
LPS-induced TLR4 neuroinflammatory signaling in CHME-5 microglial cells


Abstract
Aim: In the field of neuroinflammation, identifying specific effects of pharmacological agents and other factors is problematic given the relative difficulty and expense in obtaining and culturing primary microglia. Immortalized microglial cell lines are very useful, but only a limited number have been characterized for inflammatory signaling. Therefore, characterization of lipopolysaccharide (LPS)-induced toll-like receptor 4 (TLR4) signaling in CHME-5, a microglial cell line, is expected to be of value as an experimental model of inflammatory signaling in the central nervous system (CNS).
Methods: It was recently suggested that CHME-5 cells are of rat origin, not human, hence, verification of this claim using short tandem repeat genotype sequencing, along with immunoblotting, reverse transcription-polymerase chain reaction, and immunocytochemistry techniques to validate that CHME-5 retain morphological, phenotypical, and functional characteristics of primary microglia were undertaken.
Results: LPS induced inhibitor kappa B-alpha and nuclear factor-kappa B (NF-κB) p65 activation, NF-κB p65 binding activity, and tumor necrosis factor alpha gene expression. Additionally, results also confirmed the maintenance of microglial phenotype as seen with increased cluster of differentiation 68 gene and protein expression, immunofluorescence, and the absence of glial fibrillary acidic protein-immunoreactivity. TLR4 gene expression and immunofluorescence were significantly increased after LPS treatment.
Conclusion: These data demonstrate that CHME-5 cells are not human, but are indeed a beneficial tool for studying microglial inflammatory signaling.
Keywords
Introduction
The innate immune response is instrumental in combatting infection and in response to stress and physical injury. One family of receptors expressed on innate immune cells is toll-like receptors (TLRs). TLRs can recognize a host of patterns produced by bacteria, viruses, and fungi, known as pathogen-associated molecular patterns, or self-products released from apoptotic cells, called damaged-associated molecular patterns[1]. To date, 10 TLRs have been identified in humans, 12 in rodents[2,3].
TLR4 was discovered as the receptor responsible for detection of Gram-negative bacterial lipopolysaccharide (LPS), which leads to production of pro-inflammatory cytokines and up-regulation of co-stimulatory molecules necessary for initiation of the adaptive immune response[4-6]. TLR4 recognizes LPS with the aid of several accessory proteins, including LPS-binding protein (LBP), cluster of differentiation 14, and myeloid differentiation 2. Activation of TLR4 leads to initiation of 2 distinct signaling pathways, MyD88-dependent and TRIF-dependent pathways[3,7].
Until the past decade the central nervous system (CNS) has been generally considered as an "immune privileged" site because of the extensive defense and regulatory mechanisms available to protect this organ from foreign cells and pathogens[8]. But we now know there are cells expressed in the CNS that detect infection or injury and respond accordingly.
Microglia, the primary immune cells, are also known as "macrophages of the CNS". Microglia interact with other glia, namely astrocytes, to support neuronal function, survey/patrol for and clear foreign or harmful particles, and regulate neuroinflammation through pro-inflammatory mediators[9,10]. Microglial activation is characterized by morphological changes, proliferation, and up-regulation of receptors including scavenger, complement, cytokine/chemokine and pattern recognition receptors. Recognition of pathogens or insult leads to activation and release of pro-inflammatory and neurotoxic factors including tumor necrosis factor-α (TNFα), interleukin-1β, interferon gamma inducible protein-10 (CXCL10), nitric oxide (NO) and reactive oxygen species (ROS)[9,11,12]. Microglial activation has been implicated in numerous neurodegenerative diseases including multiple sclerosis, Alzheimer’s disease, and Parkinson’s disease[13-16].
TLR4 is the most widely studied TLR, and its expression is more abundant in microglia than in any other resident cell in the CNS[17,18]. TLR4 signaling is well defined in peripheral immune cells, but less clear in the CNS. Here, we characterized LPS-induced TLR4 inflammatory signal transduction in the CNS using a fetal-derived microglial cell line, CHME-5. To our knowledge, this is the first report to validate the finding that CHME-5 is not a human cell line, and show that microglial responses in CHME-5 cells are comparative to human primary microglia. Therefore, we demonstrate that CHME-5 can be used as an experimental tool for microglial research and neuroinflammatory signaling.
Methods
Cell culture
CHME-5 cells were originally developed by Janabi et al.[19] and were gifted to our lab by Dr. Pierre Talbot, Quebec, Canada. CHME-5 growth media consisted of Dulbecco’s modified eagle medium (DMEM) with 4.5 g/L glucose and sodium pyruvate without L-glutamine, 10% fetal bovine serum, 200 mmol/L L-glutamine, penicillin-streptomycin (100 U/mL potassium penicillin, 100 μg/mL streptomycin sulfate), and 250 μg/mL amphotericin B. CHME-5 cells were maintained in growth media at 37 °C, with 5% CO2. For experimental assays, growth medium was aspirated from cells and replaced with serum-free media (SFM) for no less than 16 h at 37 °C.
LPS treatment
Lipopolysaccharide from Escherichia coli O55:B5 (#L2880, Sigma-Aldrich, St. Louis, MO, USA) was purified by phenol extraction. The lyophilized powder was reconstituted in HyPure cell culture grade water and sterile filtered to a stock concentration of 1.2 mg/mL. Cells were stimulated with 1.0 μg/mL LPS unless otherwise noted. For dose-response studies, 0.001-10 μg/mL was used for stimulation.
STR genotyping
PowerPlex 21 System (Promega-#DC8902) was used to validate Short tandem repeats (STR) regions in CHME-5 cells. Reactions were set up using PowerPlex 21 5× Master Mix [5.0 µL/reaction (rxn)], PowerPlex 21 5× Primer Pair Mix (5.0 µL/rxn), DNA template (0.5 ng), control DNA (0.5 ng), and water (up to 25 µL). Thermocycler settings were as follows: 96 °C-1 min, (94 °C-10 s, 59 °C-1 min, 72 °C-1 min for 30 cycles), 60 °C-10 min. Results were analyzed using GeneMapper-ID Software (Applied Biosystems).
Protein extraction
Depending on the experiment, either whole cell lysate or cytoplasmic/nuclear extractions were isolated for subsequent assessment of protein expression. For whole cell lysates, cells were washed with ice-cold phosphate-buffered saline (PBS) and then 400 μL of cell Lysis Buffer (CBL, Cell Signaling) was added to each 100-mm dish. Following a 5-min incubation at 4 °C, lysates were collected and centrifuged at 14,000 × g (4 °C) for 10 min. The supernatant was collected and stored at -20 °C. For cytoplasmic and nuclear extracts, cells were washed with and collected in 1 mL ice-cold PBS. Cells were centrifuged at 129 × g (4 °C) for 5 min. The supernatant was aspirated and combined with 400 μL Lysis buffer 1 [10 mmol/L (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) (pH 7.9), 10 mmol/L KCl, 0.10 mmol/L ethylenediaminetetraacetic acid (EDTA), 0.10 mmol/L ethylene glycol-bis (β-aminoethyl ether)-tetraacetic acid (EGTA), 1 mmol/L dithiothreitol (DTT), 0.5 mmol/L phenylmethylsulfonyl fluoride (PMSF), 10 μg/mL leupeptin, and 10 μg/mL aprotinin] and vortexed vigorously for 10 s, followed by incubation on ice for 15 min. Next, 100 μL 5.4% igepal was added, samples vortexed for 10 s, and then centrifuged at 14,000 × g (4 °C) for 5 min. The supernatant was collected and stored at -20 °C; then 100 μL Lysis buffer 2 (20 mmol/L HEPES, 400 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT, 1 mmol/L PMSF, 1 mmol/L leupeptin, 10 μg/mL aprotinin) was added to the remaining pellet. The samples were then vortexed for 15 min at 4 °C and then centrifuged at 14,000 × g (4 °C) for 22 min. The supernatant was collected and stored at -20 °C.
