
Role of neuroinflammation in ischemic stroke

Abstract
Ischemic stroke causes the depletion of energy and induce excitotoxicity and neuroinflammation in the brain that results from thrombotic blockage. Neuroinflammation occurs initially depending on activated resident microglia that has the same function as the macrophage. Activated microglia participates in the neuroinflammatory process by phagocytosing the injured brain cells and producing the pro- and anti-inflammatory mediators. In this review, the authors present an overview of the role of microglia in mediating neuroinflammation in ischemic stroke.
Keywords
Introduction
Stroke is an acute episode of focal dysfunction of the brain, retina or spinal cord lasting longer than 24 h, or for any duration if imaging (computed tomography or magnetic resonance imaging) or autopsy show focal infarction or hemorrhage relevant to the symptoms. Stroke is comprised of ischemic stroke (most common at approximately 85%) causing cerebral, retinal, and spinal infarction and hemorrhagic stroke (15%) that may result from intracerebral hemorrhage and subarachnoid hemorrhage [Figure 1]. Almost 90% of strokes are attributable to risk factors such as hypertension, regular physical inactivity, high apolipoprotein, insufficient diet quality, psychosocial factors, current smoking, cardiac causes, high alcohol consumption, and/or diabetes mellitus.[1]
Figure 1. Stroke is comprised of ischemic stroke (85%) and hemorrhagic stroke (15%) (intracerebral hemorrhage and subarachnoid hemorrhage)
Ischemic stroke is caused by arterial embolism and in situ small vessel diseases. Embolism in brain results in oxygen and glucose deprivation, leading to brain damage and neurologic deficit. The cellular and molecular mechanisms underlying ischemic stroke-induced brain damage have been extensively investigated. Excitotoxicity, oxidative stress, and inflammation have been considered as major contributors to ischemic neuronal injury.[2] Cerebral ischemia induces large release of glutamate that causes over-activation of NMDA receptors and large inflow of Ca2+, leading to excitotoxicity-induced cell death.[3-7] The process of ischemia-reperfusion induces the production of superoxide and nitric oxide from damaged neurons and astrocytes and depletes glutathione, a primary antioxidant to protect against reactive oxygen species-mediated DNA damage.[8-10] Inflammation occurs after ischemia-reperfusion injury, which is caused by the dying cells and debris in the absence of microbes.[11,12]
There is an increasing evidence to showing complex role of the immune system in the pathophysiological changes that occur following ischemic stroke.[13] For example, brain injury activates neutrophils and macrophage/microglia,[14] as well as the lectin pathway of complement activation and the toll-like receptors (TLRs) that are the sensors in the innate immune system,[15,16] which leads to amplification of the inflammatory cascades. The immune system is closely involved in all the stages of ischemic stroke-induced brain damage and tissue repair by the parenchymal processes.[17,18] When activated, the adaptive immune system is intervened by lymphocyte populations that include T - B cells and regulatory T cells.[19] Additionally, stroke induces the deleterious antigen-specific autoreactive responses, but it also has beneficial effects.[20] The ischemic brain can act through the autonomic nervous system to have suppressive effect that can induce intercurrent infections and contribute to the morbidity and mortality after stroke.[21-23] Therefore, immune system-mediated inflammation is critically involved in determining the fate of the brain following ischemic stroke.[24-26] Understanding the mechanisms underlying role of neuroinflammation in ischemic stroke would provide important targets for the development of therapy in ischemic stroke.
The aim of this review is to offer an overview of the current knowledge about the immune system and the neuroinflammatory processes in ischemic stroke. We focus on how the neuroinflammatory processes are triggered by ischemic stroke, and how microglia cells play a role in neuroinflammation after ischemic stroke.
