
Inflammation mediates the pathogenesis of cerebral aneurysm and becomes therapeutic target


Abstract
The treatment of cerebral aneurysms (CAs) is of social importance, because poor outcomes result in subarachnoid hemorrhages after rupture. However, there is currently no medical treatment available to prevent the progression and rupture of CAs, which results in a large number of patients without receiving treatment. Recent studies using human samples have revealed the presence of inflammatory responses in lesions and also the possible correlation of inflammation with CA progression or rupture. Furthermore, experimental studies using animal models of CAs have supported the notion from human studies and have clarified the crucial contribution of inflammation to the pathogenesis. In this process, a vicious cycle/positive feedback loop includes the nuclear factor-kappa B (NF-κB) activation, which plays a role in amplifying inflammatory responses to the point of chronicity. In addition, the infiltration of macrophages via NF-κB-mediated monocyte chemotactic protein 1 induction expands inflammation in whole arterial walls and contributes to the degeneration of media by producing various cytokines and tissue-destructive proteases. These series of studies have provided an important insight - antiinflammatory drugs can be therapeutically significant in the treatment of CAs. Indeed, in animal models, some drugs with an antiinflammatory effect effectively suppressed CA formation and progression, which supports this hypothesis. In addition, in human cases, some case-control studies have reported the preventive effect of statins and nonsteroidal antiinflammatory drugs on CA rupture. Therefore, the development of novel medical treatment for preventing the progression and rupture of CAs is needed in the near future. In this literature review, articles were selected by performing a PubMed search using the key words “cerebral aneurysm” and “inflammation”.
Keywords
Introduction
Cerebral aneurysms (CAs) have a great impact on society because of their high incidence and subsequent subarachnoid hemorrhages after rupture.[1,2] In recent times, a large cohort study in Japan reported that the annual rate of CA rupture was 0.95%, and the risk was increased according to the size of CA.[3] Since subarachnoid hemorrhages have a high mortality rate of up to 50%, the prevention of CA rupture and enlargement are of considerable significance to society.[4] Many CAs are detected through brain examinations before the rupture, so there is a chance for preventative treatment. Among these incidentally detected CAs, only those with a higher probability of rupture (e.g. CAs with a large size or irregular shape) are selected for surgical treatment.[1-3,5] Importantly, the remaining portion of CAs, more than half,[3] receive no treatment, except for the treatment of risk factors related with rupture, and are only followed-up with monitoring rupture and enlargement. Considering that there is a high incidence of CAs in the general population and poor outcomes resulting from subarachnoid hemorrhages despite intensive treatment, the development of a new drug therapy for unruptured CAs is indispensable. Therefore, the mechanisms underlying the formation and progression of CAs need to be clarified.
Inflammation and cerebral aneurysm formation, progression and rupture
Over the past couple of decades, a large number of studies have examined the underlying mechanism of CA formation, progression and rupture by investigating human CA specimens. These series of studies have identified the presence of the inflammatory responses in CA lesions and have suggested the role of the inflammatory processes in the pathogenesis of CAs. For example, the expression and induction of pro-inflammatory factors such as tumor necrosis factor alpha (TNF-α), infiltration of inflammatory cells in the CA lesions (mainly macrophages), and the change in cell population during CA progression or rupture were identified.[6-13] Furthermore, comprehensive gene expression analyses have revealed the induction of pro-inflammatory genes in lesions such as TNF-α and the up-regulation of the inflammation-related pathways through bioinformatics analyses such as antigen processing, immune responses, and responses to outward stimuli, which indicated a significant contribution to the pathogenesis of CAs.[14-16] Comprehensive gene expression analyses have also identified an increase in extracellular matrix turnover.[17,18] By linkage analyses, pro-inflammatory genes or extracellular matrix-related genes positively correlate with CAs.[19-22] However, a considerable limitation is present in studies using human samples because of the variety of background characteristics such as the genetics and clinical history. Furthermore, we cannot examine the exact association of each inflammation-related factor with CA progression through pharmacological inhibition or genetic modification. However, the establishment and use of animal CA models[23-27] has overcome these intrinsic limitations associated with human samples and has greatly advanced our understanding of the mechanisms that regulate CA formation, progression, and rupture.