Immunoblot analysis
Protein extracts were subjected to 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and immunoblot analysis. Briefly, protein (50 μg) in loading dye (0.25 mmol/L Tris, pH 6.8, 10 mmol/L DTT, 30% glycerol, 10% SDS, 0.05% bromophenol blue, and 50 μL/mL β-mercaptoethanol) was boiled for 10 min and then loaded into gels. Electrophoresis was performed in running buffer (250 mmol/L glycine, 25 mmol/L Tris, 0.1% SDS) at 100 V for 15 min, and then at 125 V for 145 min for an overall running time of 2 h. Proteins were transferred onto polyvinylidene fluoride (PVDF) membranes in transfer buffer (195 mmol/L glycine, 25 mmol/L Tris, pH 8.0, 20% methanol) at 100 V for 90 min. After transfer, PVDF membranes were rinsed and blocked in bovine serum albumin (BSA) in 1× Tris-buffered saline-Tween (TBST) (150 mmol/L NaCl, 25 mmol/L Tris, pH 8.0, 2 mmol/L KCl, 0.1% Tween-20-TBST) for 2 h at 25 °C with rocking. For cluster of differentiation 68 (CD68), 2% BSA was used and 5% BSA was used for all other antibodies. Primary antibodies diluted in blocking buffer (1:500-1:2,000), were added to membranes and rocked overnight at 4 °C. Membranes were then washed 3 times with TBST for 5 min with each wash. Alkaline phosphatase-linked secondary antibodies diluted in blocking buffer (1:1,000-1:5,000) were added to membranes and rocked for 2 h at 25 °C and then washed 3 times with TBST for 20 min. Enhanced Chemifluorescence (ECF) substrate (#45-000-947, GE Healthcare Amersham) was used to image blots using the Typhoon Scanner 9410. Image J software (National Institutes of Health) was used to obtain the mean grey intensity for the bands of interest.
MTT assay
The MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay was performed to determine cell viability after treatment. Fresh SFM (1 mL) was added to each well followed by addition of 111 μL MTT. Cultures were then incubated at 37 °C for 45 min. Media was aspirated from each well and 1.5 mL dimethyl sulfoxide added followed by rocking at 25 °C for 30 min. Absorbance was read at 492 nm with the Synergy 2 plate reader (Biotek Instruments).
NF-κB p65 binding assay
Nuclear factor-kappa B (NF-κB) activation was measured using the NF-κB p65 transcription factor kit (#89859, Thermo Scientific) per manufacturer’s instructions. Briefly, binding buffer (50 μL) was added to each well, which was pre-coated with the NF-κB binding consensus sequence. Then, 10 μL of nuclear extract was added to each well, in duplicate. Following incubation for 1 h at 25 °C with mild agitation, wells were washed 3 times with 200 μL of wash buffer, then 100 μL of primary antibody (1:1,000) was added to each well, followed by a 1-h incubation at 25 °C, without agitation. Wells were washed as described above and 100 μL of secondary antibody (1:10,000) was added to each well, followed by 1-h incubation at 25 °C, without agitation. Finally, wells were washed 4 times with 200 μL of wash buffer, and 100 μL of chemiluminescent substrate was added to each well. Chemiluminescence was measured immediately with a Nikon plate reader.
RNA extraction
Following cell stimulation, cells were washed 3 times with ice-cold PBS, incubated in Trizol (1 mL/100 mm dish) at 25 °C for 5 min, and then lysates were collected in nuclease-free tubes. Chloroform (0.2 mL) was added to lysates followed by manual shaking of tubes for 15 s. Lysates were then incubated at 25 °C for 3 min, centrifuged at 12,000 × g for 15 min (4 °C) and then the upper aqueous phase was collected in fresh microcentrifuge tubes. Isopropanol (0.5 mL) was added to the aqueous phase, followed by incubation at 25 °C for 10 min, and then centrifuged at 12,000 × g for 10 min (4 °C). Next, the supernatant was removed and RNA pellets washed with 70% ethanol, and centrifuged at 7,500 × g for 2 min (4 °C). This ethanol wash step was repeated twice. RNA pellets were dried at 25 °C for 15 min, followed by addition of 35 μL nuclease-free water. RNA samples were incubated in a 65 °C water bath for 10 min and then stored at -80 °C. To obtain RNA concentrations, samples were thawed on ice and ng/mL was determined with a Nanodrop Spectrophotometer 1000 (Thermo Scientific), and validated with the 260/280 ratios between 1.8 and 2.0.
RNA integrity
RNA gels were prepared with NorthernMax-Gly Reagents (Ambion) to determine RNA integrity. A 1% agarose gel was prepared with NorthernMax-Gly buffer. To prepare samples, 3 μL RNA, 3 μL nuclease-free water and 6 μL glyoxal sample loading dye were incubated at 50 °C for 30 min. Samples were loaded and electrophoresed at 100 V for 45 min. RNA gels were stained with SYBR Safe (3 μL/50 mL in NorthernMax-Gly buffer) and rocked on a Multimixer (Lab-Line Instruments, LLC-Melrose Park, IL) for 30 min. To visualize the 28S and 18S bands, RNA gels were imaged on Typhoon Scanner 9410 (GE Healthcare Life Sciences) at 450 V.
Real-time-polymerase chain reaction
RNA was reverse transcribed using the SuperScript IV VILO Master Mix (Invitrogen, #11766050) and cDNA (100 ng) was used to perform real-time polymerase chain reaction (RT-PCR) for genes of interests. The primer for CD68 was designed using rat CD68 messenger RNA (mRNA) from the NIH National Center for Biotechnology Information website and obtained from IDT Technologies. The rat primers for TLR4, TNFα, and β-actin were obtained from IDT Technologies. RT-PCR mix included 2× PowerUp SybGreen, 0.5 μmol/L forward primer, 0.5 μmol/L reverse primer, and nuclease-free water up to 15 μL. Thermocycler settings were as follows: 50 °C-2 min, 95 °C-2 min, (95 °C-15 s, 60 °C-1 min for 40 cycles), 95 °C-15 s, 60 °C-1 min, 95 °C-15 s. Primer sequences used were as follows: TLR4-forward: 5’-GAA GCT ATA GCT TCA CCA ATT TCT CAC AA-3’, 60.2 °C; reverse: 5’-GAT AGG GTT TCC TGT CAG TAC CAA GGT TG-3’, 60.1 °C; CD68-forward: 5’-CTC AGC AGC TCT ACC ATG AGG TTC-3’, 59 °C; reverse: 5’-CTT CCG GTG GTT GTA GGT GTC TC-3’, 59.2 °C; TNFα-forward: 5’-CAG ATC ATC TTC TCA AAA CTC GAG TGA CA-3’, 60.3 °C; reverse: 5’-GTT GGT TGT CTT TGA GAT CCA TGC CAT TG’-3’, 60.1 °C; and β-actin-forward: 5’-GAA GGA TTC CTA TGT GGG CGA CGA-3’, 60.5 °C; reverse: 5’-GAG CCA CAC GCA GCT CAT TGT AG-3’, 60.3 °C.
Immunocytochemistry
Cells were exposed to SFM with or without LPS (1 μg/mL), for 10 min at 37 °C. After stimulation, cells were gently washed 3 times with ice-cold PBS and fixed with 4% paraformaldehyde (PFA) for 10 min at 25 °C. Cells were washed 3 times with ice-cold PBS and permeabilized with 0.01% Triton-X in PBS for 10 min at 25 °C. Next, cells were washed as described above and primary antibodies mouse-TLR4 (1:1,000) or rabbit-CD68 (1:1,000) in PBS added to respective wells and incubated overnight at 4 °C. The next day, cells were washed 3 times with TBST and incubated with fluorescently labeled secondary antibodies, anti-rabbit-AlexaFluor-647 (1:1,000) or anti-mouse-AlexaFluor-555 (1:1,000) while rocking for 2 h at 25 °C. Negative control cells were unstimulated cells only incubated with secondary antibodies. Cells were washed 3 times with TBST and labeled with [4’-6-diamidino-2-phenylindole (DAPI)-PBS)] for 10 min, rocking at 25 °C. Finally, cells were washed 3 times with TBST, cover slips removed from wells, and mounted on slides using anti-fade gold mounting media (Invitrogen). Slides were dried overnight and then imaged.
Epifluorescence microscopy
TLR4 and CD68 immunofluorescence was imaged with an epifluorescence microscope (Olympus BX51) using Spot 5.1 software. Images were captured with the 20× objective using fluorescent channels for DAPI (350 nm), TLR4 (TRITC-555 nm), and CD68 (Cy5-647 nm). Images were processed with CellProfiler cell image analysis software[20], and quantified in GraphPad Prism 7.0.
