-peripheral immune dialogue between central microglia and systemic lymphocytes is not properly regulated,[8] the resulting regulatory imbalance sustains a harsh environment by chronic neuroinflammation and causes secondary cell death and adverse neurological deficits. In stroke and TBI, physical trauma to the BBB activates an innate immune response, but the consequences of such mechanical damage extend beyond if not properly mitigated. Peripheral immune cells infiltrate the brain via the compromised BBB and thus exacerbate any existing central neuroinflammation.
Stroke and chronic neuroinflammation
The chronic “degenerative” stage of stroke involves BBB disruption associated with infarction of the parenchyma and cerebral vasculature.[14] At this point, invasion of immune cells and serum proteins through the damaged endothelial cell barrier precipitates adverse physiological consequences such as propagation of neuroinflammation, increased cerebral pressure and increased cellular death,[7] thus, the initial brain damage caused by stroke is exacerbated by this secondary BBB destruction. Ischemic stroke was found to induce an autoimmune response to neuronal antigens that can possibly potentiate or ameliorate long-term neuroinflammation.[15]
TBI and chronic neuroinflammation
Similarly, peripheral immune cells enter the TBI brain through the damaged BBB, continuing to release pro-inflammatory cytokines, attract more immune cells, and activate microglia, rendering a cycle of extended inflammation in the brain. A decrease in hippocampal neurons and decline in cell proliferation in the ipsilateral subventricular zone and the subgranular zoneconsistent with TBI pathology was also observed, indicating the deleterious effects of chronic inflammation.[12] Low graft survival of stem cells has been documented in the TBI brain during investigational cell therapy treatments, which may be attributed to the harsh environment caused by this secondary neuroinflammatory response.[16] Secondary neuronal damage caused by chronic inflammation of activated microglial cells appears to be the link between TBI and Alzheimer's disease (AD) neuropathology.[5] Several neuropathological hallmarks of Alzheimer’s were observed in brains of chronic TBI patients, namelyamyloid-beta (AB) plaques and neurofibrillary tangles.[17] AB42 aggregation has been attributed to aging microglia’s reduced phagocytic capacity and therefore decline in microglial clearance of AB plaques.[5] Further, post mortem analysis of TBI patient brains showed senile AB plaques present across all age groups, including children, suggesting that TBI is indeed the cause of AD in these patients.[5] Chronic inflammation and subsequent neuronal degeneration makes patients vulnerable to neurological deficits. In addition to Alzheimer’s disease, TBI is strongly associated with several other neurologic disorders 6 months or more after injury.[18] Uryu et al.[19] characterized multiple proteins implicated in neurodegenerative diseases in post-mortem TBI brains, formed within 4 h to 5 weeks of injury. AB plaques co-accumulated with amyloid precursor protein, beta-secretase, and presenilin 1 and the presence of alpha synuclein was confirmed all within the axonal bulbs.[19] Alpha-synuclein is a presynaptic neuronal protein that aggregates to form toxic protofibrils which are then released from dying neurons to contribute to pathogenesis and also accumulated in the CSF following TBI in infants and children.[20,21] Synucleinopathy additionally links TBI and Alzheimer’s to Parkinson’s disease (PD). PD displays the same active contribution of reactive microglia to loss of dopaminergic neurons.[4] This “reactive gliosis” upregulates pro-inflammatory cytokines in both the brain and CSF of PD patients.[4] Finally, microglia are the first to respond to a traumatic spinal cord injury and were found to remain activated for at least 6 months post-injury in humans.[22] Intraspinal neurons and astrocytes contribute by producing pro-inflammatory cytokine IL-1B.[22]
The next section of this paper discusses the sources of inflammation after TBI and stroke, and how inflammation contributes to the pathogenesis of these disorders.
Sources of inflammation: central and peripheral
Central and peripheral sources of neuroinflammation
There are both central and peripheral sources contributing to neuroinflammation of neurological disorders [Figure 1]. Traditionally, chronically activated microglia has been the targets of therapeutic treatment but research suggests another viable option. Stem cells were shown to preferentially migrate to the spleen following ischemic stroke,[3] and splenectomies following stroke or TBI reduced neuronal damage.[23-26] This supports the concept of an existing dialogue between the local CNS and systemic immune system, because the spleen is the primary source of systemic inflammation, it has been the focus of investigation in the “brain-spleen inflammatory coupling” associated with stroke, TBI, and other neurological disorders.[18] Therefore, sequestration of inflammation to the spleen in order to attenuate chronic neuroinflammation and improve the efficacy of stem cell therapy provides a promising therapeutic approach to stroke and TBI treatment.