In a rodent model, CAs are induced at the bifurcation sites of intracranial arteries through an increase in hemodynamic stress, which is also a trigger of CA formation in human,[28-30] and is achieved by performing one-sided carotid ligation and inducing systemic hypertension through salt over-loading.[23-25] Because CAs induced in models share common pathological features with human cases (e.g. disrupted internal elastic lamina and degenerative changes of the media, including the loss of medial smooth muscle cells) and also spontaneous rupture, they highly mimic human CAs and are presumably suitable for examining the mechanisms underlying CA formation and progression.[23,24] In some models, elastase is injected into the basal cistern to degenerate internal elastic lamina in intracranial arteries and to facilitate CA formation and progression in combination with induced systemic hypertension by angiotensin II infusion.[26,27] In this model, induced CAs in mice can rupture at a higher rate than that in former models; therefore, they can be used to examine the mechanisms regulating CA rupture.[27]
Recent experimental studies mainly using animal models of CAs[23-26] have clarified the involvement of inflammatory responses in the pathogenesis of CA formation and progression[22,31-41] and have supported the notion that inflammation in arterial walls contributes to the pathogenesis in human cases. Nuclear factor-kappa B (NF-κB) is a master transcription factor regulating the induction of various pro-inflammatory genes through the activation of responses to nociceptive stimuli.[42]Experimental studies have revealed the crucial role of NF-κB in the pathogenesis of CAs by triggering and regulating the inflammatory processes in lesions[43][Figure 1]. During CA formation, many NF-κB-activated cytokines/mediators such as interleukin (IL)-1β,[38] prostaglandinE2,[34] TNF-α,[37,40,41] and reactive oxygen species[44] are induced, and they significantly contribute to CA formation and progression. Furthermore, NF-κB-targeted pro-inflammatory genes, including matrix metalloproteinase-9,[26,32] cyclooxygenase-2 (COX-2),[34] inducible nitric oxide synthase,[36,39] monocyte chemoattractant protein-1 (MCP-1),[31,35] TNF-α,[37,40,41] and IL-1β[38] are induced and likewise contribute to CA formation and progression. In addition, the critical contribution of NF-κB to the pathogenesis of CA is demonstrated in the deficiency of the NF-κB p50 subunit in mouse or the inhibition of the NF-κB transcriptional activity by decoy oligonucleotides treatment in the rat, which both significantly suppress CA formation and progression by inhibiting NF-κB-mediated inflammatory responses in lesions.[33] Importantly, vicious cycles are formed around NF-κB activation in lesions; for example, TNF-α activates NF-κB and then NF-κB transcriptionally induces TNF-α. Similarly, NF-κB forms the positive feedback loop of the COX-2-prostaglandinE2-EP2-NF-κB pathway[34,45][Figure 1], and COX-2 is induced by hemodynamic stress loaded on the arterial walls at bifurcation sites of the intracranial arteries producing prostaglandinE2. Then synthesized prostaglandinE2 acts on one of its specific receptor subtypes, EP2, and further activates NF-κB. Because NF-κB transcriptionally induces the COX-2 expression, once hemodynamic stress activates COX-2, another positive feedback loop between the prostaglandin system and NF-κB is formed[34,45][Figure 1]. The presence of this vicious cycle and positive feedback loops amplifies and prolongs the triggered inflammatory responses. Along with the amplified inflammation, the infiltration of macrophages and other major inflammatory cells in the CA walls, which are recruited via the NF-κB-induced MCP-1 expression, contributes to the further expansion of inflammation in the whole arterial walls[31,35,43][Figure 1]. MCP-1 is first induced in the endothelial cells during CA formation and recruits macrophages in the arterial walls.[31] Then recruited macrophages produce various cytokines and tissue-destructive proteinases such as MMP-9 that contribute to the expansion of inflammation and tissue degeneration observed in CAs[31,35][Figure 1]. The critical contribution of MCP-1 mediated macrophages recruitment/infiltration in the pathogenesis is clearly shown by recent experimental reports in which the deficiency of MCP-1, administration of the dominant negative form of MCP-1 (7-ND), or depletion of macrophages by clodronate liposome all significantly suppressed CA formation and progression.[31,35]