CellProfiler
All images of nuclei were processed in Image J, with global contrast settings to clean-up and define nuclear boundaries for CellProfiler’s automatic nuclear recognition algorithm. A CellProfiler pipeline was created for each fluorescent channel image set to define global pixel intensity thresholds. All image sets were then processed for high-throughput quantification in CellProfiler to measure area and mean grey intensity. High-throughput quantification can process hundreds of images while standardizing the recognition and measurements of each cell in every image. This process increases sample size while removing inherent subjectivity of hand tracing.
Confocal microscopy
The Leica SPE Scanhead RYBV confocal microscope was used to capture images for brightfield, and immunofluorescence and all images were processed in Leica Application Suite Advanced Fluorescence. For brightfield imaging, the 488 laser (55%) was used to capture images with the 100× oil objective in unstimulated and LPS-stimulated cells, which were fixed with 4% PFA for 10 min at 25 °C. For fluorescent imaging, cells were prepared as described in the immunocytochemistry section and images captured with the 100× oil objective using fluorescent channels for DAPI (350 nm), TLR4 (Rhodamine-555 nm), and CD68 (Texas Red-647 nm). TLR4 channel (red) gain and laser was set to 925 and 33%, CD68 channel (green) gain and laser was set to 975 and 33%, and DAPI channel (blue) gain and laser was set to 839 and 15%. These settings were applied to all confocal images. One random field-of-view (FOV) was obtained for each group at a resolution of 1024 × 1024 pixels. Eighty images were then taken through the depth (Z) of 6.63 µm for each FOV. This value of 6.63 μm was used to provide equal X, Y, and Z dimensions, providing standardization of collecting depth image sets.
Three dimensional reconstruction
Confocal image sets for each fluorescent channel were converted to 8-bit and processed in image J using Isosurface within the Bone J plugin (http://bonej.org/). TLR4 three dimensional (3D) mesh was produced with a resample rate of 2 and a threshold of 20. CD68 3D mesh was generated with a resample rate of 2 and threshold of 23. DAPI 3D mesh was constructed with a resample rate of 2 and a threshold of 30. 3D meshes for all fluorescent channels were imported into Blender (blender.org) and scaled uniformly. Artifacts due to apparent non-specific and non-cellular labeling were deleted in some channels for better imaging. Lighting, cameras and mesh-materials were created to image each scene. The respective brightfield image for each FOV was then placed at the base of each 3D reconstruction and scaled to fit. Images were rendered using Blender Cycles Render at 3840 × 3840 pixel resolution.
Statistical analysis
Image J Software was used to obtain the mean grey intensity of all immunoblots and the mean was taken for each time point. GraphPad Prism 7.0 was used for transformation, quantification, and graphing of all data. For statistical analysis of mRNA and protein expression, binding activity, cell viability, and immunofluorescence, one-way analysis of variance (ANOVA) with Dunnett’s or Tukey’s multiple comparison tests were used, unless otherwise stated in the results section. Significance was determined at P < 0.05.
Results
CHME-5 cells are not of human origin (STR genotyping of CHME-5 cells)
STR Genotyping was performed to validate the claim that CHME-5 cells are not of human origin. STR genotyping confirmed that CHME-5 cells are devoid of any human genomic DNA [Supplementary Figure 1].

Supplementary Figure 1. STR genotyping for CHME-5 cells. PowerPlex 21 system was used to analyze CHME-5 cells for human genomic DNA. A: electropherogram of CHME-5 sample; B: electropherogram of human DNA sample (positive control). STR: short tandem repeat
LPS-induced activation of NF-κB p65 in CHME-5 cells
Activation of NF-κB p65 is a crucial step for nuclear translocation and transcription of pro-inflammatory mediators. Phosphorylation of NF-κB p65 was used as a measure of NF-κB activation. Immunoblot analysis indicated that LPS increased nuclear levels of phospho-NF-κB p65 [Figure 1A]. Further analysis with a Student’s t-test revealed that the increase was statistically significant, P < 0.02 [Figure 1B].
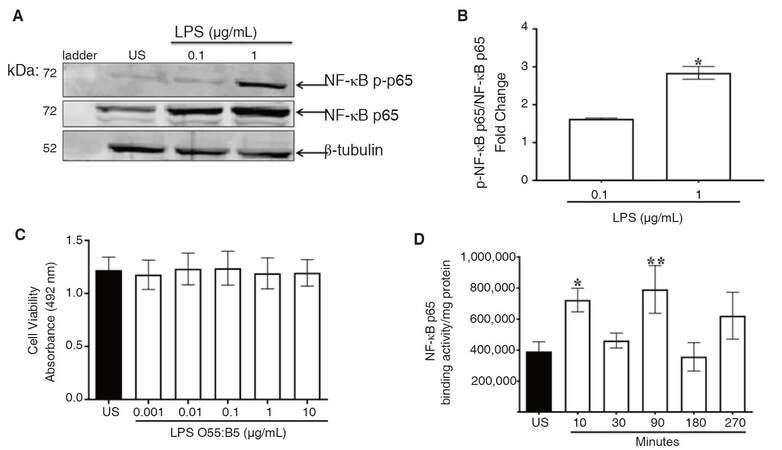
Figure 1. LPS-induced NF-κB p65 activation. CHME-5 cells were exposed to LPS (0.1-1 μg/mL) or media alone at 37 °C for 30 min. A: nuclear lysates were subjected to SDS-PAGE electrophoresis and immunoblotted with NF-κB p-p65 (1:1,000), NF-κB p65 (1:1,000), and β-tubulin (1:1,000) antibodies; B: data is normalized to US and expressed as fold change, integrated density, (*P < 0.02) vs. 0.1 μg/mL, images are representative of 3 independent experiments (n = 3) for each treatment group; C: CHME-5 cells were exposed to LPS (0.001-10 μg/mL) or media alone at 37 °C for 30 min, cell viability absorbance was read at 492 nm, 3 independent experiments were performed in duplicate (n = 3) for each treatment group; D: CHME-5 cells were stimulated with LPS (1 μg/mL) at 37 °C for 10-270 min, nuclear extracts were analyzed for NF-κB p65 binding activity, (*P < 0.04, **P < 0.01) vs. US. Image is representative of 5 independent experiments (n = 5) for each treatment group. Bars for all groups are presented as mean ± SEM. LPS: lipopolysaccharide; NF-κB: nuclear factor-kappa B; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; US: unstimulated
LPS is not toxic
The MTT assay was used to determine the extent to which LPS stimulation affects cell viability. ANOVA revealed that LPS (0.001-10 μg/mL) at 30 min did not significantly (P = 1.00) affect viability of CHME-5 cells [Figure 1C].
LPS-induced NF-κB p65 binding activity
For transcription of pro-inflammatory mediators to occur, the NF-κB p65 subunit must bind to nuclear consensus sequences. NF-κB p65 activation was also determined by assessing NF-κB p65 binding activity in nuclear extracts. Tubulin and histone 3 expression were measured as internal controls to confirm cellular compartmentalization in cytoplasmic and nuclear fractions [Supplementary Figure 2]. ANOVA and uncorrected Fisher’s LSD tests indicated that NF-κB p65 binding activity was significantly increased at 10 (P < 0.04) and 90 min (P < 0.01) [Figure 1D]. Binding activity at 30 and 180 min remained at basal levels.
