
Cerebral aneurysms and inflammation

Abstract
Multiple inflammatory factors, playing a crucial role in cerebral aneurysm formation, have been identified. Tumor necrosis factor-alpha (TNF-α) has been revealed to have a close connection with several risk factors that affect aneurysm formation. Remarkable expression in aneurysm walls of mRNA for TNF-α has been observed in humans. Possible therapeutic interventions to reduce the formation of cerebral aneurysms may include the inhibition of mediators of inflammation.
Keywords
Introduction
Most unruptured aneurysms, which are detected incidentally show stable clinical courses, exhibit fewer inflammatory or degenerative changes in the walls of affected blood vessels, and have a low risk of rupture. However, some unruptured aneurysms show significant changes in size and shape, and the rupture risk seems to be high. These aneurysms may rupture in the early phase of development or enlarge in a short time due to thinning of vessel walls resulting from the advancement of degenerative changes. It is of clinical relevance to accurately estimate the rupture risk of cerebral aneurysms, but no definitive methods exist to distinguish rupture-prone aneurysms from rupture-resistant ones. Recently, the rupture risk of unruptured cerebral aneurysms was reported. The 5-year cumulative rupture rates for aneurysms located in the internal carotid artery, anterior communicating artery, anterior cerebral artery, or middle cerebral artery in patients without a history of subarachnoid hemorrhage were 0%, 2.6%, 14.5%, and 40% for aneurysms < 7 mm, 7-12 mm, 13-24 mm, and 25 mm or greater, respectively.[1] By comparison, the rupture rates of aneurysms involving the posterior circulation and the posterior communicating artery were 2.5%, 14.5%, 18.4%, and 50%, respectively, for the same size categories. In one study, the annual rupture rate of unruptured cerebral aneurysms in a Japanese cohort was 0.95%.[2] In another, the average annual risk of rupture associated with small unruptured aneurysms was 0.54% overall, 0.34% for single aneurysms and 0.95% for multiple aneurysms.[3] The molecular mechanisms leading to the occurrence, development, and rupture of cerebral aneurysms have been experimentally investigated. Ruptured aneurysms manifest significant endothelial damage, structural changes in vessel walls, and inflammatory cell invasion compared to unruptured aneurysms.[4] The walls of ruptured aneurysms are fragile, possibly because macrophage infiltration into the aneurysm wall results in the loss of smooth muscle cells and degeneration of matrix proteins. In this manuscript, we discuss the molecular mechanisms of cerebral aneurysm development, focusing on inflammatory processes.
Inflammation and abdominal aortic aneurysms
The crucial role of inflammatory reactions can be seen in the formation of abdominal aortic aneurysms (AAAs). Important histological features of vessel walls with AAAs include chronic inflammatory cell infiltration of the adventitia and media, elastin fragmentation, degeneration, and attenuation of the media. Collagen in the media and adventitia provides tensile strength to the aortic wall. Collagen synthesis increases during the early stages of aneurysm formation, suggesting a repair process.[5] Inflammation-related mediators in aneurysm growth include matrix-degrading proteinases, proinflammatory cytokines, and chemokines.[6] In later stages, degeneration of collagen exceeds its synthesis, and when accompanied by excessive degradation of other extracellular matrix macromolecules such as elastin, ultimately favors AAA rupture. Indeed, AAAs exhibit increased local production of enzymes capable of degrading the extracellular matrix proteins collagen and elastin.[7-9] Oxidative stress and elevation of hemodynamic stress lead to degeneration of elastin or collagen. The activation of Th1 cytokines via interferon gamma and interleukin-6 polymorphism is accelerated.[10,11] The combination of inflammatory reactions and inherited vascular fragileness, along with environmental factors like advanced age or smoking, contributes to the formation and augmentation of AAAs.