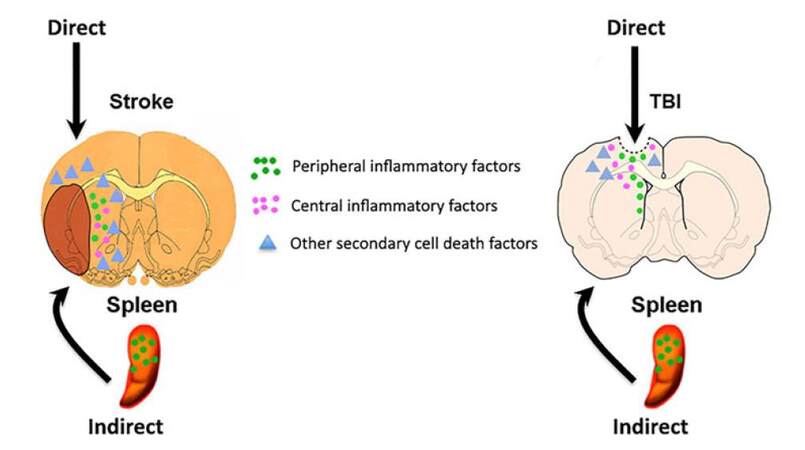
Figure 1. Central and peripheral sources of inflammation. Following CNS injury, such as stroke and TBI, the traditional concept entails a robust inflammatory response within the brain, but equally compelling recent evidence has demonstrated an active inflammatory response, especially from the spleen, contributing to the progression of the disease. Together with other secondary cell death factors, both central and peripheral inflammation exacerbate CNS injury. CNS: central nervous system; TBI: traumatic brain injury
We have chosen to separate the body’s response into two categories, central inflammation and peripheral inflammation, in an attempt to show the peripheral immune response’s contribution to the cognitive decline following TBI and stroke, and elucidate the potential for research into novel therapies. By central inflammation we are referring to the role of resident cells of the CNS in inflammation, and by peripheral inflammation we are referring to the contribution of the systemic immune response to neuroinflammation after traumatic brain injury or stroke.
Central source of neuroinflammation
Traditionally, the immune system is thought to be non-existent in the CNS. Accumulating evidence now suggests that the CNS has its own immune system, and in addition, the peripheral immune system may play a role in neuroinflammation.[27-29] As mentioned in the previous section, the negative outcomes of stroke and TBI are exacerbated by the body’s reaction to the injury. The body’s inflammatory response, which protects against infection, also induces a chronic state of deterioration in the CNS, exacerbating the neurological deficits caused by the initial injury.
Glia
When talking about the CNS and immunity, it is important to highlight the role of glia. Glia are non-neuronal cells that maintain homeostasis; two common glia are astrocytes and microglia. Astrocytes make up the blood brain barrier, which separates the CNS from the rest of the body, including the peripheral immune system. To prevent entry of peripheral immune cells and counter otherwise widespread cerebral inflammation, the BBB forms a physical boundary with a specialized microvasculature consisting of endothelial cells connected by adherent and tight junctions.[30] This allows control of cerebral homeostasis via selective transport of molecules and cells.[31] When the BBB is compromised and microglia are activated, inflammation of the brain ensues.
Microglia
Microglias are the innate immune cells of the CNS. They are cells of myeloid lineage that populate the CNS during embryogenesis, and thus act similarly to the peripherally acting cells of the myeloid lineage: macrophages and dendritic cells.[32] Resident microglia are capable of clearing foreign pathogens or mediating a local, innate immune response in the brain. Microglia can be observed along a continuum of three idealized phenotypic states: resting, activated non-phagocytic (antigen presenting cell like), and activated phagocytic, depending on their level of activation, which is dictated by the amount and type of cytokines in the surrounding microenvironment.[33-35] Microglia sense the microenvironment, and mount a protective immune response after TBI, however the response is excessive and prolonged, and ends up leading to further degeneration instead of regeneration and repair.[11] Like macrophages, microglia can polarize into two subcategories, M1 or M2.[6] M1 is predominantly pro-inflammatory, and secretes high levels of pro-inflammatory cytokines like IL-1, IL12, and IFN-γ, and low levels of anti inflammatory cytokines like IL-10.[6] M2 is typically anti-inflammatory, and acts to clear debris and promote regeneration.[3] Pro-inflammatory M1 macrophages predominate after CNS injury.[6,11]
Central immune cells contribute to diffuse axonal injury
As mentioned in the previous section, current research shows that neuroinflammation is a major source of secondary cell death after TBI and stroke.[36-38] The major players in neuroinflammation are immune cells, microglia, cytokines, and chemokines that altogether exacerbate neuronal cell death after initial injury,[39] and lead to a phenomenon known as diffuse axonal injury, which leads to extensive lesions of cerebral white matter over a widespread area, outside of the initial lesion.[27,40] Insult to the CNS, either TBI or stroke, generates a neuroprotective immune response to prevent infection and stimulate neuronal repair. After injury to the CNS, neurons, astrocytes, and microglial cells all respond to play a role in the inflammatory response that ensues.[27] Glutamate release after TBI causes hyperactivity of neurons, leading to prolonged levels of intracellular calcium, and eventually cell death, this is known as excitotoxicity.[28] Both astrocytes and microglia contribute to the inflammatory response by producing chemokines; chemokines then attract monocytes to the site of injury.[27]
Microglial cell function and CNS injury
Microglia has several distinctive properties that allow them to participate in the pathological neurodegenerative processes after CNS injury. Upon activation, microglia undergo morphological changes, proliferation, and expression of major histocompatibility complex (MHC) class II molecules.[35] They are capable of phagocytosis of damaged and fragmented neuronal elements, antigen presentation to T lymphocytes, and production of soluble factors that at sufficient concentrations can induce further tissue injury and gliosis.[6,35] Pro-inflammatory cytokines, such as these released after neuronal injury, are strong activators of microglia.[41-43] Once activated, microglia produce more pro-inflammatory cytokines such as IL-1β, which ultimately leads to an extensive chronic proinflammatory state in the CNS.[11,44]
Both TBI and stroke is characterized by an initial insult to the CNS, compromising the blood brain barrier and causing microglial activation.[6] Prolonged microglial activation leads to a chronic inflammatory response that causes excitotoxicity, oxidative stress, mitochondrial dysfunction, blood brain barrier disruption, and inflammation.[45-49] Inflammation activates microglia, which then release more pro-inflammatory cytokines such as TNFα and IL-1β which cause upregulation of cell adhesion molecules in the surrounding vasculature and lead to a further increase in blood brain barrier permeability, and allows systemic involvement in neuroinflammation.[6,11,44,50]
M1 microglia afford neurodegenerative effects
As mentioned before, microglia can polarize into either M1 or M2 when activated. After CNS injury, both types of microglia are present, but type M1 tends to predominate and persist, leading to neurodegeneration instead of repair.[6,11] The capacity of the microglia to drive the response to CNS injury towards either further damage or repair exemplifies its role as a key player in central immunity, and is the reason it has become the target in studying the cognitive decline due to neuroinflammation after traumatic brain injury and stroke.