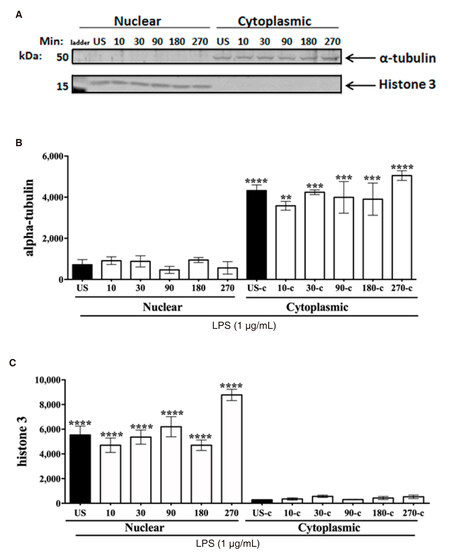
Supplementary Figure 2. α-tubulin and histone 3-immunoreactivity in CHME-5 cellular fractions. CHME-5 cells were sitmulated with LPS (1 μg/mL) and incubated at 37 °C for a time course (0-270 min). A: cytoplasmic and nuclear extracts were subjected to SDS-PAGE electrophoresis and immunoblotted with α-tubulin (1:1,000) or histone 3 (1:2,000) antibodies; B: integrated of α-tubulin cytoplasmic fractions, (**P = 0.003, ***P = 0.0004, ****P < 0.0001); C: similarly, histone 3-immunoreactivity was significantly increased in nuclear fractions, (****P < 0.0001). Image is representative of 3 individual experiments, n = 3. Data points are given as mean ± SEM. LPS: lipopolysaccharide; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresi
LPS-mediated IκBα phosphorylation in CHME-5 cells
Inhibitor kappa b alpha (IκBα) is the negative regulator for NF-κB and thus must be activated for release of NF-κB into the nucleus. Phosphorylation of IκBα in cytoplasmic lysates was used as a measure of IκBα activation [Figure 2A]. ANOVA and Dunnett’s multiple comparison tests revealed LPS significantly increased IκBα activation at 10 min following stimulation, (P < 0.02) [Figure 2B]. At 30-270 min, stimulation levels of p-IκBα were similar to control cells. At early time points, IκBα did not undergo degradation in CHME-5 cells [Supplementary Figure 3].
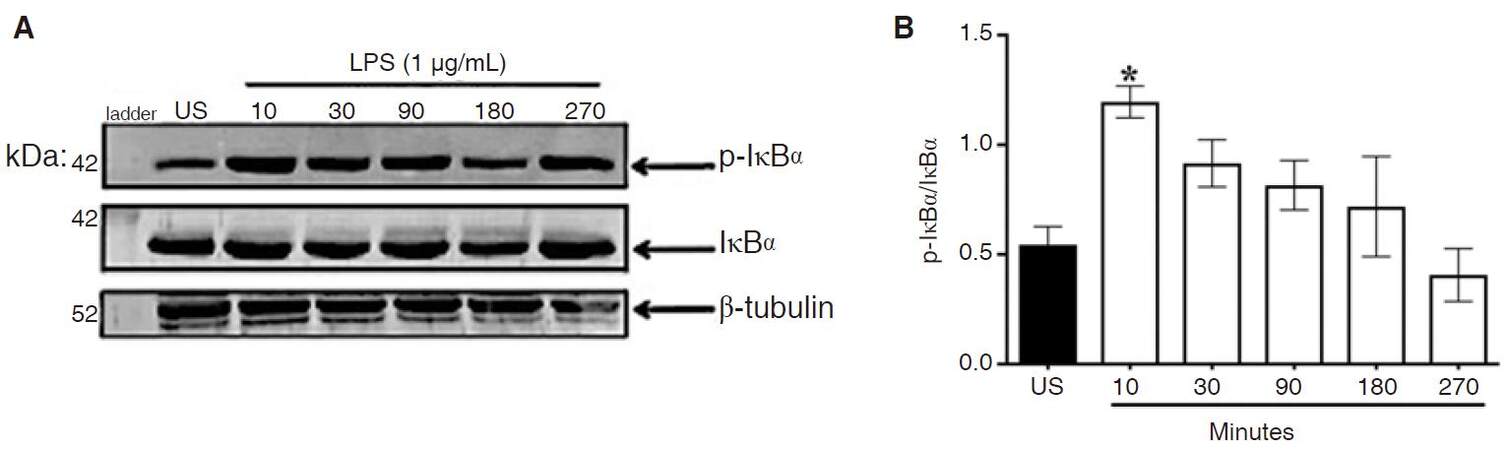
Figure 2. LPS-induced IκBα activation. CHME-5 cells were stimulated with LPS (1 μg/mL) at 37 °C for 10-270 min. A: cytoplasmic lysates were subjected to SDS-PAGE electrophoresis and immunoblotted with p-IκBα (1:1,000), IκBα (1:1,000), and β-tubulin (1:1,000) antibodies; B: integrated density, *P < 0.02 vs. US. Image is representative of 4 independent experiments (n = 4) for each treatment group. Bars for all groups are presented as mean ± SEM. LPS: lipopolysaccharide; IκBα: inhibitor kappa b alpha; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; US: unstimulated
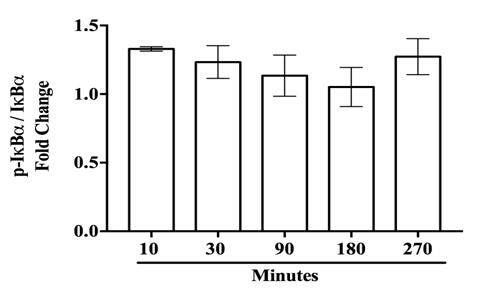
Supplementary Figure 3. IκBα does not undergo degradation at early timepoints in CHME-5 cells. CHME-5 cells were stimulated with LPS (1 μg/mL) and incubated at 37 °C for 10-270 min. Integrated density bands for IκBα/β-tubulin were normalized to US, P = 0.624. Image is representative of 4 individual experiments, n = 4. Data points are given as mean ± SEM. IκBα: inhibitor kappa b alpha; LPS: lipopolysaccharide
LPS-induced TNFα gene expression
Once NF-κB p65 is phosphorylated and translocated into the nucleus, it binds to pro-inflammatory consensus sequences for initiation of gene transcription mediators, such as TNFα. Compared to unstimulated cells, stimulation with LPS (1 μg/mL) significantly increased TNFα gene expression at 3 (P < 0.002), 6 (P < 0.02), and 18 (P < 0.003) h, as indicated by Kruskal-Wallis and Dunn’s multiple comparison tests [Figure 3A]. As an internal control, RNA integrity was verified by visualizing the 28S and 18S ribosomal RNA (rRNA) bands [Supplementary Figure 4].
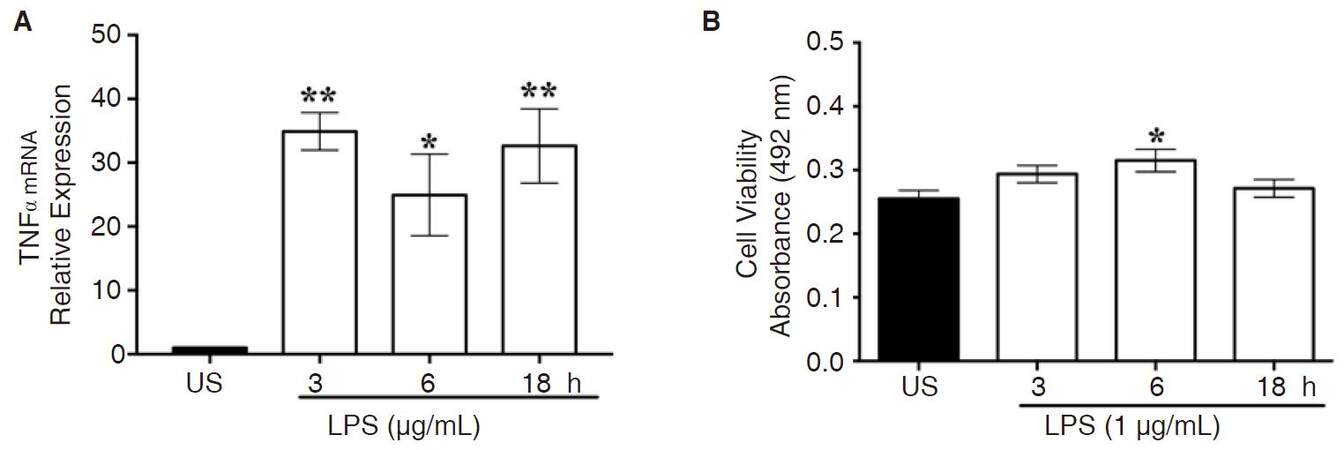
Figure 3. LPS-induced TNFα gene expression. A: CHME-5 cells were treated with LPS at 37 °C for 3, 6, and 18 h. TNFα mRNA expression was analyzed by RT-PCR analysis with β-actin as housekeeping gene, 3 h (**P < 0.002), 6 h (*P < 0.02), and 18 h (**P < 0.003) vs. US. Image is representative of 3 independent experiments (n = 3) for each treatment group; B: cell viability absorbance was read at 492 nm, *P < 0.01 vs. US. Experiments were carried out in duplicate (n = 3) for each treatment group. Bars for all groups are presented as mean ± SEM. LPS: lipopolysaccharide; TNFα: tumor necrosis factor-α; RT-PCR: real-time polymerase chain reaction; US: unstimulated

Supplementary Figure 4. Ribosomal RNA (rRNA) bands. CHME-5 cells were stimulated with LPS (1 μg/mL) and incubated at 37 °C for 3, 6, and 18 h. 28S and 18S rRNAs verified integrity of RNA. Image is representative of 3 individual experiments, n = 3. LPS: lipopolysaccharide
LPS is not toxic at later time points
The MTT assay revealed that LPS was not cytotoxic, rather LPS treatment resulted in a minimal increase in cell viability at 6 h, as indicated by ANOVA and Dunnett’s multiple comparison tests (P < 0.01) [Figure 3B].