Neuroinflammation
Neuroinflammation, an inflammatory response in the brain, occurs in a variety of acute brain diseases.[27,28] The non-diseased brain is separated by the blood brain barrier (BBB) from periphery.[29] The BBB prevents immune cells that are in the blood from entering brain tissue.[30] Brain is an independent immune-privileged organ with the innate. Neuroinflammation is regulated by the production of reactive oxygen species (ROS), cytokines and chemokines.[31] Once neuroinflammation happens, it enhances the release of several cytokines in the brain.[32,33] It also involves the reaction of innate immune cells (i.e. the microglia) in the parenchyma, the infiltration of myeloid cells and the adaptive immune cells (i.e. lymphocytes).[34] But the own innate immune system of brain operates mainly dependent on microglia, astrocyte and the expression of TLRs on these glia as well as the release of interleukins.[35,36]
Microglia is an innate immune cell that is well-characterized as the resident macrophage of the brain.[37] Astrocyte is important mediator of homeostasis in the brain.[38] These two cells are key players in the multicellular response to central nervous system (CNS) trauma and disease, including the immune reactions.[39,40] TLRs, the well-defined pattern recognition receptors of the immune system,[41] can initiate an immune response upon exposure to harmful microorganisms[42] and play a key role in macrophage activation. Neuronal TLR’s play a central role in connecting the interactions between the immune system and the nervous system.[42] Interleukin’s act as essential innate immune modulators and conduct an array of biological processes.[43]
The neuroinflammation process is decided by the scene, duration and course of the neurological insult.[44] Neuroinflammation can perform function that are either supportive or destructive by which is determined by the immune signals relayed to the CNS. The nature of neuroinflammatory function can depend on the conditions and the intensity and duration of inflammation.[45] The positive role associated with neuroinflammation is only present for a brief, controlled inflammatory situations and responses and this can be considered as performing a protective function to the host organism.[46-48] For example, during low transient inflammation that may occur during infections, the immune cell signals to the brain by increasing the expression of interleukin (IL)-1 cytokine, this then increasing the ‘survellience’ role of glia cells in the brain if infected.[49,50] The transient inflammation of traumatic CNS injury, following the expression of IL-4, has been shown to promote injury recovery and axonal regrowth.[51,52] On the contrary, the negative aspects of neuroinflammation mainly represent maladaptive inflammatory responses.[53,54] The common characteristics of this aspect is increasing, supraphysiological production of cytokines [IL-1 and tumor necrosis factor (TNF)], ROS, and other inflammatory mediators including inducible nitric oxide synthase.[55] These markers are highly evident in the high traumatic CNS, giving rise to collateral damage.[56] Following the acute phase of CNS trauma, the IL-1 and IL-6 drive a low-level and chronic inflammatory response, leading to cognitive impairments and reduced neuronal plasticity.[57]
Microglia and neuroinflammation
Microglia are the innate immune cells of the CNS, and are key modulators of the immune response in the brain.[37] Microglia is considered as the resident macrophage in the brain and the initial responders to tissue damage.[58] Microglia express receptors that respond to various stimuli that may as a consequence result in there activation.[59] A large number of studies indicate that microglia expresses different proteins and cytokines that display different role to express different function.[60] Activated microglia have two phenotypes: classically activated (M1) and alternatively activated (M2).[61] The M1 microglia are pro-inflammatory and thus secrete cytokines and oxidative metabolites such as IL-1β, TNF, IL-6 and nitric oxide,[62] whereas M2 microglia contributes to recovery after brain injury. M2 microglia expresses anti-inflammatory mediators, such as IL-10, IL-4 and give out various neurotrophic factors, which prevent inflammation and improve injury [Figure 2].[63] M1 microglia tends to induce neuronal cell death. Recent research has demonstrated that the M1 phenotype microglia can be switched to the M2 phenotype.[64] One study has shown that HIV-associated dementia initiates and maintains M1 phenotype microglia in the CD40 ligation by CD40L and TNFα. These microglia may later switch microglia to the M2 phenotype via up-regulation of CD45.[65] In a pathological condition, the corresponding stimuli may active microglia and cause them to change their shape and function and initiate phagocytosis.[66] Microglia works in close association with astrocytes to release cytokines that lead to a cascade of events which can modulate the neuroinflammatory respond. Meanwhile, the microglia cells produce and release excitotoxic metabolites that can damage surrounding tissue. Sometimes a short-term neuroinflammatory response is likely good for recovering the damages or infected tissue.[67] On the contrary, a long period of time neuroinflammatory process may damage the surrounding brain tissue.[68]
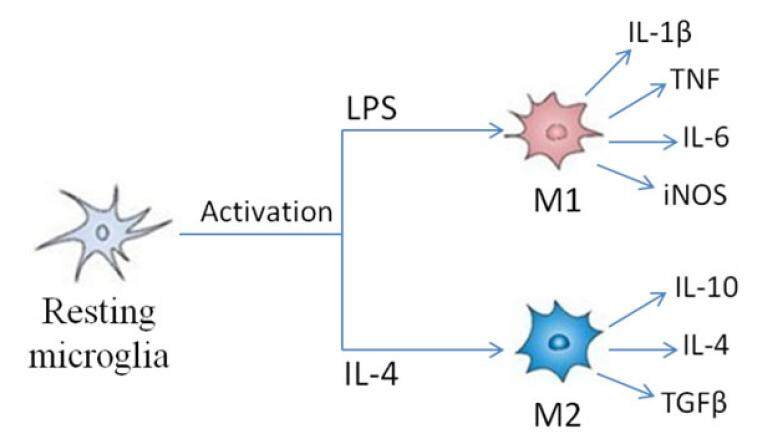
Figure 2. Activated microglia has two activation phenotypes: classically activated (M1) and alternatively activated (M2). M1 microglia is considered as pro-inflammatory, activated by LPS which produces pro-inflammatory cytokines and oxidative metabolites such as IL-1β, TNF, IL-6 and nitric oxide. M2 microglia contributes to recovery after brain injury activated by IL-4 and express anti-inflammatory mediators, such as IL-10, IL-4, TGFβ. LPS: lipopolysaccharide; IL: interleukin; TNF: tumor necrosis factor; TGF: transforming growth factor