Pathological processes involving microglia
In addition to recruiting other immune cells to the site of injury, microglia contributes directly to neuronal damage through several pathological processes. When highly activated, microglia are capable of phagocytosis.[35] In the case of CNS damage, activated microglia phagocytose neuronal elements. Activated microglia also produce reactive oxygen species and reactive nitrogen species.[6] These are highly reactive molecules that increase the oxidative stress, and lead to destruction of neuronal cell membranes through lipoperoxidation.[51] Cell membranes allow the cell to maintain homeostasis; once the membrane is compromised the cell can die.
Chronic activation of microglia
The secondary inflammatory damage after insult to the CNS can be observed as cognitive decline days and even up to years after initial injury.[11] In animal models it has been shown that microglia can be active for up to one year after TBI.[6,52-55] Post mortem studies have shown activated microglia up to 17 years after TBI in adult humans.[6,55] The observed cognitive decline, in conjunction with the presence of activated microglia after injury suggests a persistent chronic inflammatory stage mediated by microglia that exacerbates TBI and stroke pathology.
Peripheral source of neuroinflammation
The immune system consists of a network of cells, tissues, and organs that coordinate to protect the body from foreign pathogens, and promote tissue healing and regeneration. When the body is injured, cell death leads to leakage of nuclear or cytosolic proteins or protein fragments into the extracellular space. These intracellular fragments are pro-inflammatory signals called damage associated molecular patterns (DAMPS). DAMPS are recognized by pattern recognition receptors on dendritic cells, macrophages, and other cells such as vascular cells, epithelial cells, and fibroblasts, and elicit a pro-inflammatory response from these cells.[56] Once an immune response is mounted, it can either persist as chronic inflammation, or move towards resolution and tissue healing.
Peripheral immune cells are recruited after CNS insult
The inflammatory response activates the complement system to recruit immune cells to the intrathecal compartment;[48] neutrophils, monocytes and lymphocytes all cross the blood brain barrier and chemotax towards the site of injury.[57] Once these cells have reached the site of injury, they are activated to secrete free radicals, pro-inflammatory cytokines, prostaglandins and other inflammatory mediators, resulting in recruitment of more immune cells and microglia to the site of injury.[57,58] A great deal of research has been done on microglia, highlighting it as the key player in coordinating the immune response after an insult to the CNS.
Systemic immune response
The role of resident immune cells in the CNS after TBI is only part of the story. To get a full picture of the inflammatory response to TBI we must also look at the peripheral immune system. Multi-organ damage following TBI can lead to a more robust immune response in the brain,[28] highlighting the possibility that systemic inflammation could play a role in neuroinflammation. Because the BBB is compromised in a CNS injury, circulating inflammatory cells and cytokines can access the brain and contribute to the pathogenesis of TBI.[28,59] Leakage of chemokines and other inflammatory molecules through the compromised blood brain barrier into systemic circulation can attract peripheral immune cells to the site of injury.[28] This can potentially lead to an overactive inflammatory response known as systemic immune response syndrome.[28] Negative feedback to systemic inflammation is provided by the hypothalamus-pituitary-adrenal axis and sympathetic nervous system efferents.[28] In TBI, an imbalance between systemic immune response and negative feedback can lead to either excessive organ damage or susceptibility to infections and lack of regeneration.[28]
Role of peripheral chemokines
Cytokines and chemokines are very important to the pathogenesis of TBI. Although their exact role is unclear, data suggests that cytokines play a pivotal role in the body’s response to TBI. After insult to the CNS, upregulation of the following cytokines occurs: TNFα, IL-1β, IL-2, IL-6, IL-8, IL-4, IL-18.[60-62] One important peripherally secreted chemokine chemokine (C-C motif) ligand 20 (CCL20) is upregulated after TBI, and interacts with CC chemokine receptor 6 (CCR6) to induce chemotaxis of T cells, B cells, and dendritic cells.[28] These cells can be found in the spleen, and are known to contribute to the pathogenesis of TBI.[28] Other peripheral cells are found at the site of injury, and contribute to the inflammatory process. It is known that the concentration of neutrophils peaks around 3-24 h after injury, and the concentration of monocytes peaks around 1-2 days after spinal cord injury.[63]
Synergistic central and peripheral inflammation
In the case of chronic neuroinflammation, both central and peripheral sources of inflammation work together to create a hyperactive immune response that ultimately leads to further damage rather than repair of neural tissue.[27,28]
It is well known that CCL20 acts as a chemokine for CCR6 expressing cells. In an experimental autoimmune encephalomyelitis (EAE) model, which is an animal model for brain inflammation similar to multiple sclerosis in humans, researchers have observed that CCL20 acts as a ligand for CCR6, allowing homing of lymphocytes, and other leukocytes to neural tissue.[64] In this specific case, it allows trafficking of Th17 or Th1 CD4+ Th cells, which release pro-inflammatory cytokines that can cause chronic inflammation.[64,65] CCL20 expression in the choroid plexus allows passage of CCR6+ Th cells to enter the CNS in the uninflamed brain, which then allows a CCR6 independent pathway of recruitment of T cells to the brain parenchyma in the EAE model.[64] CCL20 expression is upregulated by proinflammatory cytokines IL6, and IL17.[65]