Molecular biology of cerebral aneurysm formation
Multiple inflammatory factors have been identified that play a crucial role in cerebral aneurysm formation.[12] Inflammatory cells such as macrophages, monocytes, and T lymphocytes have been found in aneurysm walls.[4,13] The infiltration of leukocytes is related to the impairment or elimination of collagen fibers. The plasma levels of cytokines, collagenase, and elastase are elevated in patients with cerebral aneurysms.[14,15] Recently, nuclear factor-kappa B (NF-κB) and tumor necrosis factor-alpha (TNF-α) have been widely investigated as potentially key molecules in the inflammatory process. NF-κB is a transcription factor that is known to be closely related to inflammation. NF-κB is activated in endothelial cells at the site of arterial bifurcation in the early stages of aneurysm formation, which induces hemodynamic stress.[16] This activation is attributable to hemodynamic stress in the affected endothelial cells. It is thought that activated NF-κB incites several downstream inflammation-related genes at the transcriptional level. Monocyte chemoattractant protein-1 (MCP-1) is one target of NF-κB and is an indispensable factor for the migration of macrophages to the lesion site. The transcription of MCP-1 is controlled by NF-κB at the cerebral aneurysmal wall. MCP-1 is secreted from the endothelial cell layer in the early stages of aneurysm formation and from all layers of the arterial wall in later stages. Macrophage infiltration can be suppressed by the use of MCP-1 knockout mice or MCP-1 inhibitor, and leads to inhibition of aneurysm formation.[17] This evidence highlights the significance of MCP-1 and macrophage infiltration. Other factors controlled by NF-κB are inducible nitric oxide synthase (iNOS) and interleukin-1 beta (IL-1β), which are known as apoptosis-inducing factors. An investigation of experimentally induced cerebral aneurysms in rats revealed that apoptosis occurs in smooth muscle cells located within the medial layer of affected vessels, and is associated with inflammation.[18] iNOS activity results in the production of nitric oxide, which is an important factor involved in inflammatory reactions and the preservation of arterial regulation. iNOS is principally expressed in inflammatory cells such as macrophages, and may impair arterial wall integrity or induce apoptosis. The expression of iNOS is facilitated in the media and adventitia during the early phase of aneurysm formation. The incidence of experimentally induced cerebral aneurysms in iNOS knockout mice is the same as that in control mice, however the aneurysm size is significantly smaller.[19] This suggests that iNOS contribute to aneurysmal augmentation by promoting apoptosis in medial smooth muscle cells. IL-1β is an inflammatory cytokine which is activated by cleaved caspase 1. IL-1β is also produced in the early phase of aneurysm formation, mainly by medial smooth muscle cells. In IL-1β knockout mice, the progression of aneurysm development is significantly impaired.[20] This means that inflammatory reactions in the arterial wall contribute to aneurysm enlargement and that IL-1β is a significant mediator of this process.
Tumor necrosis factor-alpha
Tumor necrosis factor-alpha has been revealed to have a close connection with several risk factors that affect aneurysm formation. Remarkable expression in aneurysm walls of mRNA for TNF-α has been observed in humans.[21,22] In addition, therapeutic administration of a TNF-α inhibitor significantly reduced aneurysm formation in rats.[23] There has been some investigation into the relationship between TNF-α expression and aneurysm formation or rupture. Inflammation induced by expression of TNF-α leads to the degeneration of endothelial cells, the internal elastic lamina, and medial smooth muscle. Cerebral aneurysms are stabilized when the expression of TNF-α is reduced, or expression of anti-inflammatory cytokines increases, however continuous expression of TNF-α induces aneurysmal rupture.[24] TNF-α also increases the permeability of the aneurysm wall via cytokine cascades and induces the migration of macrophages or neutrophils to inflamed endothelial cells. In addition, TNF-α plays a role in other pathological manifestations such as modulation of the blood-brain barrier, fluid accumulation, and regulation of intracranial blood flow in aneurysmal subarachnoid hemorrhage.[25] In transgenic mice that are deficient for TNF-α or TNF receptors, susceptibility to nitric oxide is notably increased.[26,27] In mice with congenital TNF-α receptor deficiency, the deleterious effects of oxidative stress are increased by traumatic or ischemic loading, which indicates that stimulation of antioxidant pathways by TNF-α may provide protection against odixative damage.[28]
Mechanisms of cerebral aneurysm formation in humans
Aneurysm formation has been shown to occur at sites of constant hemodynamic stress both in humans and in experimentally induced models of cerebral aneurysm. This means that hemodynamic stress initiates early stage aneurysm formation. Aneurysm formation progresses when degenerative changes exceed vessel repair due to vascular remodeling. In addition to inflammation and apoptosis, extracellular matrix decomposition and endothelial dysfunction play critical roles in aneurysm formation. It has been found that gene expression in cerebral aneurysms may be linked to specific genetic regions. Furthermore, genetic regions that promote cerebral aneurysm formation are also linked to AAAs.[29] The factor analysis which affected to cerebral aneurysmal formation in human would progress in the future.
Histopathology of ruptured aneurysms
Loss of the internal elastic lamina and degeneration of medial vascular smooth muscle are common histopathological features of cerebral aneurysms. These degenerative changes occur in the aneurysm wall and can lead to aneurysmal rupture. Following rupture, the wall around the ruptured site is immensely thin and covered by a thrombosed fibrin plug. Infiltration of inflammatory cells such as neutrophils, lymphocytes, and macrophages into the adventitia also occurs. In addition, complement and immunoglobulin deposits form between the medial vascular smooth muscle and adventitia.[13] Loss of vascular smooth muscle cells, thinning of collagen fibers, and inflammation are more prominent in the walls of ruptured aneurysms compared to unruptured ones.[30] Therefore, these factors seem to play an important role in the weakening of aneurysmal walls [Figure 1].