Role of microglia in neuroinflammation after stroke
Neuroinflammation occurs in different types of brain injuries including ischemic stroke. Ischemic stroke mediated brain injury results in necrosis and apoptosis.[69-71] The damaged cells and debris induces neuroinflammation in areas in and around the ischemic injury in the brain.[72] Ischemia-induced cell debris and increased ROS lead to neuroinflammation by activating resident microglia and astrocytes as well as attracting infiltrating leukocytes from circulating blood.[73] These cells increase major histocompatibility complex class II molecules and cytokines.[74-76] Following activation of microglia, the release of pro-inflammatory mediators from these microglia favor the permeability of the BBB. Together with the secretion of chemokines, this promotes the successive entry of systemic leukocytes including neutrophils, macrophages and lymphocytes, which share several functional features with microglia.[77,78]
Microglia is the resident macrophage of the brain and a key modulator of immunologic responses after ischemic stroke. Under normal conditions, microglia is primarily involved in activity-dependent synaptic pruning and repair.[37] When ischemic stroke occurs, the native microglia undergoes morphological transformation from a ramified resting state in preparation for the forthcoming immune response.[79,80] Once reperfusion beginning, microglia come to be activated to an active, characterized by many branching processes in the penumbra, motile amoeboid state.[81] These activated microglia start to engulf endothelial cells via phagocytosis, which allows the entrance of blood serum components.[82] Active microglia phagocytoses foreign organisms as well as injured brain cells.[60,83] In ischemic stroke, activation of microglia is the early stages of the neuroinflammation process even within minutes.[83-85] Several reports have demonstrated that defective microglial activation increased the infarction and apoptosis after ischemic stroke.[86]
Microglial activation following ischemic stroke can promote activated microglia to migrate toward the ischemic hemisphere of the cerebral cortex.[87] It is suggested that active microglia have predominantly harmful effects in the acute stages of ischemic stroke and most beneficial effects appear in delayed stages.[62,88] Microglia morphology is changed either to M1, the typically activated phenotype, or to M2, an alternatively activated phenotype, after stroke.[61,89,90] M1 microglia activated by LPS and the pro-inflammatory cytokine interferon-gamma (IFN-γ) shows harmful effect after stroke.[91] In contrast, M2 phenotype microglia contribute to stroke recovery through anti-inflammatory cytokines such as IL-4.[92] In ischemic stroke, the M2 phenotype is dominant in both local microglia and newly recruited macrophages at earlier stages. The M1 phenotype increases progressively in peri-infarct regions. Thus, ischemic neuron induces changes towards the M2 phenotype in microglia and macrophages.[62] Considering the opposing roles of microglia phenotypes in ischemic stroke, it is critical to develop therapeutic strategy by restraining the morphological transformation and promoting the beneficial of microglia.
Roles of cytokines in cerebral ischemia
IFN-γ
IFN is a type cytokines that plays a key role in the immune system. The IFN family cytokines are divided into two types. Type I IFNs constitute by a largest IFN class and comprise the IFN-α, -β, -ε, -κ, and -ω, type that share notable sequence homology and are produced by most cell types. IFN-γ is a unique member of the type II IFN.[93,94] IFN-γ is principally secreted by monocytes, macrophages, T cells, natural killer (NK) cells, dendritic cells and B lymphocytes. IFN-γ is a critical regulator of immune function and provides a robust first-line of defense against invading pathogens. Additionally, IFN-γ has plenty of biological functions including regulation of several aspects of the immune responses, stimulation of antigen presentation via upregulating class I and class II major histocompatibility complex (MHC) molecules on the surface of macrophages and T cells. IFN-γ when bound to its cognate receptor can activate a variety of downstream signaling pathways, particularly the Janus kinase (JAK)/signal transducer and activator of transcription (STAT).[95,96] All of these characteristics potentially influence the process of atherogenesis. Numerous lines of evidence have indicated that IFN-γ is highly expressed in atherosclerotic lesions and believed to have a critical role in the atherogenesis.[97] Stroke is the main atherosclerosis disease.[98] Under inflammatory conditions, MHC class II specific CD4+ cells will be activated. Activated CD4+ cells easily infiltrate through BBB into the CNS following cerebral I/R.[99] Therefore, microglia have the opportunity to retain and further stimulate CD4+ cells already primed to differentiate into T helper 1 (TH1) cells producing proinflammatory cytokines (IL-2, IFN-γ, TNF-α) or into T helper 2 (TH2) cells producing cytokines that support antibody-mediated responses (IL-4, IL-5, IL-10, lL-13).[100] IFN-γ is thought to have a key role in the polarization of microglia. TH1 cells produces proinflammatory cytokines IFN-γ that can return to activation microglia into M1 phenotype, shows pro-inflammatory response, and produces pro-inflammatory cytokines and oxidative metabolites.