CD68 gene expression in CHME-5 cells
CD68 is an established microglial marker, which is up-regulated during activation. Quantification of CD68 mRNA revealed a significant up-regulation of CD68 gene expression at 3 (P < 0.0001) and 6 (P < 0.01) h compared to unstimulated cells, as indicated by ANOVA and Dunnett’s multiple comparison tests, while expression returned to basal levels by 18 h [Figure 4A].
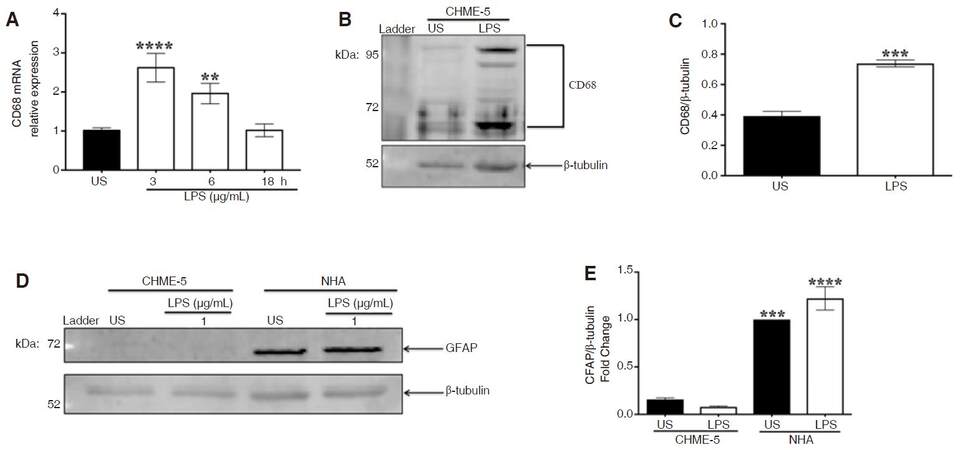
Figure 4. CHME-5 cells are CD68-positive and GFAP-negative. CHME-5 cells were stimulated with LPS (1 μg/mL) at 37 °C for 3, 6, and 18 h. A: CD68 mRNA expression was analyzed using RT-PCR analysis with β-actin as housekeeping gene, 3 h (****P < 0.0001) and 6 h (**P < 0.01) vs. US. Image is representative of 3 independent experiments (n = 3) for each treatment group; B: CHME-5 cells were stimulated with LPS (1 μg/mL) at 37 °C for 10 min, whole cell lysates were subjected to SDS-PAGE electrophoresis and immunoblotted with CD68 (1:500) and β-tubulin (1:1,000) antibodies; C: integrated density, ***P < 0.002 vs. US. Image is representative of 4 independent experiments (n = 4) for each group; D: CHME-5 and NHA cells were stimulated with LPS (1 μg/mL) at 37 °C for 10 min, whole cell lysates were subjected to SDS-PAGE electrophoresis and immunoblotted with GFAP (1:2,000) and β-tubulin (1:1,000) antibodies; E: integrated density, (***P < 0.002, ****P < 0.0001) vs. CHME-5. Image is representative of 3 independent experiments (n = 3) for each treatment group. Bars are presented as mean ± SEM. CD68: cluster of differentiation 68; GFAP: glial fibrillary astrocytic protein; LPS: lipopolysaccharide; RT-PCR: real-time polymerase chain reaction; US: unstimulated; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis
CD68 protein expression in CHME-5 cells
CD68 protein expression was analyzed after LPS stimulation in whole cell lysates [Figure 4B]. CD68 expression was significantly greater in LPS-stimulated cells at 10 min compared to unstimulated cells, as indicated by Student’s t-test, (P < 0.0002) [Figure 4C]. Multiple CD68 protein bands were detected in LPS-stimulated cells, most prominently at 68, 90, and 110 kilodaltons, which reflect the glycosylated form of CD68 protein[21].
CHME-5 cells are GFAP negative
To further confirm CHME-5 cells are microglia and not astrocytes, glial fibrillary astrocytic protein (GFAP) was measured at 10 min using whole cell lysates from normal human astrocytes (NHA) as a positive control [Figure 4D]. ANOVA and Tukey's multiple comparison tests revealed that GFAP expression in unstimulated and LPS-stimulated NHA was significantly greater than in CHME-5 cells (US-P < 0.0002, LPS-stimulated-P < 0.0001); indeed GFAP-immunoreactivity in CHME-5 cells was negligible [Figure 4E].
TLR4 gene and protein expression in CHME-5 cells
LPS induces the innate immune response through binding TLR4 and initiating intracellular signal transduction, which occurs rapidly, especially if proteins are not made de novo, and therefore, protein expression was analyzed as early as 10 min. On the other hand, RNA transcription is a lengthy process and therefore we chose to look at later time points (3, 6, 18 h). ANOVA and Dunnett’s multiple comparison tests revealed that TLR4 mRNA expression was significantly elevated 3 h (P < 0.0001) and 6 h (P < 0.006) after LPS treatment; by 18 h, expression was similar to unstimulated control [Figure 5A]. We also determined the extent to which LPS modulates TLR4 protein expression in CHME-5 cells. ANOVA and Dunnett’s post hoc analysis of immunoblots revealed significant increases in TLR4 protein expression at 90 (P < 0.005) and 270 (P < 0.01) min after LPS treatment [Figure 5B and C].
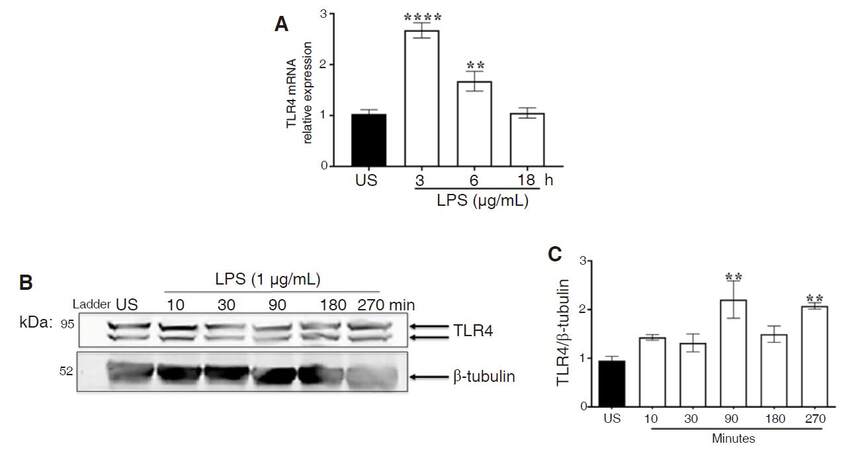
Figure 5. LPS-induced TLR4 expression. CHME-5 cells were stimulated with LPS (1 μg/mL) at 37 °C for 3, 6, and 18 h. A: TLR4 mRNA expression was analyzed using RT-PCR analysis with β-actin as housekeeping gene, 3 h (****P < 0.0001) and 6 h (**P < 0.006) vs. US. Image is representative of 3 independent experiments (n = 3) for each treatment group; B: CHME-5 cells were stimulated with LPS (1 μg/mL) at 37 °C for 10-270 min, whole cell lysates were subjected to SDS-PAGE electrophoresis and immunoblotted with TLR4 (1:1,000) and β-tubulin (1:1,000) antibodies; C: integrated density, 90 min (**P < 0.005) and 270 min (**P < 0.01) vs. US. Images are representative of 5 independent experiments (n = 5) for each treatment group. Bars are presented as mean ± SEM. LPS: lipopolysaccharide; TLR4: toll-like receptor 4; RT-PCR: real-time polymerase chain reaction; US: unstimulated; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis
Quantitative analysis of CD68 and TLR4 immunocytochemistry
Epifluorescence microscopy was employed to visualize and quantify CD68- and TLR4-immunofluorescence in unstimulated and LPS-stimulated CHME-5 cells [Figure 6A and B]. Epifluorescence microscopy revealed that CD68 protein expression is constitutively expressed in unstimulated CHME-5 cells. Analysis with CellProfiler revealed a significant increase in CD68-immunofluorescence, in each cell, compared to unstimulated and negative control cells, as assessed with Kruskal-Wallis and Dunn’s multiple comparison tests, (P < 0.0001) [Figure 6C]. Kruskal-Wallis and Dunn’s multiple comparison tests also revealed a significant increase in TLR4-immunofluorescence, in each cell, in LPS-stimulated cells compared to unstimulated cells and negative controls, (P < 0.0001) [Figure 6D].