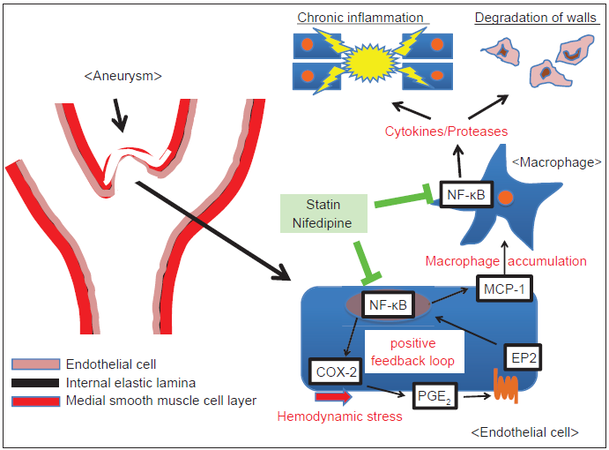
Figure 1. Schematic drawing of the mechanisms that regulate aneurysmal formation and progression. PGE2: prostaglandin E2; COX-2: cyclooxygenase-2; MCP-1: monocyte chemoattractant protein-1; NF-kB: nuclear factor-kappa B
The remaining question to be solved is whether the processes are regulating the initiation and progression of CAs are different. This important issue remains to be elucidated. As an initiation, as well as the progression of CAs, can be suppressed by inhibiting the inflammatory processes in lesions, these two steps of the pathogenesis presumably share the same underlying mechanisms in terms of inflammation. However, because the hemodynamic status surrounding CA lesions is completely different (e.g. a high hemodynamic status at the prospective site of the initiation[46,47] but a low hemodynamic status in the dome of the enlarging CAs[48,49]), there must be some differences in the processes that regulate the initiation and progression of CAs, and this is worthy of investigation.
In summary, based on the recent studies on CAs, long-lasting inflammatory responses in arterial walls play a crucial role in CA formation and progression, and NF-κB mediates this inflammation as a major transcription factor that regulates inflammation. In addition, the presence of a vicious cycle/positive feedback loop (i.e. NF-κB activation and macrophage infiltration via the NF-κB-induced MCP-1 expression) seems to be two major mechanisms that contribute to the amplification, expansion, and chronicity of inflammatory responses.
This recent experimental evidence on the role of inflammation in CAs may be useful in developing of therapeutic drugs for CA treatment. Recent experimental studies in human and rodent models have greatly advanced our understanding of the pathogenesis of CAs, making it more likely that the current treatment of CAs will be improved.
Potential of antiinflammatory drugs for treating cerebral aneurysms in animal models
As discussed, a long-lasting inflammatory response is detected in CA lesions, which plays a crucial role in the pathogenesis of CAs. Recent findings in rodent models have amassed evidence indicating the therapeutic effect of antiinflammatory drugs on the further enlargement or rupture of CAs and have proposed the potential of these drugs for treating CA.[31,33-36,50-58] Among these drugs that have a suppressive effect on CAs in animal models, statin,[50,51,55] nifedipine,[52] and emedastine difumarate[53] are already used in humans with clinical indications. Therefore, these drugs are good candidates for treating CAs in humans to prevent rupture or enlargement. We summarize the effect of these drugs on CAs in animal models.
Statin, a hydroxyl-3-methylglutaryl coenzyme A reductase inhibitor, is widely used as a cholesterol-lowering drug. In addition, it has an antiinflammatory and an anti-NF-κB effect, which are well known as “pleiotropic effects of statins”, although the precise mechanisms underlying its antiinflammatory effect remain to be elucidated.[59-61] Different kinds of statins orally administered to rat all effectively suppress inflammatory responses in CA lesions, which is evident by the activation of NF-κB and the expression of pro-inflammatory factors.[50,51] Furthermore, simvastatin orally given to rats exerts a protective effect on the endothelial cells in CA lesions.[51] Thereby, statins significantly suppress CA formation and enlargement of preexisting CAs in the rat model through their pleiotropic antiinflammatory effect.[50,51,55] Statins are prescribed to many patients with hypercholesterolemia, and since its safety is well established, statins may be promising drugs for treating patients with CAs and hypercholesterolemia [Figure 1].