In a lateral fluid percussion model of TBI, Das shows that CCL20 expression is upregulated in the thymus and spleen 24 h after TBI, and upregulated in the cortex and hippocampus 48 h after TBI.[27] Based on the evidence obtained from the EAE model, this suggests a mechanism for peripheral involvement in neuroinflammation.[27,28,64] The fact that CCL20 is expressed in the spleen and thymus after TBI, before it is expressed in the brain, and brain CCL20 expression is reduced in rat’s who’s spleens have been removed suggests a peripheral mechanism of activation for CCL20 expression in the CNS.[27] It also speaks to the role that CCL20 plays in neuroinflammation after TBI. In other words, CCL20 upregulation in the spleen and thymus after TBI could indicate a peripheral signal that drives neuronal degeneration.[27]
In stroke, we see a similar peripheral involvement in chronic inflammation after insult. Nguyen and colleagues characterized the cytokine profiles in mice after ischemic CNS infarct, and showed a polarized T cell response based on the type of mouse used.[66] C57BL/6 mice had a Th1 polarized response, and BALB/c mice had a Th2 polarized response.[66] This suggests that the chronic inflammatory response in stroke patients could follow different courses, depending on the individual afflicted.[66]
In all of these instances, peripheral involvement in neuroinflammation acts in addition to the central inflammation perpetuated by microglia and other inflammatory mediators. In summary, injury to the CNS leads to a peripheral and central response that act together to cause inflammation, which eventually leads to a chronic inflammatory state that causes neural degeneration rather than repair and resolution after insult.
Although a strong connection between CNS injury and the immune system has been shown, little research has been directed at exploring the role of the thymus in TBI. Recent studies have shown upregulation of CCL20 in the thymus after TBI.[27] Other studies have shown that the liver may play a role in exacerbating the neuronal degeneration after TBI (Campbell et al.[67]). Depletion of hepatic Kupffer Cells reduced ED-1 positive macrophage and neutrophil migration into an IL-1β injected brain.[67] As a reservoir of peripheral immune cells, the spleen has been shown to play a major role in traumatic brain injury.[28]
Role of the spleen as a major peripheral inflammatory contributor to CNS injury
Function of the spleen
It has been shown that the spleen initiates an immune response that exacerbates the pathology of stroke and TBI, however the connection between brain injury and a splenic response has yet to be fully elucidated. The spleen has several functions in the body. It is a major lymphatic organ that lies within the peritoneal cavity; it actively monitors the body’s circulation and filters blood.[68,69] In humans, the spleen plays a role in the mononuclear phagocyte system, recycles iron from old red blood cells, and mounts a defense against blood borne pathogens.[69,70]
The spleen as a reservoir of systemic immune cells
The spleen is also a reservoir of platelets, peripheral macrophages, and other immune cells.[28,70,71] Scientists used to think that the majority of monocytes patrolled the circulation, and irreversibly differentiated into macrophages and dendritic cells upon extravasation and tissue entry.[70] It is now known that the spleen actually acts as a reservoir for undifferentiated monocytes, and monocytes in the spleen outnumber monocytes patrolling the circulation.[68,70] This means that a majority of undifferentiated monocytes reside in the spleen, waiting to be deployed. Monocytes, distinct from macrophages and dendritic cells, cluster in the cords of the subcapsular red pulp of the spleen.[70]
Splenic immune cells home to injuries throughout the body
Spleen has the ability to rapidly deploy this cohort of undifferentiated monocytes.[68,70,72] Splenic monocytes have been shown to exit the spleen and accumulate in the heart after myocardial infarction to participate in immunological processes such as wound healing.[70] In the context of CNS injury, several aspects of the splenic response have been observed. One study has shown that the number of T cells in the spleen decreases 1-2 days after traumatic brain injury.[72] In a study that induced middle cerebral artery occlusion (MCAO) on mice, researchers observed both splenic contraction and a reduction in the number of splenic cells after stroke was induced.[68] In that same experiment, splenic contraction coincided with a decrease in monocytes in the spleen, and a concurrent increase in the same subsets of monocytes in the ischemic brain.[68]
Spleen and CNS injuries
Since research has shown that the immunologic response to TBI and stroke can in fact exacerbate the damage from the initial injury, and further research has suggested that the spleen plays a role in mounting an immune response to the injured CNS, researchers tried knocking out the function of the spleen to observe the effect on TBI and stroke. Ajmo et al.[73] showed that removal of the spleen two weeks before permanent MCAO significantly reduced the infarction volume. In another study, researchers showed that removal of the spleen just before temporary MCAO caused a reduction in the accumulation of monocytes in the brain, but did not significantly change the infarct size.[68] In addition, splenectomy in rats immediately after traumatic brain injury reduced circulating levels of pro-inflammatory cytokines, decreased mortality, and increased cognitive functioning.[74] Furthermore, it has been shown that splenectomy immediately after mild TBI in rats attenuated CCL20 chemokine expression and neurodegeneration in the brain.[27]
The spleen and cognitive deficits
Although splenectomy is probably not advisable in human patients that have received a traumatic brain injury or stroke, these studies highlight the importance of the spleen in CNS injury. The splenectomy studies, in conjunction with the studies that show a loss of immune cells from the spleen and the appearance of the same subset of cells in infarcted brain tissue after stroke, lead researchers to believe that the spleen is bolstering the immune response in CNS injury. This data suggests that the spleen plays a role in the secondary wave of neurodegeneration after TBI and stroke, leading to more severe cognitive deficits.