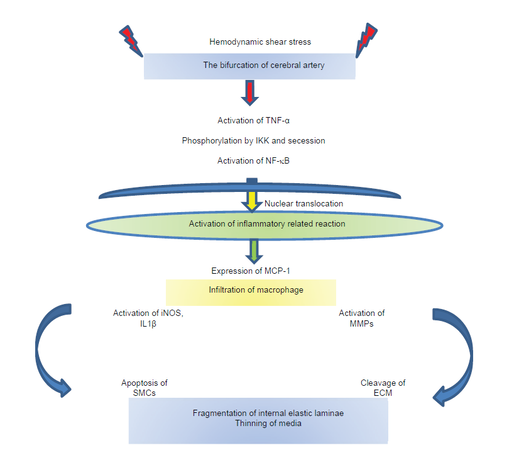
Figure 1. Factors associated with the formation of cerebral aneurysms. TNF-α: tumor necrosis factor-alpha; IKK: inhibitor of kappa kinase; NF-κB: nuclear factorkappa B; MCP-1: monocyte chemoattractant protein-1; iNOS: inducible nitric oxide synthase; IL-1β: interleukin-1b; MMPs: matrix metalloproteinases; SMCs: smooth muscle cells; ECM: extracellular matrix
Conclusion
Inflammatory cells have been found in the walls of cerebral aneurysms, and several inflammatory factors are reported to play crucial roles in cerebral aneurysm formation. Possible therapeutic interventions to reduce the formation of cerebral aneurysms may include the inhibition of mediators of inflammation.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Wiebers DO, Whisnant JP, Huston J 3rd, Meissner I, Brown RD Jr, Piepgras DG, Forbes G, Thielen K, Nichols D, O'Fallon W, Peacock J, Jaeger L, Kassell N, Kongable-Beckman G, Torner JC; International Study of Unruptured Intracranial Aneurysms Investigators. Unruptured intracranial aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet 2003;362:103-10.
2. UCAS Japan Investigators, Morita A, Kirino T, Hashi K, Aoki N, Fukuhara S, Hashimoto N, Nakayama T, Sakai M, Teramoto A, Tominari S, Yoshimoto T. The natural course of unruptured cerebral aneurysms in a Japanese cohort. N Engl J Med 2012;366:2474-82.
3. Sonobe M, Yamazaki T, Yonekura M, Kikuchi H. Small unruptured intracranial aneurysm verification study: SUAVe study, Japan. Stroke 2010;41:1969-77.
4. Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke 1999;30:1396-401.
5. Satta J, Haukipuro K, Kairaluoma MI, Juvonen T. Aminoterminal propeptide of type III procollagen in the follow-up of patients with abdominal aortic aneurysms. J Vasc Surg 1997;25:909-15.
6. Schouten O, van Laanen JH, Boersma E, Vidakovic R, Feringa HH, Dunkelgrün M, Bax JJ, Koning J, van Urk H, Poldermans D. Statins are associated with a reduced infrarenal abdominal aortic aneurysm growth. Eur J Vasc Endovasc Surg 2006;32:21-6.
7. Dobrin PB, Mrkvicka R. Failure of elastin or collagen as possible critical connective tissue alterations underlying aneurysmal dilatation. Cardiovasc Surg 1994;2:484-8.
8. Knox JB, Sukhova GK, Whittemore AD, Libby P. Evidence for altered balance between matrix metalloproteinases and their inhibitors in human aortic diseases. Circulation 1997;95:205-12.
9. Thompson RW, Parks WC. Role of matrix metalloproteinases in abdominal aortic aneurysms. Ann N Y Acad Sci 1996;800:157-74.
10. Basso F, Lowe GD, Rumley A, McMahon AD, Humphries SE. Interleukin-6 -174G>C polymorphism and risk of coronary heart disease in West of Scotland coronary prevention study (WOSCOPS). Arterioscler Thromb Vasc Biol 2002;22:599-604.
11. Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 2006;26:987-94.
12. Hashimoto T, Meng H, Young WL. Intracranial aneurysms: links among inflammation, hemodynamics and vascular remodeling. Neurol Res 2006;28:372-80.
13. Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery 1999;45:1137-46.