IL-1β
IL-1β belongs to the family IL-1. IL-1β is a key immunoregulatory and proinflammatory cytokine that affects almost all cell types. IL-1β is produced following the formation of a inflammasome; such as monocytes and macrophage/microglia.[101] After Ischemic stroke, IL-1β can activate nuclear factor (NF)-kB via the activation of TLRs allowing NF-kB to transactivate genes associated with cytokines, chemokines and other proinflammatory mediators.[102] In a pathological condition, IL-1β also connects with the activation and proliferation of astrocytes and microglia. After Ischemic stroke, the microglia will be activated, the M1 phenotype of microglia can express IL-1β which act as a proinflammatory cytokines to play neurotoxic effect.[62] In addition, IL-1β can prime the endothelium for increased leukocyte adherence and edema formation.[103] At supraphysiological levels IL-1β can be neurotoxic, however, IL-1β can also promote astrocytes to secrete survival promoting factors.[104] IL-1β when bound to its cognate receptor the IL-1 receptor (IL-1R) can also result in IL-1R-dependent increase in NF-kB pathways. However, if the levels of IL-1β are increased above a specific threshold, it can result in the increase of greater amounts of the IL-1 receptor antagonist (IL-1Ra). It is this balance between IL-1β and its antagonist the IL-1Ra that is more important for its global effect and role than just the IL-1β itself.[105] Thus, we predict that balance of IL-1β and IL-1Ra might be good predictor for patient outcome following ischemic stroke. However, few clinical studies have made use of their level as stroke biomarkers. IL-1β levels mostly were associated with poor long-term functional outcome in study,[106] while IL-1Ra levels have shown to be predictive of the development of post-stroke infections.[107]
Transforming growth factor beta
Transforming growth factor beta (TGF-β) proteins are multifunctional cytokines with pleiotropic functions.[108] TGF-β can regulate various biological processes, including hematopoiesis, angiogenesis, cell proliferation, differentiation, migration and apoptosis. TGF-β also plays an important role in the regulation of the immune system. TGF-β is a superfamily, including inhibins, activins, growth differentiation factors (GDFS), bone morphogenetic proteins (BMPs), TGF-β isoforms, and glial cell derived factors.[109] The main research object is TGF-β isoforms. TGF-β exists in at least three isoforms: TGF-β1, TGF-β2, and TGF-β3.[110] In the TGF-β superfamily, only TGF-β1, produced by activated microglia, and TGF-β2, produced by astrocytes and neurons.[111] TGF-β1 and TGF-β2 increased prominently after ischemic stroke. After Ischemic stroke, TGF-β produced by activated M2 phenotype macrophage, plays an anti-inflammatory role and contributes to recovery after brain injury.[63] TGF-β reduces microglial activation and thus reduces the potential harmful effects associated with activated microglia. TGF-β decreases the expression of other poisonous cytokines and suppresses the release of oxygen and nitrogen derived products. TGF-β can also stimulate the release of IL-1Ra and promote angiogenesis.[112] Its protective effects, however, are limited to the peri-infarcted area, as TGF-β can inhibit apoptosis but not necrosis.[113]
IL-4
IL-4, its congener of IL-13, a product of select immune cells that has highly polyfunctional properties. IL-4 is known to regulate a variety of immune and inflammatory responses, including T cell differentiation and IgE class in B cells.[114] IL-4 is primary produced by TH2 cells.[115] During CD4+ cellular activation, cytokines are through T cell receptor mediated signaling and co-stimulation. For instance, IL-4 mediated activation of the signal transducer and activator of transcription 6 plays an important role during TH2 cell differentiation.[116] IL-4 have an unique properties as it polarizes macrophages/microglia toward the M2 phenotype which is anti-inflammatory phenotype.[117] M2 macrophages/microglia expresses anti-inflammatory mediators and give out various neurotrophic factors that aid in the resolution of inflammation via increased trophic input and the augmentation of phagocytosis and proteolysis of dead, diseased cells/proteins, ultimately paving the way for tissue repair.[118] Consequently, IL-4 may have a neuroprotective function to promote tissue repair and may act as a therapeutic factor.
Stroke-associated infection and neuroinflammation
Infection frequently occurs in both and after stroke that can induce immune and neuroinflammatory responses.[119-122] The characteristics of post-stroke infections include immune suppression, elevation of IL-6, decreases in TNF-α levels and inflammation are among the factors. Along with stroke-associated infection, inflammatory responses are the defense mechanism against infection and it can also be a pathogenic mechanism that precipitates stroke and neurological sequelae.[123] It is generally recognized that stroke-associated infection may be a source of inflammation and autoimmunity as infection facilitates the maturation of APCs into potent immunostimulatory cells.[124] Stroke-associated infection is mostly induced by virus.[125-127] Virus enters the CNS through two pathways: (1) hematogenous dissemination through BBB;[125] (2) neuronal retrograde dissemination.[126] It also suggested that virus can replicate in macrophage and CCR5+ T cells in the CNS.[127]
Conclusion
The role of neuroinflammation in ischemic stroke has drawn increasing attention. In this review, we summarize the relevance of inflammation in the nervous system and introduce the neuroinflammatory cells and mediators that occur following ischemic stroke. Microglia is the resident macrophages of the brain. After ischemic stroke, the M1 and M2 phenotype of microglia play different roles at different times. The M1 phenotype tends to induce neuronal cell death, but M2 microglia contributes to the recovery after brain injury. Down-regulation of M1 phenotype and up-regulation of M1 phenotype are considered to be the potential strategy to counteract ischemic brain injury. Recent research has demonstrated that the M1 phenotype can be switched to the M2 phenotype. But the underlying mechanisms remain unclear. Thus, understanding how and why the M1 phenotype is down-regulated and the M2 phenotype up-regulated are important current and next steps to improve our understanding of the differing role of microglia post-stroke. Probing the mechanisms of M1-M2 switch could provide new approach to protect against ischemic neuronal death. Properly controlling the transformation of microglia is an important task in the treatment of ischemic stroke.
Declarations
Authors’ contributionsConcept and design: Q. Wan
Data analysis, manuscript preparation and editing: R. Liu
Literature search: M.X. Pan, J.C. Tang, Y. Zhang, H.B. Liao, Y. Zhuang, D. Zhao
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentNo patients were involved.