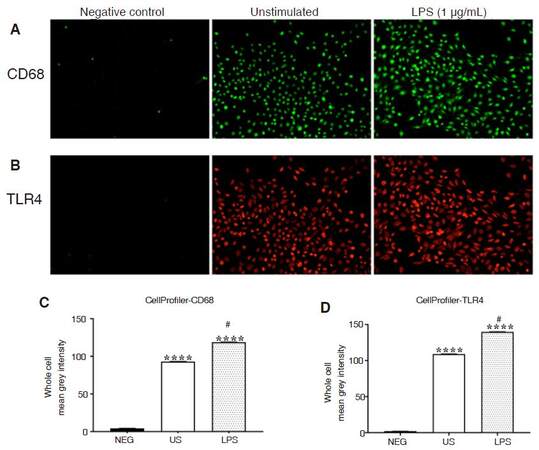
Figure 6. Quantitative analysis of CD68 and TLR4 immunofluorescence. CHME-5 cells were stimulated with LPS (1 μg/mL) at 37 °C for 10 min. Cells were fixed and stained. A: CD68 (1:1,000) and anti-rabbit-Alexa Fluor 647 (1:1,000); B: TLR4 (1:1,000) and anti-mouse-Alexa Fluor 555 (1:1,000) antibodies; A-B: epifluorescence microscopy-40× objective; C-D: CellProfiler analysis for mean grey intensity, (****P < 0.0001) vs. NEG and (#P < 0.0001) vs. US. Bars are presented as mean ± SEM. CD68: cluster of differentiation 68; TLR4: toll-like receptor 4; LPS: lipopolysaccharide; NEG: negative control (only incubated with secondary antibody not primary), US: unstimulated
Qualitative analysis of CD68 and TLR4 immunocytochemistry/3D reconstruction
Next, we captured images of unstimulated and LPS-stimulated cells with brightfield and fluorescent imaging, using confocal microscopy, to observe cellular morphology and protein expression, respectively. Overall, unstimulated cells displayed elongated cellular bodies with longer processes, while LPS-stimulated cells exhibited rounded or swollen bodies with shorter processes [Figure 7A]. TLR4 immunofluorescence was observed in both unstimulated and LPS-stimulated cells [Figure 7B]. Similarly, CD68 immunofluorescence was also expressed in unstimulated and LPS-stimulated cells [Figure 7C]. Both CD68 and TLR4 immunofluorescence was merged with DAPI [Figure 7D]. A chosen FOV superimposed onto brightfield images showed TLR4 and CD68 in 3D reconstruction [Figure 7E]. 3D reconstruction images of a close-up side view displayed CD68 and TLR4 immunofluorescence in punctate form, in both experimental treatment groups [Figure 7F].
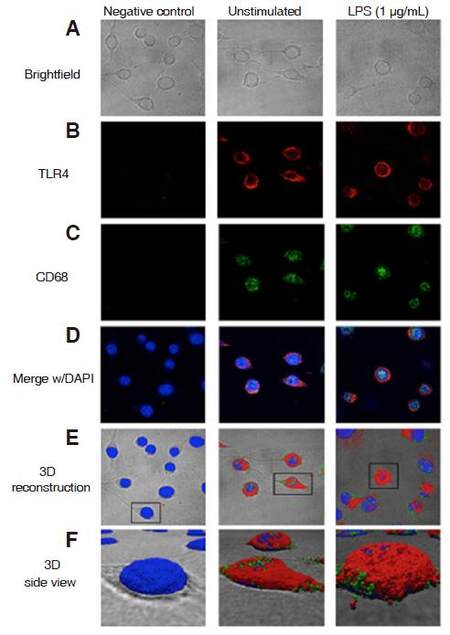
Figure 7. Visualization and 3D reconstruction of CD68 and TLR4 immunofluorescence. CHME-5 cells were stimulated with LPS (1 μg/mL) at 37 °C for 10 min. A: brightfield images were captured with 100× oil objective, cells were fixed and labeled; B: TLR4 (1:1,000) and anti-mouse-Alexa Fluor 555 (1:1,000); C: CD68 (1:1,000) and anti-rabbit-Alexa Fluor 647 (1:1,000) antibodies and DAPI (blue); B-D: confocal microscopy-100× oil objective; D: images merged with DAPI; E: 3D reconstruction with overlay on brightfield images; F: 3D reconstruction of close-up side view. CD68: cluster of differentiation 68; TLR4: toll-like receptor 4; LPS: lipopolysaccharide; DAPI: 4'-6-diamidino-2-phenylindole
Discussion
Microglia are considered the macrophages of the CNS and are central to inflammation in the brain[22]. Microglia are of monocytic lineage and take residence in the CNS during the first and second trimesters of embryonic development[22,23]. Much of what is known about microglial cells is derived from in vitro studies using primary or transformed cell lines of mouse or rat origin. The establishment of a microglial cell line, CHME-5, was an important advancement for investigating microglia. CHME-5 cells were previously immortalized and validated to have similar morphological and functional properties of primary microglia[19,24].
Interestingly, a recent publication[25] suggested that CHME-5 cells are of rat origin, not human. In the study, they performed genotyping by macrosatellite analysis, and investigated the CYCT1 gene expression in CHME-5 cells using both human and rat primers, and showed rat CYCT1 gene expression, but not the human counterpart in these cells[25]. The CHME-5 cell line currently being used in numerous labs is apparently of non-human origin, but to our knowledge this has yet to be independently verified. In the present study, human STR genotyping was performed on CHME-5 cells, which confirmed that these cells are not of human origin.
Importantly, we have used CHME-5 cells to advance our understanding of TLR4-mediated signaling in microglial cells. Conversely, during our review of the literature we identified an article characterizing primary human microglia in which data revealed large inconsistencies in microglial and inflammatory genes between primary human microglia and human microglial cell lines, including CHME-5 cells[26]. Knowing what we do now about the non-human origin of CHME-5 cells from Garcia-Mesa et al.[25] 2017, the inability to detect gene expression might have been due to the use of human primers. Taking this into consideration, we characterized TLR4 neuroinflammatory signaling in CHME-5 cells, as a rat cell line, validated that these cells are not of human origin, and demonstrated that CHME-5 cells remain a viable tool to study microglial-like inflammatory responses.
Brightfield imaging revealed morphological characteristics of "resting" vs. "activated" microglia. Unstimulated and LPS-treated cells displayed morphological signatures of microglia, as seen in previous studies[22,27-29], which included smaller bodies and elongated processes, or amoeboid-shaped and rounder cellular bodies, respectively. This demonstrates that CHME-5 cells retain characteristics that define their role as microglial cells.