Nifedipine is a Ca2+ channel blocker and a widely used antihypertensive drug. In addition to its antihypertensive effect, recently published in vitro experiments demonstrated its suppressive effect on the activation/induction of pro-inflammatory factors such as NF-κB and MCP-1 in cultured cells.[62-64] Consistently, nifedipine subcutaneously injected into rat with CAs inhibits the activation of NF-κB and suppresses the infiltration of macrophages via the NF-κB-mediated MCP-1 induction.[52] The CA formation and the enlargement of preexisting CAs are inhibited due to the reduction of the inflammatory response.[52] Since hypertension is a major risk factor for rupture of preexisting CAs, nifedipine may be a promising drug candidate for preventing CA rupture through its synergistic preventive effects on hypertension and inflammation [Figure 1]. Similarly, imidapril, a widely used angiotensin-converting enzyme inhibitor, exerts the potent suppressive effect on CAs induced in rat models through its anti-MMP-9 effect, which is independent of its antihypertensive effect.[54] Therefore, imidapril may also be a good drug candidate for treating CAs.
The mast cell is a major cell type that regulates allergic inflammation through the release of histamines from its granules.[65] Since it is a tissue-dwelling cell and contains a variety of cytokines and proteinases in its granules, it can rapidly respond to outward inflammatory stimuli and trigger inflammation.[65] Indeed, in inflammation-related diseases, such as vascular disease, atherosclerosis and aortic aneurysm, mast cells contribute to their pathogenesis by triggering and regulating inflammation through the release of cytokines and proteinases such as IL-6 and chymase.[66-70] In human CAs, the presence of mast cells has been identified,[11] and the increasing number of infiltrated mast cells in ruptured CAs indicates this cell’s role in the pathogenesis of CAs.[11] The significant contribution of mast cells to CAs was shown by a recent study[53] in which there was an increase in mast cells during CA formation and progression. Further, the pharmacological inhibition of the degranulation in mast cells by emedastine difumarate or tranilast, which are widely used antiallergy drugs in humans, effectively suppresses CA formation and progression by inhibiting the inflammatory responses of lesions.[53] These findings suggest the potential of the degranulation inhibitor of mast cells as drugs for treating CAs in humans.
Potential of antiinflammatory drugs for treating cerebral aneurysms in humans
In human cases, the beneficial effect of statins and nonsteroidal antiinflammatory drugs (NSAIDs) on CA rupture has been demonstrated, especially through prospective intervention trials.
A recent hospital-based case-control study implicated the potential of statins for preventing CA rupture in humans.[71] They enrolled 117 cases (patients with subarachnoid hemorrhage due to CA rupture) and 304 controls (patients with unruptured CAs) from 15 institutions in Japan, and the use of statins in each group was statistically compared. The patients’ background characteristics including age were similar.[71] As expected from previous reports, the size of the CAs and the patients’ current smoking status were properly selected as factors that correlated with rupture;[3,5,72,73] therefore, this study’s results seemed reliable. Stains were used in 9.4% of cases (11/117 cases) and 26.0% of the controls (79/304 controls), and the ratio of statins used between the groups was statistically different (P < 0.001).[71] Furthermore, after stratifying the data by the serum cholesterol level, administration of a statin was still inversely correlated with the risk of CA rupture in patients with serum cholesterol levels > 130 mg/dL.[71] According to logistic regression analysis, the use of any statins, independent of the type of statins, was inversely correlated with CA rupture with an adjusted odds ratio of 0.30.[71] Therefore, statins can be promising drugs for preventing CA rupture in humans with hypercholesterolemia. However, statins have a potent cholesterol-lowing effect and can sometimes decrease the serum cholesterol level below the normal limit even in patients without hypercholesterolemia. Although hypercholesterolemia was not a risk factor for CAs, and about 74% patients were without hypercholesterolemia in this study, the safety of statins on patients without hypercholesterolemia should be considered, and further randomized placebo-controlled study is warranted.