14. Gaetani P, Grazioli V, Tancioni F, Casari E, Tartara F, Rodriguezy Baena R. Abnormalities of collagen cross-linkage in posterior communicating artery aneurysms: a preliminary study. Neurol Res 1996;18:541-5.
15. Gaetani P, Rodriguez y Baena R, Tartara F, Messina AL, Tancioni F, Schiavo R, Grazioli V. Metalloproteases and intracranial vascular lesions. Neurol Res 1999;21:385-90.
16. Aoki T, Kataoka H, Shimamura M, Nakagami H, Wakayama K, Moriwaki T, Ishibashi R, Nozaki K, Morishita R, Hashimoto N. NF-kappaB is a key mediator of cerebral aneurysm formation. Circulation 2007;116:2830-40.
17. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Egashira K, Hashimoto N. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke 2009;40:942-51.
18. Kondo S, Hashimoto N, Kikuchi H, Hazama F, Nagata I, Kataoka H. Apoptosis of medial smooth muscle cells in the development of saccular cerebral aneurysms in rats. Stroke 1998;29:181-8.
19. Sadamasa N, Nozaki K, Hashimoto N. Disruption of gene for inducible nitric oxide synthase reduces progression of cerebral aneurysms. Stroke 2003;34:2980-4.
20. Moriwaki T, Takagi Y, Sadamasa N, Aoki T, Nozaki K, Hashimoto N. Impaired progression of cerebral aneurysms in interleukin-1beta-deficient mice. Stroke 2006;37:900-5.
21. Jayaraman T, Berenstein V, Li X, Mayer J, Silane M, Shin YS, Niimi Y, Kiliç T, Gunel M, Berenstein A. Tumor necrosis factor alpha is a key modulator of inflammation in cerebral aneurysms. Neurosurgery 2005;57:558-64.
22. Ruigrok YM, Tan S, Medic J, Rinkel GJ, Wijmenga C. Genes involved in the transforming growth factor beta signalling pathway and the risk of intracranial aneurysms. J Neurol Neurosurg Psychiatry 2008;79:722-4.
23. Yokoi T, Isono T, Saitoh M, Yoshimura Y, Nozaki K. Suppression of cerebral aneurysm formation in rats by a tumor necrosis factor-a inhibitor. J Neurosurg 2014;120:1193-200.
24. Jayaraman T, Paget A, Shin YS, Li X, Mayer J, Chaudhry H, Niimi Y, Silane M, Berenstein A. TNF-alpha-mediated inflammation in cerebral aneurysms: a potential link to growth and rupture. Vasc Health Risk Manag 2008;4:805-17.
25. Sibson NR, Blamire AM, Perry VH, Gauldie J, Styles P, Anthony DC. TNF-alpha reduces cerebral blood volume and disrupts tissue homeostasis via an endothelin- and TNFR2-dependent pathway. Brain 2002;125:2446-59.
26. Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg F, Mattson M. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med 1996;2:788-94.
27. Turrin N, Rivest S. Tumor necrosis factor alpha but not interleukin 1 beta mediates neuroprotection in response to acute nitric oxide excitotoxicity. J Neurosci 2006;26:143-51.
28. Shohami E, Ginis I, Hallenbeck JM. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev 1999;10:119-30.
29. Ruigrok YM, Elias R, Wijmenga C, Rinkel GJ. A comparison of genetic chromosomal loci for intracranial, thoracic aortic, and abdominal aortic aneurysms in search of common genetic risk factors. Cardiovasc Pathol 2008;17:40-7.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Yokoi T, Saito M, Yoshimura Y, Tsuji K, Nozaki K. Cerebral aneurysms and inflammation. Neurosciences 2015;2:55-8. http://dx.doi.org/10.4103/2347-8659.153977
AMA Style
Yokoi T, Saito M, Yoshimura Y, Tsuji K, Nozaki K. Cerebral aneurysms and inflammation. Neuroimmunology and Neuroinflammation. 2015; 2: 55-8. http://dx.doi.org/10.4103/2347-8659.153977
Chicago/Turabian Style
Yokoi, Toshihiro, Makoto Saito, Yayoi Yoshimura, Keiichi Tsuji, Kazuhiko Nozaki. 2015. "Cerebral aneurysms and inflammation" Neuroimmunology and Neuroinflammation. 2: 55-8. http://dx.doi.org/10.4103/2347-8659.153977
ACS Style
Yokoi, T.; Saito M.; Yoshimura Y.; Tsuji K.; Nozaki K. Cerebral aneurysms and inflammation. Neurosciences. 2015, 2, 55-8. http://dx.doi.org/10.4103/2347-8659.153977
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.