Microglia up-regulate several activation markers in response to damage, disease, or loss of homeostatic conditions, such as CD68, which is a microglial activation marker expressed in endolysosomes and in the plasma membrane[30-32]. CD68 gene and protein expression were increased after LPS treatment during early time points, consistent with the rapid responsiveness of microglia as first responders in the CNS[33,34], and, aligns with their role of surveying and returning their environment to homeostasis[9,35]. The expression of CD68, along with morphological attributes observed with immunocytochemistry, indicates that these cells retain phenotypic properties of microglia. To ensure that our culture was not contaminated with astrocytes, GFAP-immunoreactivity was assessed in CHME-5 cells, and as expected, cells did not express GFAP. CHME-5 cells, unlike NHA, showed no GFAP-immunoreactivity, which confirmed CHME-5 cells are not astrocytes. These cells do indeed express CD68, and importantly are GFAP-negative, which eliminates our concern that CHME-5 may be astrocytes or that the culture is contaminated with astrocytes.
We demonstrated that LPS induced inflammatory signaling without inducing cytotoxicity. This finding is consistent with other studies[36], and indicates that results obtained throughout these experiments were not due to LPS toxicity, but rather to specific LPS-induced inflammatory responses.
To investigate LPS-induced TLR4 neuroinflammatory signaling, we used Escherichia coli LPS O55:B5 to model an inflammatory state in the CNS and studied several signaling proteins that are constitutively expressed and activated in the TLR4 pathway. Analysis of 2 crucial intracellular TLR4 signaling proteins, IκBα and NF-κB, were chosen to investigate neuroinflammatory signaling in CHME-5 cells. LPS-induced NF-κB p65 activation was consistent with other microglial cell lines[28,37-39]. Given the fact that many studies investigating microglial responses used the same dose of LPS (1 μg/mL), we are confident that CHME-5 cells were adequately treated to observe inflammatory responses[36,40,41]; unlike the use of a lower dose of LPS (1 ng/mL), which failed to activate and release nitric oxide in CHME-5 cells[42]. Additionally, other studies using human primary microglia and CHME-5 cells have used similar LPS dose range (0.1-1 μg/mL) and time points (6 h for RT-PCR) to investigate inflammatory responses [26,36,37,40,41].
The ability of p65 to be activated, translocate into the nucleus, and bind to NF-κB consensus sequences, is crucial in mediating inflammatory responses in a timely manner. Here, we demonstrated a second, more functional mean of NF-κB activation. LPS-induced NF-κB binding activity was exhibited in 2 waves, at 10 and 90 min. This LPS-induced biphasic activity, has been previously shown in macrophages and is attributed to several possibilities: (1) platelet activating factor (PAF), which is up-regulated during inflammatory stimuli, is involved in NF-κB nuclear translocation, which in turn produces pro-inflammatory cytokines, ultimately up-regulating PAF again in a feedback loop or (2) the release of p65 is not only attributed to IκBα, but IκBβ as well[43,44]. NF-κB is first released from IκBα and then from IκBβ, causing a biphasic response[43]. In fact, we also observed that the NF-κB target gene, TNFα, displayed similar biphasic-like expression in CHME-5 cells following LPS treatment, which was also seen in HeLa cells[45].
LPS-induced IκBα activation as demonstrated by phosphorylation is an early (within 10 min), transient event in CHME-5 cells. Phosphorylation was assessed in cytoplasmic fractions; thus it is presumable that after 10 min IκBα translocated into the nucleus or underwent proteasomal degradation[46]. Additionally, even though the data shows a trend towards IκBα degradation, the analysis revealed that this was not the case during early events in CHME-5 cells. It may be that in CHME-5 cells, evaluation in whole cell lysates is needed to get a detailed assessment of IκBα degradation, as research indicates that IκBα is also present in the nucleus[46,47]. IκBα signal-dependent degradation only occurs in response to ubiquitination of lysine residues[47,48], therefore further analysis of ubiquitination in CHME-5 cells is warranted to elucidate these events.
TLR4, a transmembrane glycoprotein, is part of the innate immune response, and is expressed in the CNS, primarily in microglia[17,18]. Consequently, in addition to assessing LPS-induced signaling, we were interested in the effects of LPS on TLR4 expression. Increased TLR4 gene expression following LPS treatment was consistent with reports showing increases in TLR4 expression in whole blood cells and monocytes as early as 2 and 3 h, respectively[49,50]. The increase observed in TLR4 gene expression in CHME-5 cells following LPS treatment has been reported to occur through binding of the master regulator PU.1 to TLR4 promoter regions, in response to endotoxin[51]. The expression of TLR4 as early as 3 h may be attributed to TLR4 being an early or middle phase gene that peaks at 1 h and 3 h[45]. LPS-induced TLR4 protein expression increased compared to unstimulated cells as seen in both immunoblot analysis and immunocytochemistry. Activation at 270 min may also be due to late-phase NF-κB activation, which is attributed to TRIF-dependent signaling[3]. On the other hand, the lack of TLR4 protein expression at 180 min may be due to negative regulation. There are several negative regulators that control TLR4 inflammatory signaling at different stages in the signaling pathway, such as selective androgen receptor modulator, RP105, and ST2L, which can be induced as early as 10 min, as in the case of IRAK-M[52,53]. Further investigation is warranted to determine mechanisms for regulation of LPS-induced TLR4 signaling in CHME-5 cells.
We provide novel images of CHME-5 cells, showing TLR4 and CD68 immunofluorescence. Epifluorescence imaging revealed that CD68 and TLR4 are constitutively expressed and are robustly up-regulated following LPS stimulation. Understandably, up-regulation of CD68 is expected due to its state of activation in response to LPS. Moreover, confocal imaging provided novel visualization of the expression of TLR4 and CD68 in unstimulated and LPS-stimulated cells. Furthermore, we provided a 3D representation of these proteins in CHME-5 cells, in response to LPS. The ability to reconstruct cells and observe protein expression in a 3D setting provides spatial awareness based on fluorescent intensity. Together, these imaging studies provide new, qualitative information about CD68/TLR4 expression in CHME-5 cells.
In summary, understanding microglial inflammatory responses is very important given the instrumental role of these cells in the innate CNS immune response and mounting data makes clear that microglia have diverse roles in the CNS. It is generally difficult and expensive to obtain human primary microglia and experiments are often challenging due to limited cell numbers. Cell lines are therefore essential to advance the field of neuroinflammation, in particular, inflammation exacerbating neurodegenerative diseases. Relative to other cell types, availability of microglial cell lines is limited, thus, it is important to maximize our understanding of those tools that are available. Here, we provide novel insights into CHME-5 cells by characterizing TLR4 neuroinflammatory signaling, which aligned with responses seen in other microglial cell lines, such as BV2, HAPI, and human primary microglia. We have also validated very recent findings suggesting that subsets of CHME-5 cells, currently in use, are of a rat, not human origin. With this present research, it is our expectation that CHME-5 will remain a useful tool in the study of microglial cells, particularly as related to neuroinflammation.
Declarations
AcknowledgmentsWe would like to acknowledge “Cancer Sucks” for their contribution to the purchase of the BioRad Thermocycler.
OSU-Center for Health Sciences, Department of Pharmacology and Physiology: We would like to thank Daniel Buck-Clay, Kelly McCracken, and Joni Finfrock for technical assistance with this project.
OSU Center for Health Sciences, Department of Forensics: We would like to thank Jun Fu, Ph.D. and Robert Allen, Ph.D. for performing the STR genotyping on CHME-5 sample.
Authors’ contributionsConcept, design, literature search, experimental studies, data acquisition, data analysis, statistical analysis, manuscript preparation, and manuscript editing: L. Figueroa-Hall
Data acquisition and data analysis for fluorescence studies, manuscript review: M. Anderson
Design, data analysis, manuscript editing: S. Das
Experimental design, manuscript editing, manuscript review: C.W. Stevens
Concept, definition of intellectual content, manuscript editing and review: R.L. Davis
Financial support and sponsorshipOklahoma Center for the Advancement of Science & Technology-HR14-007.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2017.
REFERENCES
1. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011;34:637-50.
4. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998;282:2085-8.
5. Hoebe K, Janssen EM, Kim SO, Alexopoulou L, Flavell RA, Han J, Beutler B. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol 2003;4:1223-9.
6. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem 1999;274:10689-92.
7. Lee CC, Avalos AM, Ploegh HL. Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol 2012;12:168-79.
8. Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol 2015;36:569-77.
9. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005;308:1314-8.
11. Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev 2004;17:942-64.
12. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell 2010;140:918-34.