Blood flow and microglial cytokines
Quantifying blood flow to the spleen after injury is important to understand the role of the spleen as a mediator in the immune process. Several ways of measuring blood flow to the spleen have been described. The control of blood supply to the spleen involves several aspects. It has been shown that IL-1 increases splenic blood flow by affecting the sympathetic vasoconstrictor tonus. In order for the spleen to remain perfused, resident macrophages must produce IL-1β to counteract noradrenergic vasoconstriction.[75] Sympathetic tone reduces perfusion, whereas inflammatory mediators such as IL-1β increase perfusion.
Blood flow after CNS insult and spleen
Blood flow to the spleen after TBI shows a biphasic hemodynamic pattern. In a study by Yuan et al.,[76] blood flow measurements were taken at 5 min, 15 min, 30 min, and 60 min after injury. Fluid percussion brain injury produced an immediate systemic hypertension followed by hypotension and low cardiac output. Immediately after TBI, blood flow to all organs either remained the same or increased for 30 min, then gradually decreased.[76] Blood flow to the kidney and spleen were decreased the most after TBI, which was attributed to sympathetic activity because of the high amount of sympathetic vasoconstrictor fibers running to those organs.[76] The resulting hyperactive sympathetic response, similar to what happens after TBI, is characterized by a widespread vasoconstriction that is also selective; flow is decreased through kidneys and splanchnic organs such as the spleen but not decreased to the heart.[76] It will be interesting if a similar phenomenon characterized by blood flow alterations in the spleen accompanies stroke.
Brain-spleen inflammatory coupling in CNS injuries
Lastly, it has been shown that immune cells in the spleen respond to cholinergic input. Studies have shown that there is a correlation between brain injury and autonomic release of pro-inflammatory cytokines from splenic macrophages. In a concept known as “brain-spleen inflammatory coupling”, researchers have hypothesized that the changes in autonomic input after CNS injury lead to systemic responses, including a response from the spleen. In increase in pro-inflammatory cytokines in the brain after CNS injury stimulates the posterior hypothalamus to increase sympathetic tone, leading to catecholamine release from the adrenal glands and peripheral vasoconstriction.[71] It has been shown that macrophages in the liver respond to adrenergic/cholinergic input, and thus can respond to changes in autonomic tone.[71,73,77] The body responds to CNS injury by increasing sympathetic tone, and immune cells in the spleen respond to this adrenergic input by producing large amounts of the pro-inflammatory cytokines TNF-α and IL-1β.[71,78] It is hypothesized that this systemic inflammatory response to TBI and stroke exacerbates TBI pathology.
Whereas elevated sympathetic tone increases the pro-inflammatory response from the spleen, increased parasympathetic tone has been shown to decrease the pro-inflammatory response from the spleen.[71] Macrophages in the spleen express a nicotinic catecholamine receptor α7nAChR which responds to parasympathetic input by reducing production of the pro-inflammatory cytokine TNFα.[71,79] Selectively activating this receptor after stroke in rats was shown to reduce infarct size and improve survival.[80] Other studies have shown improved neurological outcomes in animal models for stroke by either direct or indirect stimulation of this receptor.[71] This evidence suggests that sympathetic and parasympathetic tone affect the splenic response to CNS injury, and targeting this response has the potential for novel therapeutic strategies.
A paradigm-shift in our understanding of CNS inflammation
Many neurological disorders, including traditionally considered acute injuries such as stroke and TBI, have now been recognized as being plagued by neuroinflammation, which significantly contributes to the disease progression and is associated with secondary cell death reminiscent of chronic neurodegeneration. A worsening prognosis of stroke and TBI has implicated massive inflammation, arising not just from the injured but equally robustly detected from the peripheral organs, specifically the spleen. By investigating the sources and mechanisms of neuroinflammation, in particular the role of spleen-mediated inflammatory response, novel cell death pathways as well as innovative therapeutic targets are identified for interrupting this inflammatory process and providing avenues for ameliorating the secondary cell death of stroke and TBI. Recognizing that both the CNS and the peripheral immune system play important roles in the inflammatory process is a key to deciphering the cellular, molecular, and genetic pathways of neuroinflammation, and broadens our scope for developing new anti-inflammation-based treatments. As a major secondary lymphoid organ, the spleen intimately participates in the peripheral immune response that can exacerbate neuroinflammation and the subsequent chronic neurodegeneration. Accordingly, when contemplating with an anti-inflammatory strategy for stroke and TBI, an in-depth examination of this accumulating preclinical evidence suggesting the involvement of central and systemic sources of inflammation will be critical to a better understanding of the pathology and treatment of the secondary cell death that closely approximates the disease progression of stroke and TBI.