Ethics approvalNot applicable.
REFERENCES
1. Feigin VL, Krishnamurthi R. Stroke is largely preventable across the globe: where to next? The Lancet 2016;388:733-4.
2. Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron 2010;67:181-98.
3. Lucas DR, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol 1957;58:193-201.
4. Lerea LS, McNamara JO. Ionotropic glutamate receptor subtypes activate c-fos transcription by distinct calcium-requiring intracellular signaling pathways. Neuron 1993;10:31-41.
5. Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 1993;260:181-6.
6. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999;22:391-7.
7. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 2003;4:552-65.
8. Brennan-Minnella AM, Won SJ, Swanson RA. NADPH oxidase-2: linking glucose, acidosis, and excitotoxicity in stroke. Antioxid Redox Signal 2015;22:161-74.
9. Won SJ, Kim JE, Cittolin-Santos GF, Swanson RA. Assessment at the single-cell level identifies neuronal glutathione depletion as both a cause and effect of ischemia-reperfusion oxidative stress. J Neurosci 2015;35:7143-52.
10. Andrabi SA, Kang HC, Haince JF, Lee YI, Zhang J, Chi Z, West AB, Koehler RC, Poirier GG, Dawson TM, Dawson VL. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat Med 2011;17:692-9.
11. Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol 2012;8:401-10.
12. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med 2011;17:796-808.
13. Candeias SM, Gaipl US. The immune system in cancer prevention, development and therapy. Anticancer Agents Med Chem 2016;16:101-7.
14. Misra DP, Agarwal V. Innate immune cells in the pathogenesis of primary systemic vasculitis. Rheumatol Int 2016;36:169-82.
15. d'Onofrio A. Spatiotemporal effects of a possible chemorepulsion of tumor cells by immune system effectors. J Theor Biol 2012;296:41-8.
16. Odendall C, Kagan JC. Activation and pathogenic manipulation of the sensors of the innate immune system. Microbes Infect 2017;19:229-37.
17. Amantea D, Micieli G, Tassorelli C, Cuartero MI, Ballesteros I, Certo M, Moro MA, Lizasoain I, Bagetta G. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front Neurosci 2015;9:147.
18. Magnus T, Wiendl H, Kleinschnitz C. Immune mechanisms of stroke. Curr Opin Neurol 2012;25:334-40.
19. den Haan JMM, Arens R, van Zelm MC. The activation of the adaptive immune system: cross-talk between antigen-presenting cells, T cells and B cells. Immunology Letters 2014;162:103-12.
20. Gu Y, Zhao W, Meng F, Qu B, Zhu X, Sun Y, Shu Y, Xu Q. Sunitinib impairs the proliferation and function of human peripheral T cell and prevents T-cell-mediated immune response in mice. Clin Immunol 2010;135:55-62.
21. Dorrance AM, Fink G. Effects of stroke on the autonomic nervous system. Compr Physiol 2015;5:1241-63.
22. Micieli G, Cavallini A. The autonomic nervous system and ischemic stroke: a reciprocal interdependence. Clin Auton Res 2008;18:308-17.
23. Kanai M, Kubo H, Kitamura Y, Izawa KP, Ono K, Ando H, Nozoe M, Mase K, Shimada S. Difference in autonomic nervous activity in different subtypes of noncardioembolic ischemic stroke. Int J Cardiol 2015;201:171-3.
25. Becker KJ, Buckwalter M. Stroke, inflammation and the immune response: dawn of a new era. Neurotherapeutics 2016;13:659-60.
26. Mcdonough A, Weinstein JR. Neuroimmune response in ischemic preconditioning. Neurotherapeutics 2016;13:748-61.
27. Weiner HL, Selkoe DJ. Inflammation and therapeutic vaccination in CNS diseases. Nature 2002;420:879-84.
28. Schwartz M. Macrophages and microglia in central nervous system injury: are they helpful or harmful? J Cereb Blood Flow Metab 2003;23:385-94.
29. Schultzberg M, Lindberg C, Aronsson AF, Hjorth E, Spulber SD, Oprica M. Inflammation in the nervous system -- physiological and pathophysiological aspects. Physiol Behav 2007;92:121-8.
30. Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008;57:178-201.
31. Mayer AM, Clifford JA, Aldulescu M, Frenkel JA, Holland MA, Hall ML, Glaser KB, Berry J. Cyanobacterial Microcystis aeruginosa lipopolysaccharide elicits release of superoxide anion, thromboxane B(2), cytokines, chemokines, and matrix metalloproteinase-9 by rat microglia. Toxicol Sci 2011;121:63-72.
32. Pascual M, Baliño P, Aragón CMG, Guerri C. Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: role of TLR4 and TLR2. Neuropharmacology 2015;89:352-9.
33. Na KS, Jung HY, Kim YK. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2014;48:277-86.
34. Ip CW, Kroner A, Groh J, Huber M, Klein D, Spahn I, Diem R, Williams SK, Nave KA, Edgar JM, Martini R. Neuroinflammation by cytotoxic T-lymphocytes impairs retrograde axonal transport in an oligodendrocyte mutant mouse. PLoS One 2012;7:e42554.