13. Diestel A, Aktas O, Hackel D, Hake I, Meier S, Raine CS, Nitsch R, Zipp F, Ullrich O. Activation of microglial poly(ADP-ribose)-polymerase-1 by cholesterol breakdown products during neuroinflammation: a link between demyelination and neuronal damage. J Exp Med 2003;198:1729-40.
14. Aktas O, Ullrich O, Infante-Duarte C, Nitsch R, Zipp F. Neuronal damage in brain inflammation. Arch Neurol 2007;64:185-9.
15. Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord 2000;14 Suppl 1:S47-53.
16. Gao HM, Liu B, Zhang W, Hong JS. Novel anti-inflammatory therapy for Parkinson's disease. Trends Pharmacol Sci 2003;24:395-401.
17. Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol 2002;61:1013-21.
18. Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J 2001;15:155-63.
19. Janabi N, Peudenier S, Héron B, Ng KH, Tardieu M. Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. Neurosci Lett 1995;195:105-8.
20. Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 2006;7:R100.
21. Kurushima H, Ramprasad M, Kondratenko N, Foster DM, Quehenberger O, Steinberg D. Surface expression and rapid internalization of macrosialin (mouse CD68) on elicited mouse peritoneal macrophages. J Leukoc Biol 2000;67:104-8.
22. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011;91:461-553.
23. Monier A, Adle-Biassette H, Delezoide AL, Evrard P, Gressens P, Verney C. Entry and distribution of microglial cells in human embryonic and fetal cerebral cortex. J Neuropathol Exp Neurol 2007;66:372-82.
24. Peudenier S, Hery C, Montagnier L, Tardieu M. Human microglial cells: characterization in cerebral tissue and in primary culture, and study of their susceptibility to HIV-1 infection. Ann Neurol 1991;29:152-61.
25. Garcia-Mesa Y, Jay TR, Checkley MA, Luttge B, Dobrowolski C, Valadkhan S, Landreth GE, Karn J, Alvarez-Carbonell D. Immortalization of primary microglia: a new platform to study HIV regulation in the central nervous system. J Neurovirol 2017;23:47-66.
26. Melief J, Sneeboer MA, Litjens M, Ormel PR, Palmen SJ, Huitinga I, Kahn RS, Hol EM, de Witte LD. Characterizing primary human microglia: A comparative study with myeloid subsets and culture models. Glia 2016;64:1857-68.
27. Lee S, Lee J, Kim S, Park JY, Lee WH, Mori K, Kim SH, Kim IK, Suk K. A dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J Immunol 2007;179:3231-41.
28. Cheepsunthorn P, Radov L, Menzies S, Reid J, Connor JR. Characterization of a novel brain-derived microglial cell line isolated from neonatal rat brain. Glia 2001;35:53-62.
29. Sheng W, Zong Y, Mohammad A, Ajit D, Cui J, Han D, Hamilton JL, Simonyi A, Sun AY, Gu Z, Hong JS, Weisman GA, Sun GY. Pro-inflammatory cytokines and lipopolysaccharide induce changes in cell morphology, and upregulation of ERK1/2, iNOS and sPLA2-IIA expression in astrocytes and microglia. J Neuroinflammation 2011;8:121.
30. Saito N, Pulford KA, Breton-Gorius J, Massé JM, Mason DY, Cramer EM. Ultrastructural localization of the CD68 macrophage-associated antigen in human blood neutrophils and monocytes. Am J Pathol 1991;139:1053-9.
31. Pulford KA, Sipos A, Cordell JL, Stross WP, Mason DY. Distribution of the CD68 macrophage/myeloid associated antigen. Int Immunol 1990;2:973-80.
32. Ramprasad MP, Terpstra V, Kondratenko N, Quehenberger O, Steinberg D. Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc Natl Acad Sci U S A 1996;93:14833-8.
33. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci 1996;19:312-8.
34. Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev 1995;20:269-87.
36. Lindberg C, Crisby M, Winblad B, Schultzberg M. Effects of statins on microglia. J Neurosci Res 2005;82:10-9.
37. Lee SC, Liu W, Dickson DW, Brosnan CF, Berman JW. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J Immunol 1993;150:2659-67.
38. Wu CF, Bi XL, Yang JY, Zhan JY, Dong YX, Wang JH, Wang JM, Zhang R, Li X. Differential effects of ginsenosides on NO and TNF-alpha production by LPS-activated N9 microglia. Int Immunopharmacol 2007;7:313-20.
39. Horvath RJ, Nutile-McMenemy N, Alkaitis MS, Deleo JA. Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J Neurochem 2008;107:557-69.
40. Atanassov CL, Muller CD, Dumont S, Rebel G, Poindron P, Seiler N. Effect of ammonia on endocytosis and cytokine production by immortalized human microglia and astroglia cells. Neurochem Int 1995;27:417-24.
41. Jana M, Pahan K. Gemfibrozil, a lipid lowering drug, inhibits the activation of primary human microglia via peroxisome proliferator-activated receptor β. Neurochem Res 2012;37:1718-29.
42. Lisi L, Laudati E, Miscioscia TF, Dello Russo C, Topai A, Navarra P. Antiretrovirals inhibit arginase in human microglia. J Neurochem 2016;136:363-72.
43. Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, Ghosh S. I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappa B. Cell 1995;80:573-82.
44. Han SJ, Ko HM, Choi JH, Seo KH, Lee HS, Choi EK, Choi IW, Lee HK, Im SY. Molecular mechanisms for lipopolysaccharide-induced biphasic activation of nuclear factor-kappa B (NF-kappa B). J Biol Chem 2002;277:44715-21.
45. Tian B, Nowak DE, Brasier AR. A TNF-induced gene expression program under oscillatory NF-kappaB control. BMC Genomics 2005;6:137.
46. Arenzana-Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay RT, Virelizier JL, Dargemont C. Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J Cell Sci 1997;110:369-78.
47. Krappmann D, Wulczyn FG, Scheidereit C. Different mechanisms control signal-induced degradation and basal turnover of the NF-kappaB inhibitor IkappaB alpha in vivo. EMBO J 1996;15:6716-26.
48. Majdalawieh A, Ro HS. Regulation of IkappaBalpha function and NF-kappaB signaling: AEBP1 is a novel proinflammatory mediator in macrophages. Mediators Inflamm 2010;2010:823821.
49. Bosisio D, Polentarutti N, Sironi M, Bernasconi S, Miyake K, Webb GR, Martin MU, Mantovani A, Muzio M. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-gamma: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood 2002;99:3427-31.
50. Armstrong L, Medford AR, Hunter KJ, Uppington KM, Millar AB. Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp Immunol 2004;136:312-9.
51. Pedchenko TV, Park GY, Joo M, Blackwell TS, Christman JW. Christman, Inducible binding of PU.1 and interacting proteins to the Toll-like receptor 4 promoter during endotoxemia. Am J Physiol Lung Cell Mol Physiol 2005;289:L429-37.
52. Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel SN, Belkaid Y, Kurt-Jones EA, Karp CL. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol 2005;6:571-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Figueroa-Hall LK, Anderson MB, Das S, Stevens CW, Davis RL. LPS-induced TLR4 neuroinflammatory signaling in CHME-5 microglial cells. Neurosciences 2017;4:219-31. http://dx.doi.org/10.20517/2347-8659.2017.38
AMA Style
Figueroa-Hall LK, Anderson MB, Das S, Stevens CW, Davis RL. LPS-induced TLR4 neuroinflammatory signaling in CHME-5 microglial cells. Neuroimmunology and Neuroinflammation. 2017; 4: 219-31. http://dx.doi.org/10.20517/2347-8659.2017.38
Chicago/Turabian Style
Figueroa-Hall, Leandra K., Michael B. Anderson, Subhas Das, Craig W. Stevens, Randall L. Davis. 2017. "LPS-induced TLR4 neuroinflammatory signaling in CHME-5 microglial cells" Neuroimmunology and Neuroinflammation. 4: 219-31. http://dx.doi.org/10.20517/2347-8659.2017.38
ACS Style
Figueroa-Hall, LK.; Anderson MB.; Das S.; Stevens CW.; Davis RL. LPS-induced TLR4 neuroinflammatory signaling in CHME-5 microglial cells. Neurosciences. 2017, 4, 219-31. http://dx.doi.org/10.20517/2347-8659.2017.38
About This Article
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.