Authors’ contributions
Conceptualized the topic: C.V. Borlongan
Drafted, wrote, and approved the final version of this paper: P. Marcet, N. Santos, C.V. Borlongan
Acknowledgments
The authors thank the staff of the USF Borlongan Neural Transplantation Laboratory for the conceptual discussion of topics covered in this manuscript.
Financial support and sponsorship
C.V. Borlongan is supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke 1R01NS071956, 1R01NS090962, 1R21NS089851, 1R21NS094087 and 1R21NS094908, Department of Defense W81XWH-11-1-0634, James and Esther King Foundation for Biomedical Research Program, SanBio Inc., KM Pharmaceuticals, NeuralStem Inc, and Karyopharm Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the sponsors.
Conflicts of interest
C.V. Borlongan has patents and patent applications relating to stem cell therapy for stroke and TBI.
Patient consent
There is no patient data involved.
Ethics approval
Not applicable.
REFERENCES
1. Borlongan CV, Burns J, Tajiri N, Stahl CE, Weinbren NL, Shojo H, Sanberg PR, Emerich DF, Kaneko Y, van Loveren HR. Epidemiological survey-based formulae to approximate incidence and prevalence of neurological disorders in the United States: a meta-analysis. PLoS One 2013;8:e78490.
2. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics -- 2015 update: a report from the American Heart Association. Circulation 2015;131:e29-322.
3. Acosta SA, Tajiri N, de la Pena I, Bastawrous M, Sanberg PR, Kaneko Y, Borlongan CV. Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson's disease. J Cell Physiol 2015;230:1024-32.
4. Acosta SA, Tajiri N, Hoover J, Kaneko Y, Borlongan CV. Intravenous bone marrow stem cell grafts preferentially migrate to spleen and abrogate chronic inflammation in stroke. Stroke 2015;46:2616-27.
5. Giunta B, Obregon D, Velisetty R, Sanberg PR, Borlongan CV, Tan J. The immunology of traumatic brain injury: a prime target for Alzheimer's disease prevention. J Neuroinflammation 2012;9:185.
6. Hernandez-Ontiveros DG, Tajiri N, Acosta S, Giunta B, Tan J, Borlongan CV. Microglia activation as a biomarker for traumatic brain injury. Front Neurol 2013;4:30.
7. Borlongan CV, Glover LE, Sanberg PR, Hess DC. Permeating the blood brain barrier and abrogating the inflammation in stroke: implications for stroke therapy. Curr Pharm Des 2012;18:3670-6.
8. Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci 2006;9:268-75.
9. Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab 2012;32:1677-98.
10. Beschorner R, Schluesener HJ, Gözalan F, Meyermann R, Schwab JM. Infiltrating CD14+ monocytes and expression of CD14 by activated parenchymal microglia/macrophages contribute to the pool of CD14+ cells in ischemic brain lesions. J Neuroimmunol 2002;126:107-15.
11. Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat 2015;11:97-106.
12. Acosta SA, Tajiri N, Shinozuka K, Ishikawa H, Grimmig B, Diamond DM, Sanberg PR, Bickford PC, Kaneko Y, Borlongan CV. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS One 2013;8:e53376.
13. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 2010;87:779-89.
15. Ortega SB, Noorbhai I, Poinsatte K, Kong X, Anderson A, Monson NL, Stowe AM. Stroke induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov Med 2015;19:381-92.
16. Dela Peña I, Sanberg PR, Acosta S, Tajiri N, Lin SZ, Borlongan CV. Stem cells and G-CSF for treating neuroinflammation in traumatic brain injury: aging as a comorbidity factor. J Neurosurg Sci 2014;58:145-9.
18. Bazarian JJ, Cernak I, Noble-Haeusslein L, Potolicchio S, Temkin N. Long-term neurologic outcomes after traumatic brain injury. J Head Trauma Rehabil 2009;24:439-51.
19. Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol 2007;208:185-92.
20. Stefanis L. α-Synuclein in Parkinson's disease. Cold Spring Harb Perspect Med 2012;2:a009399.
21. Su E, Bell MJ, Wisniewski SR, Adelson PD, Janesko-Feldman KL, Salonia R, Clark RS, Kochanek PM, Kagan VE, Bayır H. α-Synuclein levels are elevated in cerebrospinal fluid following traumatic brain injury in infants and children: the effect of therapeutic hypothermia. Dev Neurosci 2010;32:385-95.
22. Hawthorne AL, Popovich PG. Emerging concepts in myeloid cell biology after spinal cord injury. Neurotherapeutics 2011;8:252-61.
23. Ajmo CT Jr, Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, Pennypacker KR. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res 2008;86:2227-34.
24. Belinga VF, Wu GJ, Yan FL, Limbenga EA. Splenectomy following MCAO inhibits the TLR4-NF-κB signaling pathway and protects the brain from neurodegeneration in rats. J Neuroimmunol 2016;293:105-13.
25. Chu W, Li M, Li F, Hu R, Chen Z, Lin J, Feng H. Immediate splenectomy down-regulates the MAPK-NF-κB signaling pathway in rat brain after severe traumatic brain injury. J Trauma Acute Care Surg 2013;74:1446-53.
26. Seifert HA, Leonardo CC, Hall AA, Rowe DD, Collier LA, Benkovic SA, Willing AE, Pennypacker KR. The spleen contributes to stroke induced neurodegeneration through interferon gamma signaling. Metab Brain Dis 2012;27:131-41.