Nonsteroidal antiinflammatory drugs are broad COX inhibitors that involute symptoms of acute inflammation such as fever, swelling etc. A recent experimental study revealed the involvement of COX-2, the inducible form of COX, in the pathway of CA formation and progression by triggering and maintaining inflammation in lesions, suggesting the therapeutic effect of NSAIDs on CA rupture and progression. Conversely, NSAIDs inhibit the production of thromboxane A2 (a prostaglandin formed by the sequential actions of COX and thromboxane synthase from arachidonic acid) and thereby exert an antiplatelet effect, creating the potential for an increase in CA rupture and exacerbation of a subarachnoid hemorrhage after rupture. Consistent with these conflicting findings on NSAID treatment, results from recently published case-control studies were controversial in terms of the preventive effect of NSAIDs (i.e. the anti-inflammatory and antiplatelet effects) on CA rupture.[74-76] In a nested case-control study that enrolled patients from the International Study of Unruptured Intracranial Aneurysms (58 cases and 213 controls), frequent aspirin usage, 3 times/week, suppressed CA rupture with an adjusted odds ratio of 0.27 (P = 0.03) according to multivariable risk factor analyses.[74] Careful attention is necessary to interpret the data, because enrolled patients had relatively large aneurysms located at the posterior circulation, which are not representative of unruptured CAs.[74] Another study also demonstrated the suppressive effect of aspirin on CA rupture.[75] This study enrolled 717 consecutive patients with CAs (30 patients were excluded due to clopidogrel and/or warfarin use) and 897 CAs. During the follow-up, 274 patients presented with aneurysmal subarachnoid hemorrhage. The rate of CA rupture (subarachnoid hemorrhage) was significantly different between the groups (P = 0.016) with 40% of patients not using aspirin and 28% using aspirin,[75] suggesting the preventive effect of aspirin on CA rupture. Notably, aspirin did not influence the overall outcome.[75] Conversely, a recently published large nested case-control study enrolled 2,065 patients with subarachnoid hemorrhages and 20,649 controls, and it showed a significant increase in the risk of subarachnoid hemorrhage with an odd ratio of 1.5.[76] In addition, since NSAIDs have a considerable side effect (i.e. gastrointestinal hemorrhage), especially in elder patients, the administration of NSAIDs should be accompanied continuous attention and careful follow-up. However, the selective COX-2 inhibitor can eliminate the side effect derived from the nonselective inhibition of endogenous prostaglandin synthesis via COX-1 activity that is seen in NSAID treatment; therefore, the selective COX-2 inhibitor is another and more promising drug candidate for treating CAs. Indeed, in animal models, celecoxib, a selective COX-2 inhibitor, effectively suppressed CA progression.[34,45] However, a recent clinical trial for colon cancer reported an increase of cardiac failure and myocardial infarction with selective COX-2 inhibitor use[77] presumably due to the impairing balance between thromboxane A2 and prostacyclin, suggesting that COX-2 inhibitors may not be therapeutic drugs for preventing CA rupture. As previously discussed, currently published case-control studies have shown the controversy over the effect of NSAIDs on CA rupture. Although NSAIDs can be drug candidates for preventing the rupture of preexisting CAs, future randomized-control studies are warranted.
Conclusion
Cerebral aneurysms are of social importance, because of the resultant subarachnoid hemorrhage after rupture. The current problem with treating CAs is the lack of medical treatment to prevent their enlargement or rupture. Recent studies on human samples and experimental models have revealed the crucial role that chronic inflammatory responses play in the pathogenesis of CAs. Some drug candidates for treating CAs have been identified through experimental and case-control studies in humans. Therefore, the development of medical treatment for CAs is more likely in the near future.