35. Allaman I, Belanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci 2011;34:76-87.
36. Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res 2007;85:2059-70.
37. El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC. Microglia, scavenger receptors, and the pathogenesis of Alzheimer's disease. Neurobiol Aging 1998;19:S81-4.
38. Kim JY, Park J, Chang JY, Kim SH, Lee JE. Inflammation after ischemic stroke: the role of leukocytes and glial cells. Exp Neurobiol 2016;25:241-51.
39. Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014;81:229-48.
40. Cekanaviciute E, Buckwalter MS. Astrocytes: integrative regulators of neuroinflammation in stroke and other neurological diseases. Neurotherapeutics 2016;13:685-701.
41. Lin J, Kakkar V, Lu X. Essential roles of Toll-like receptors in atherosclerosis. Curr Med Chem 2016;23:431-54.
42. Rietdijk CD, Van Wezel RJA, Garssen J, Kraneveld AD. Neuronal toll-like receptors and neuro-immunity in Parkinson's disease, Alzheimer's disease and stroke. Neuroimmunol Neuroinflammation 2016;3:27.
43. Neves F, Abrantes J, Almeida T, de Matos AL, Costa PP, Esteves PJ. Genetic characterization of interleukins (IL-1alpha, IL-1beta, IL-2, IL-4, IL-8, IL-10, IL-12A, IL-12B, IL-15 and IL-18) with relevant biological roles in lagomorphs. Innate Immun 2015;21:787-801.
44. Filiou MD, Arefin AS, Moscato P, Graeber MB. 'Neuroinflammation' differs categorically from inflammation: transcriptomes of Alzheimer's disease, Parkinson's disease, schizophrenia and inflammatory diseases compared. Neurogenetics 2014;15:201-12.
45. Chiu CC, Liao YE, Yang LY, Wang JY, Tweedie D, Karnati HK, Greig NH. Neuroinflammation in animal models of traumatic brain injury. J Neurosci Methods 2016;272:38-49.
46. Li C, Wang J, Fang Y, Liu Y, Chen T, Sun H, Zhou XF, Liao H. Nafamostat mesilate improves function recovery after stroke by inhibiting neuroinflammation in rats. Brain Behav Immun 2016;56:230-45.
47. Desai A, Park T, Barnes J, Kevala K, Chen H, Kim HY. Reduced acute neuroinflammation and improved functional recovery after traumatic brain injury by alpha-linolenic acid supplementation in mice. J Neuroinflammation 2016;13:253.
48. Teng SX, Katz PS, Maxi JK, Mayeux JP, Gilpin NW, Molina PE. Alcohol exposure after mild focal traumatic brain injury impairs neurological recovery and exacerbates localized neuroinflammation. Brain Behav Immun 2015;45:145-56.
49. Nagarajan UM, Sikes JD, Yeruva L, Prantner D. Significant role of IL-1 signaling, but limited role of inflammasome activation, in oviduct pathology during Chlamydia muridarum genital infection. J Immunol 2012;188:2866-75.
50. Yang H, Ko HJ, Yang JY, Kim JJ, Seo SU, Park SG, Choi SS, Seong JK, Kweon MN. Interleukin-1 promotes coagulation, which is necessary for protective immunity in the lung against Streptococcus pneumoniae infection. J Infect Dis 2013;207:50-60.
51. Fenn AM, Hall JC, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique arginase+/IL-1beta+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Ralpha after traumatic spinal cord injury. J Neurosci 2014;34:8904-17.
52. Lee SI, Jeong SR, Kang YM, Han DH, Jin BK, Namgung U, Kim BG. Endogenous expression of interleukin-4 regulates macrophage activation and confines cavity formation after traumatic spinal cord injury. J Neurosci Res 2010;88:2409-19.
53. Le Thuc O, Blondeau N, Nahon JL, Rovere C. The complex contribution of chemokines to neuroinflammation: switching from beneficial to detrimental effects. Ann N Y Acad Sci 2015;1351:127-40.
54. Ransohoff RM, Schafer D, Vincent A, Blachère NE, Bar-Or A. Neuroinflammation: ways in which the immune system affects the brain. Neurotherapeutics 2015;12:896-909.
55. Finnie JW. Neuroinflammation: beneficial and detrimental effects after traumatic brain injury. Inflammopharmacology 2013;21:309-20.
56. Anthony DC, Walker K, Perry VH. The therapeutic potential of CXC chemokine blockade in acute inflammation in the brain. Expert Opin Investig Drugs 1999;8:363-71.
57. Dursun E, Gezen-Ak D, Hanagasi H, Bilgic B, Lohmann E, Ertan S, Atasoy IL, Alaylioglu M, Araz OS, Onal B, Gunduz A, Apaydin H, Kiziltan G, Ulutin T, Gurvit H, Yilmazer S. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer's disease, mild cognitive impairment or Parkinson's disease. J Neuroimmunol 2015;283:50-7.
58. Brandenburg S, Müller A, Turkowski K, Radev YT, Rot S, Schmidt C, Bungert AD, Acker G, Schorr A, Hippe A, Miller K, Heppner FL, Homey B, Vajkoczy P. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathologica 2015;131:365-78.