27. Das M, Leonardo CC, Rangooni S, Pennypacker KR, Mohapatra S, Mohapatra SS. Lateral fluid percussion injury of the brain induces CCL20 inflammatory chemokine expression in rats. J Neuroinflammation 2011;8:148.
28. Das M, Mohapatra S, Mohapatra SS. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J Neuroinflammation 2012;9:236.
29. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest 2012;122:1164-71.
30. Shinozuka K, Dailey T, Tajiri N, Ishikawa H, Kim DW, Pabon M, Acosta S, Kaneko Y, Borlongan CV. Stem cells for neurovascular repair in stroke. J Stem Cell Res Ther 2013;4:12912.
31. Hawkins RA, O'Kane RL, Simpson IA, Viña JR. Structure of the blood-brain barrier and its role in the transport of amino acids. J Nutr 2006;136:S218-26.
32. Town T, Nikolic V, Tan J. The microglial "activation" continuum: from innate to adaptive responses. J Neuroinflammation 2005;2:24.
33. Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, Hart RP, Schwartz M. Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem 2005;92:997-1009.
34. van Landeghem FK, Stover JF, Bechmann I, Brück W, Unterberg A, Bührer C, von Deimling A. Early expression of glutamate transporter proteins in ramified microglia after controlled cortical impact injury in the rat. Glia 2001;35:167-79.
35. Williams K, Bar-Or A, Ulvestad E, Olivier A, Antel JP, Yong VW. Biology of adult human microglia in culture: comparisons with peripheral blood monocytes and astrocytes. J Neuropathol Exp Neurol 1992;51:538-49.
36. Acosta SA, Diamond DM, Wolfe S, Tajiri N, Shinozuka K, Ishikawa H, Hernandez DG, Sanberg PR, Kaneko Y, Borlongan CV. Influence of post-traumatic stress disorder on neuroinflammation and cell proliferation in a rat model of traumatic brain injury. PLoS One 2013;8:e81585.
37. Acosta SA, Tajiri N, Shinozuka K, Ishikawa H, Sanberg PR, Sanchez-Ramos J, Song S, Kaneko Y, Borlongan CV. Combination therapy of human umbilical cord blood cells and granulocyte colony stimulating factor reduces histopathological and motor impairments in an experimental model of chronic traumatic brain injury. PLoS One 2014;9:e90953.
38. Tajiri N, Acosta SA, Shahaduzzaman M, Ishikawa H, Shinozuka K, Pabon M, Hernandez-Ontiveros D, Kim DW, Metcalf C, Staples M, Dailey T, Vasconcellos J, Franyuti G, Gould L, Patel N, Cooper D, Kaneko Y, Borlongan CV, Bickford PC. Intravenous transplants of human adipose-derived stem cell protect the brain from traumatic brain injury-induced neurodegeneration and motor and cognitive impairments: cell graft biodistribution and soluble factors in young and aged rats. J Neurosci 2014;34:313-26.
39. Tajiri N, Kellogg SL, Shimizu T, Arendash GW, Borlongan CV. Traumatic brain injury precipitates cognitive impairment and extracellular Aβ aggregation in Alzheimer's disease transgenic mice. PLoS One 2013;8:e78851.
40. Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 1989;15:49-59.
41. Frei K, Siepl C, Groscurth P, Bodmer S, Schwerdel C, Fontana A. Antigen presentation and tumor cytotoxicity by interferon-gamma-treated microglial cells. Eur J Immunol 1987;17:1271-8.
42. Panek RB, Benveniste EN. Class II MHC gene expression in microglia. Regulation by the cytokines IFN-gamma, TNF-alpha, and TGF-beta. J Immunol 1995;154:2846-54.
43. Suzumura A, Mezitis SG, Gonatas NK, Silberberg DH. MHC antigen expression on bulk isolated macrophage-microglia from newborn mouse brain: induction of Ia antigen expression by gamma-interferon. J Neuroimmunol 1987;15:263-78.
44. Walker DG, Kim SU, McGeer PL. Complement and cytokine gene expression in cultured microglial derived from postmortem human brains. J Neurosci Res 1995;40:478-93.
45. Bramlett HM, Dietrich WD. Long-term consequences of traumatic brain injury: current status of potential mechanisms of injury and neurological outcomes. J Neurotrauma 2015;32:1834-48.
46. Glover LE, Tajiri N, Lau T, Kaneko Y, van Loveren H, Borlongan CV. Immediate, but not delayed, microsurgical skull reconstruction exacerbates brain damage in experimental traumatic brain injury model. PLoS One 2012;7:e33646.
47. Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 2010;31:596-604.
48. Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury -- an inflammatory disease? Brain Res Rev 2005;48:388-99.
49. Shahaduzzaman M, Acosta S, Bickford PC, Borlongan CV. α-Synuclein is a pathological link and therapeutic target for Parkinson's disease and traumatic brain injury. Med Hypotheses 2013;81:675-80.
50. Yu S, Kaneko Y, Bae E, Stahl CE, Wang Y, van Loveren H, Sanberg PR, Borlongan CV. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res 2009;1287:157-63.
51. Dasuri K, Zhang L, Keller JN. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med 2013;62:170-85.
52. Rojanathammanee L, Floden AM, Manocha GD, Combs CK. Attenuation of microglial activation in a mouse model of Alzheimer's disease via NFAT inhibition. J Neuroinflammation 2015;12:42.