Acknowledgments
I would like to express my gratitude to all the researchers, collaborators, technical assistants and secretaries contributing to our studies cited in the present manuscript. I also express my sincere gratitude to grants supporting our research works.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Wiebers DO, Piepgras DG, Brown RD Jr, Meissner I, Torner J, Kassell NF, Whisnant JP, Huston J 3rd, Nichols DA. Unruptured aneurysms. J Neurosurg 2002;96:50-1.
2. Rinkel GJ, Djibuti M, Algra A, van Gijn J. Prevalence and risk of rupture of intracranial aneurysms: a systematic review. Stroke 1998;29:251-6.
3. UCAS Japan Investigators, Morita A, Kirino T, Hashi K, Aoki N, Fukuhara S, Hashimoto N, Nakayama T, Sakai M, Teramoto A, Tominari S, Yoshimoto T. The natural course of unruptured cerebral aneurysms in a Japanese cohort. N Engl J Med 2012;366:2474-82.
5. Greving JP, Wermer MJ, Brown RD Jr, Morita A, Juvela S, Yonekura M, Ishibashi T, Torner JC, Nakayama T, Rinkel GJ, Algra A. Development of the PHASES score for prediction of risk of rupture of intracranial aneurysms: a pooled analysis of six prospective cohort studies. Lancet Neurol 2014;13:59-66.
6. Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery 1999;45:1137-46.
7. Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, Jaaskelainen J. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke 2004;35:2287-93.
8. Jayaraman T, Berenstein V, Li X, Mayer J, Silane M, Shin YS, Niimi Y, Kilic T, Gunel M, Berenstein A. Tumor necrosis factor alpha is a key modulator of inflammation in cerebral aneurysms. Neurosurgery 2005;57:558-64.
9. Laaksamo E, Tulamo R, Liiman A, Baumann M, Friedlander RM, Hernesniemi J, Kangasniemi M, Niemela M, Laakso A, Frosen J. Oxidative stress is associated with cell death, wall degradation, and increased risk of rupture of the intracranial aneurysm wall. Neurosurgery 2013;72:109-17.
10. Tulamo R, Frösen J, Junnikkala S, Paetau A, Pitkäniemi J, Kangasniemi M, Niemela M, Jaaskelainen J, Jokitalo E, Karatas A, Hernesniemi J, Meri S. Complement activation associates with saccular cerebral artery aneurysm wall degeneration and rupture. Neurosurgery 2006;59:1069-76.
11. Hasan D, Chalouhi N, Jabbour P, Hashimoto T. Macrophage imbalance (M1 vs. M2) and upregulation of mast cells in wall of ruptured human cerebral aneurysms: preliminary results. J Neuroinflammation 2012;9:222.
12. Laaksamo E, Tulamo R, Baumann M, Dashti R, Hernesniemi J, Juvela S, Niemela M, Laakso A. Involvement of mitogenactivated protein kinase signaling in growth and rupture of human intracranial aneurysms. Stroke 2008;39:886-92.
13. Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke 1999;30:1396-401.
14. Krischek B, Kasuya H, Tajima A, Akagawa H, Sasaki T, Yoneyama T, Ujiie H, Kubo O, Bonin M, Takakura K, Hori T, Inoue I. Networkbased gene expression analysis of intracranial aneurysm tissue reveals role of antigen presenting cells. Neuroscience 2008;154:1398-407.
15. Kurki MI, Häkkinen SK, Frösen J, Tulamo R, von und zu Fraunberg M, Wong G, Tromp G, Niemela M, Hernesniemi J, Jaaskelainen JE, YlaHerttuala S. Upregulated signaling pathways in ruptured human saccular intracranial aneurysm wall: an emerging regulative role of Tolllike receptor signaling and nuclear factor-kappaB, hypoxiainducible factor1A, and ETS transcription factors. Neurosurgery 2011;68:1667-75.
16. Shi C, Awad IA, Jafari N, Lin S, Du P, Hage ZA, Shenkar R, Getch CC, Bredel M, Batjer HH, Bendok BR. Genomics of human intracranial aneurysm wall. Stroke 2009;40:1252-61.