59. Huck JH, Freyer D, Bottcher C, Mladinov M, Muselmann-Genschow C, Thielke M, Gladow N, Bloomquist D, Mergenthaler P, Priller J. De novo expression of dopamine D2 receptors on microglia after stroke. J Cereb Blood Flow Metab 2015;35:1804-11.
61. Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol 2010;10:453-60.
62. Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012;43:3063-70.
63. Nair MG, Guild KJ, Artis D. Novel effector molecules in type 2 inflammation: lessons drawn from helminth infection and allergy. J Immunol 2006;177:1393-9.
64. Yu Z, Sun D, Feng J, Tan W, Fang X, Zhao M, Zhao X, Pu Y, Huang A, Xiang Z, Cao L, He C. MSX3 switches microglia polarization and protects from inflammation-induced demyelination. J Neurosci 2015;35:6350-65.
65. Salemi J, Obregon DF, Cobb A, Reed S, Sadic E, Jin J, Fernandez F, Tan J, Giunta B. Flipping the switches: CD40 and CD45 modulation of microglial activation states in HIV associated dementia (HAD). Mol Neurodegener 2011;6:3.
66. Appel SH, Zhao W, Beers DR, Henkel JS. The microglial-motoneuron dialogue in ALS. Acta Myol 2011;30:4-8.
68. Versijpt J, Debruyne JC, Van Laere KJ, De Vos F, Keppens J, Strijckmans K, Achten E, Slegers G, Dierckx RA, Korf J, De Reuck JL. Microglial imaging with positron emission tomography and atrophy measurements with magnetic resonance imaging in multiple sclerosis: a correlative study. Mult Scler 2005;11:127-34.
69. Chen WW, Zhang X, Huang WJ. Role of neuroinflammation in neurodegenerative diseases (Review). Mol Med Rep 2016;13:3391-6.
70. Sun L, Strelow H, Mies G, Veltkamp R. Oxygen therapy improves energy metabolism in focal cerebral ischemia. Brain Res 2011;1415:103-8.
71. Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 2014;115:157-88.
72. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol 2010;28:321-42.
73. Kim JY, Park J, Chang JY, Kim SH, Lee JE. Inflammation after ischemic stroke: the role of leukocytes and glial cells. Exp Neurobiol 2016;25:241-51.
74. Kuric E, Ruscher K. Dynamics of major histocompatibility complex class II-positive cells in the postischemic brain--influence of levodopa treatment. J Neuroinflammation 2014;11:145.
75. Huber S, Schramm C, Lehr HA, Mann A, Schmitt S, Becker C, Protschka M, Galle PR, Neurath MF, Blessing M. Cutting edge: TGF-beta signaling is required for the in vivo expansion and immunosuppressive capacity of regulatory CD4+CD25+ T cells. J Immunol 2004;173:6526-31.
76. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 2014;6:13.
77. Noh H, Jeon J, Seo H. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem Int 2014;69:35-40.
78. Veltkamp R, Gill D. Clinical trials of immunomodulation in ischemic stroke. Neurotherapeutics 2016;13:791-800.
79. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005;8:752-8.
80. Dunn AK, Bolay H, Moskowitz MA, Boas DA. Dynamic imaging of cerebral blood flow using laser speckle. J Cereb Blood Flow Metab 2001;21:195-201.
81. Xiong XY, Liu L, Yang QW. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog Neurobiol 2016;142:23-44.
82. Jolivel V, Bicker F, Biname F, Ploen R, Keller S, Gollan R, Jurek B, Birkenstock J, Poisa-Beiro L, Bruttger J, Opitz V, Thal SC, Waisman A, Bauerle T, Schafer MK, Zipp F, Schmidt MH. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol 2015;129:279-95.
83. Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol 2006;84:49-59.
84. Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, Allan SM. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab 2007;27:1941-53.
85. Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 2007;27:2596-605.
86. Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci 2012;32:10383-95.
87. Yu YM, Kim JB, Lee KW, Kim SY, Han PL, Lee JK. Inhibition of the cerebral ischemic injury by ethyl pyruvate with a wide therapeutic window. Stroke 2005;36:2238-43.
88. Ponomarev ED, Veremeyko T, Weiner HL. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia 2013;61:91-103.
91. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 2011;480:104-8.
92. Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 2009;4:399-418.
93. Yu XH, Jiang N, Zheng XL, Cayabyab FS, Tang ZB, Tang CK. Interleukin-17A in lipid metabolism and atherosclerosis. Clin Chim Acta 2014;431:33-9.
94. Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res 2008;79:360-76.
95. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 2004;75:163-89.
96. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 2014;32:513-45.
97. Yu XH, Zhang J, Zheng XL, Yang YH, Tang CK. Interferon-gamma in foam cell formation and progression of atherosclerosis. Clin Chim Acta 2015;441:33-43.
98. Folsom AR, Gottesman RF, Appiah D, Shahar E, Mosley TH. Plasma d-Dimer and incident ischemic stroke and coronary heart disease: the atherosclerosis risk in communities study. Stroke 2016;47:18-23.
99. Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol 1994;55:195-203.
100. Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today 1996;17:138-46.
101. Satoh T, Otsuka A, Contassot E, French LE. The inflammasome and IL-1beta: implications for the treatment of inflammatory diseases. Immunotherapy 2015;7:243-54.