53. Nagamoto-Combs K, Combs CK. Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells). J Neurosci 2010;30:9641-6.
54. Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI, McIntosh TK. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma 1997;14:715-27.
55. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 2011;70:374-83.
56. Toubai T, Mathewson ND, Magenau J, Reddy P. Danger signals and graft-versus-host disease: current understanding and future perspectives. Front Immunol 2016;7:539.
57. Fluiter K, Opperhuizen AL, Morgan BP, Baas F, Ramaglia V. Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J Immunol 2014;192:2339-48.
58. Bellander BM, Singhrao SK, Ohlsson M, Mattsson P, Svensson M. Complement activation in the human brain after traumatic head injury. J Neurotrauma 2001;18:1295-311.
59. Utagawa A, Truettner JS, Dietrich WD, Bramlett HM. Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Exp Neurol 2008;211:283-91.
60. DiPiro JT, Howdieshell TR, Goddard JK, Callaway DB, Hamilton RG, Mansberger AR Jr. Association of interleukin-4 plasma levels with traumatic injury and clinical course. Arch Surg 1995;130:1159-62.
61. Marcu AC, Paccione KE, Barbee RW, Diegelmann RF, Ivatury RR, Ward KR, Loria RM. Androstenetriol immunomodulation improves survival in a severe trauma hemorrhage shock model. J Trauma 2007;63:662-9.
63. Schwab JM, Zhang Y, Kopp MA, Brommer B, Popovich PG. The paradox of chronic neuroinflammation, systemic immune suppression, autoimmunity after traumatic chronic spinal cord injury. Exp Neurol 2014;258:121-9.
64. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 2009;10:514-23.
65. Meares GP, Ma X, Qin H, Benveniste EN. Regulation of CCL20 expression in astrocytes by IL-6 and IL-17. Glia 2012;60:771-81.
66. Nguyen TV, Frye JB, Zbesko JC, Stepanovic K, Hayes M, Urzua A, Serrano G, Beach TG, Doyle KP. Multiplex immunoassay characterization and species comparison of inflammation in acute and non-acute ischemic infarcts in human and mouse brain tissue. Acta Neuropathol Commun 2016;4:100.
67. Campbell SJ, Zahid I, Losey P, Law S, Jiang Y, Bilgen M, van Rooijen N, Morsali D, Davis AE, Anthony DC. Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology 2008;55:780-7.
68. Kim E, Yang J, Beltran CD, Cho S. Role of spleen-derived monocytes/macrophages in acute ischemic brain injury. J Cereb Blood Flow Metab 2014;34:1411-9.
70. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009;325:612-6.
71. Rasouli J, Lekhraj R, Ozbalik M, Lalezari P, Casper D. Brain-spleen inflammatory coupling: a literature review. Einstein J Biol Med 2011;27:74-77.
72. Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One 2012;7:e41892.
73. Ajmo CT Jr, Collier LA, Leonardo CC, Hall AA, Green SM, Womble TA, Cuevas J, Willing AE, Pennypacker KR. Blockade of adrenoreceptors inhibits the splenic response to stroke. Exp Neurol 2009;218:47-55.
74. Li M, Li F, Luo C, Shan Y, Zhang L, Qian Z, Zhu G, Lin J, Feng H. Immediate splenectomy decreases mortality and improves cognitive function of rats after severe traumatic brain injury. J Trauma 2011;71:141-7.
75. Rogausch H, Del Rey A, Kabiersch A, Besedovsky HO. Interleukin-1 increases splenic blood flow by affecting the sympathetic vasoconstrictor tonus. Am J Physiol 1995;268:R902-8.
76. Yuan XQ, Wade CE, Prough DS, DeWitt DS. Traumatic brain injury creates biphasic systemic hemodynamic and organ blood flow responses in rats. J Neurotrauma 1990;7:141-53.
77. Grisanti LA, Perez DM, Porter JE. Modulation of immune cell function by α(1)-adrenergic receptor activation. Curr Top Membr 2011;67:113-38.
79. Wang JW, Wang HD, Cong ZX, Zhou XM, Xu JG, Jia Y, Ding Y. Puerarin ameliorates oxidative stress in a rodent model of traumatic brain injury. J Surg Res 2014;186:328-37.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Marcet P, Santos N, Borlongan CV. When friend turns foe: central and peripheral neuroinflammation in central nervous system injury. Neurosciences 2017;4:82-92. http://dx.doi.org/10.20517/2347-8659.2017.07
AMA Style
Marcet P, Santos N, Borlongan CV. When friend turns foe: central and peripheral neuroinflammation in central nervous system injury. Neuroimmunology and Neuroinflammation. 2017; 4: 82-92. http://dx.doi.org/10.20517/2347-8659.2017.07
Chicago/Turabian Style
Marcet, Paul, Nicole Santos, Cesar V. Borlongan. 2017. "When friend turns foe: central and peripheral neuroinflammation in central nervous system injury" Neuroimmunology and Neuroinflammation. 4: 82-92. http://dx.doi.org/10.20517/2347-8659.2017.07
ACS Style
Marcet, P.; Santos N.; Borlongan CV. When friend turns foe: central and peripheral neuroinflammation in central nervous system injury. Neurosciences. 2017, 4, 82-92. http://dx.doi.org/10.20517/2347-8659.2017.07
About This Article
Special Issue
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 0 clicks
Cite This Article 0 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.