17. Li L, Yang X, Jiang F, Dusting GJ, Wu Z. Transcriptomewide characterization of gene expression associated with unruptured intracranial aneurysms. Eur Neurol 2009;62:330-7.
18. Peters DG, Kassam AB, Feingold E, Heidrich-O'Hare E, Yonas H, Ferrell RE, Brufsky A. Molecular anatomy of an intracranial aneurysm: coordinated expression of genes involved in wound healing and tissue remodeling. Stroke 2001;32:1036-42.
19. Inoue K, Mineharu Y, Inoue S, Yamada S, Matsuda F, Nozaki K, Takenaka K, Hashimoto N, Koizumi A. Search on chromosome 17 centromere reveals TNFRSF13B as a susceptibility gene for intracranial aneurysm: a preliminary study. Circulation 2006;113:2002-10.
20. Low SK, Zembutsu H, Takahashi A, Kamatani N, Cha PC, Hosono N, Kubo M, Matsuda K, Nakamura Y. Impact of LIMK1, MMP2 and TNF-α variations for intracranial aneurysm in Japanese population. J Hum Genet 2011;56:211-6.
21. Ruigrok YM, Rinkel GJ, Wijmenga C. The versican gene and the risk of intracranial aneurysms. Stroke 2006;37:2372-4.
22. Kataoka H, Aoki T. Molecular basis for the development of intracranial aneurysm. Expert Rev Neurother 2010;10:173-87.
23. Aoki T, Nishimura M. The development and the use of experimental animal models to study the underlying mechanisms of CA formation. J Biomed Biotechnol 2011;2011:535921.
24. Hashimoto N, Handa H, Hazama F. Experimentally induced cerebral aneurysms in rats. Surg Neurol 1978;10:3-8.
25. Morimoto M, Miyamoto S, Mizoguchi A, Kume N, Kita T, Hashimoto N. Mouse model of cerebral aneurysm: experimental induction by renal hypertension and local hemodynamic changes. Stroke 2002;33:1911-5.
26. Nuki Y, Tsou TL, Kurihara C, Kanematsu M, Kanematsu Y, Hashimoto T. Elastase-induced intracranial aneurysms in hypertensive mice. Hypertension 2009;54:1337-44.
27. Hosaka K, Downes DP, Nowicki KW, Hoh BL. Modified murine intracranial aneurysm model: aneurysm formation and rupture by elastase and hypertension. J Neurointerv Surg 2014;6:474-9.
28. Jou LD, Lee DH, Morsi H, Mawad ME. Wall shear stress on ruptured and unruptured intracranial aneurysms at the internal carotid artery. AJNR Am J Neuroradiol 2008;29:1761-7.
29. Takeuchi S, Karino T. Flow patterns and distributions of fluid velocity and wall shear stress in the human internal carotid and middle cerebral arteries. World Neurosurg 2010;73:174-85.
30. Shojima M, Oshima M, Takagi K, Torii R, Hayakawa M, Katada K, Morita A, Kirino T. Magnitude and role of wall shear stress on cerebral aneurysm: computational fluid dynamic study of 20 middle cerebral artery aneurysms. Stroke 2004;35:2500-5.
31. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Egashira K, Hashimoto N. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke 2009;40:942-51.
32. Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke 2007;38:162-9.
33. Aoki T, Kataoka H, Shimamura M, Nakagami H, Wakayama K, Moriwaki T, Ishibashi R, Nozaki K, Morishita R, Hashimoto N. NF-kappaB is a key mediator of cerebral aneurysm formation. Circulation 2007;116:2830-40.
34. Aoki T, Nishimura M, Matsuoka T, Yamamoto K, Furuyashiki T, Kataoka H, Kitaoka S, Ishibashi R, Ishibazawa A, Miyamoto S, Morishita R, Ando J, Hashimoto N, Nozaki K, Narumiya S. PGE(2)-EP(2) signalling in endothelium is activated by haemodynamic stress and induces cerebral aneurysm through an amplifying loop via NFκB. Br J Pharmacol 2011;163:1237-49.
35. Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N, Lawton MT, Young WL, Liang EI, Nuki Y, Hashimoto T. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke 2011;42:173-8.