102. Liao Z, Xiao HT, Zhang Y, Tong RS, Zhang LJ, Bian Y, He X. IL-1β: a key modulator in asthmatic airway smooth muscle hyper-reactivity. Expert Rev Respir Med 2015;9:429-36.
103. del Zoppo GJ. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 2009;158:972-82.
104. Albrecht PJ, Enterline JC, Cromer J, Levison SW. CNTF-activated astrocytes release a soluble trophic activity for oligodendrocyte progenitors. Neurochem Res 2007;32:263-71.
105. Rothwell N. Interleukin-1 and neuronal injury: mechanisms, modification, and therapeutic potential. Brain Behav Immun 2003;17:152-7.
106. Protopsaltis J, Kokkoris S, Korantzopoulos P, Milionis HJ, Karzi E, Anastasopoulou A, Filioti K, Antonopoulos S, Melidonis A, Giannoulis G. Prediction of long-term functional outcome in patients with acute ischemic non-embolic stroke. Atherosclerosis 2009;203:228-35.
107. Tanzi P, Cain K, Kalil A, Zierath D, Savos A, Gee JM, Shibata D, Hadwin J, Carter K, Becker K. Post-stroke infection: a role for IL-1ra? Neurocrit Care 2011;14:244-52.
108. Gonzalo-Gil E, Galindo-Izquierdo M. Role of transforming growth factor-beta (TGF) beta in the physiopathology of rheumatoid arthritis. Reumatol Clin 2014;10:174-9.
109. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-beta structure and activation. Nature 2011;474:343-9.
110. Papageorgis P. TGFβ signaling in tumor initiation, epithelial-to-mesenchymal transition, and metastasis. J Oncol 2015;2015:587193.
111. Vitkovic L, Maeda S, Sternberg E. Anti-inflammatory cytokines: expression and action in the brain. Neuroimmunomodulation 2001;9:295-312.
112. Pantoni L, Sarti C, Inzitari D. Cytokines and cell adhesion molecules in cerebral ischemia: experimental bases and therapeutic perspectives. Arterioscler Thromb Vasc Biol 1998;18:503-13.
113. Jiang Y, Sun L, Xuan X, Wang J. Impacts of N-Butylphthalide on expression of growth factors in rats with focal cerebral ischemia. Bosn J Basic Med Sci 2016;16:102-7.
114. Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci 2015;35:11281-91.
116. Le Gros G, Ben-Sasson SZ, Seder R, Finkelman FD, Paul WE. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. J Immunol 2008;181:2943-51.
117. Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor γ in microglia/macrophages. Ann Neurol 2007;61:352-62.
118. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012;122:787-95.
120. Lindsberg PJ, Grau AJ. Inflammation and infections as risk factors for ischemic stroke. Stroke 2003;34:2518-32.
122. Westendorp WF, Nederkoorn PJ, Vermeij JD, Dijkgraaf MG, van de Beek D. Post-stroke infection: a systematic review and meta-analysis. BMC Neurol 2011;11:110.
123. Emsley HC, Hopkins SJ. Acute ischaemic stroke and infection: recent and emerging concepts. Lancet Neurol 2008;7:341-53.
124. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol 2000;18:767-811.
125. Yang WX, Terasaki T, Shiroki K, Ohka S, Aoki J, Tanabe S, Nomura T, Terada E, Sugiyama Y, Nomoto A. Efficient delivery of circulating poliovirus to the central nervous system independently of poliovirus receptor. Virology 1997;229:421-8.
126. Aronsson F, Robertson B, Ljunggren HG, Kristensson K. Invasion and persistence of the neuroadapted influenza virus A/WSN/33 in the mouse olfactory system. Viral Immunol 2003;16:415-23.
127. Schnell G, Joseph S, Spudich S, Price RW, Swanstrom R. HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog 2011;7:e1002286.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Liu R, Pan MX, Tang JC, Zhang Y, Liao HB, Zhuang Y, Zhao D, Wan Q. Role of neuroinflammation in ischemic stroke. Neurosciences 2017;4:158-66. http://dx.doi.org/10.20517/2347-8659.2017.09
AMA Style
Liu R, Pan MX, Tang JC, Zhang Y, Liao HB, Zhuang Y, Zhao D, Wan Q. Role of neuroinflammation in ischemic stroke. Neuroimmunology and Neuroinflammation. 2017; 4: 158-66. http://dx.doi.org/10.20517/2347-8659.2017.09
Chicago/Turabian Style
Liu, Rui, Meng-Xian Pan, Jun-Chun Tang, Ya Zhang, Hua-Bao Liao, Yang Zhuang, Dan Zhao, Qi Wan. 2017. "Role of neuroinflammation in ischemic stroke" Neuroimmunology and Neuroinflammation. 4: 158-66. http://dx.doi.org/10.20517/2347-8659.2017.09
ACS Style
Liu, R.; Pan M.X.; Tang J.C.; Zhang Y.; Liao H.B.; Zhuang Y.; Zhao D.; Wan Q. Role of neuroinflammation in ischemic stroke. Neurosciences. 2017, 4, 158-66. http://dx.doi.org/10.20517/2347-8659.2017.09
About This Article
Special Issue
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 76 clicks
Cite This Article 76 clicks

 Like This Article 13
likes
Like This Article 13
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.