36. Fukuda S, Hashimoto N, Naritomi H, Nagata I, Nozaki K, Kondo S, Kurino M, Kikuchi H. Prevention of rat cerebral aneurysm formation by inhibition of nitric oxide synthase. Circulation 2000;101:2532-8.
37. Aoki T, Fukuda M, Nishimura M, Nozaki K, Narumiya S. Critical role of TNF-alpha-TNFR1 signaling in intracranial aneurysm formation. Acta Neuropathol Commun 2014;2:34.
38. Moriwaki T, Takagi Y, Sadamasa N, Aoki T, Nozaki K, Hashimoto N. Impaired progression of cerebral aneurysms in interleukin-1beta-deficient mice. Stroke 2006;37:900-5.
39. Sadamasa N, Nozaki K, Hashimoto N. Disruption of gene for inducible nitric oxide synthase reduces progression of cerebral aneurysms. Stroke 2003;34:2980-4.
40. Starke RM, Chalouhi N, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Wada K, Shimada K, Hasan DM, Greig NH, Owens GK, Dumont AS. Critical role of TNF-α in cerebral aneurysm formation and progression to rupture. J Neuroinflammation 2014;11:77.
41. Yokoi T, Isono T, Saitoh M, Yoshimura Y, Nozaki K. Suppression of cerebral aneurysm formation in rats by a tumor necrosis factor-a inhibitor. J Neurosurg 2014;120:1193-200.
42. Hayden MS, Ghosh S. NFkappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 2012;26:203-34.
43. Aoki T, Nishimura M. Targeting chronic inflammation in cerebral aneurysms: focusing on NF-kappaB as a putative target of medical therapy. Expert Opin Ther Targets 2010;14:265-73.
44. Aoki T, Nishimura M, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Reactive oxygen species modulate growth of cerebral aneurysms: a study using the free radical scavenger edaravone and p47phox(-/-) mice. Lab Invest 2009;89:730-41.
45. Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends Pharmacol Sci 2012;33:304-11.
46. Alfano JM, Kolega J, Natarajan SK, Xiang J, Paluch RA, Levy EI, Siddiqui AH, Meng H. Intracranial aneurysms occur more frequently at bifurcation sites that typically experience higher hemodynamic stresses. Neurosurgery 2013;73:497-505.
47. Metaxa E, Tremmel M, Natarajan SK, Xiang J, Paluch RA, Mandelbaum M, Siddiqui AH, Kolega J, Mocco J, Meng H. Characterization of critical hemodynamics contributing to aneurysmal remodeling at the basilar terminus in a rabbit model. Stroke 2010;41:1774-82.
48. Boussel L, Rayz V, McCulloch C, Martin A, Acevedo-Bolton G, Lawton M, Higashida R, Smith WS, Young WL, Saloner D. Aneurysm growth occurs at region of low wall shear stress: patient-specific correlation of hemodynamics and growth in a longitudinal study. Stroke 2008;39:2997-3002.
49. Tanoue T, Tateshima S, Villablanca JP, Viñuela F, Tanishita K. Wall shear stress distribution inside growing cerebral aneurysm. AJNR Am J Neuroradiol 2011;32:1732-7.
50. Aoki T, Kataoka H, Ishibashi R, Nakagami H, Nozaki K, Morishita R, Hashimoto N. Pitavastatin suppresses formation and progression of cerebral aneurysms through inhibition of the nuclear factor kappaB pathway. Neurosurgery 2009;64:357-65.
51. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Simvastatin suppresses the progression of experimentally induced cerebral aneurysms in rats. Stroke 2008;39:1276-85.
52. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Nifedipine inhibits the progression of an experimentally induced cerebral aneurysm in rats with associated down-regulation of NF-kappa B transcriptional activity. Curr Neurovasc Res 2008;5:37-45.
53. Ishibashi R, Aoki T, Nishimura M, Hashimoto N, Miyamoto S. Contribution of mast cells to cerebral aneurysm formation. Curr Neurovasc Res 2010;7:113-24.
54. Ishibashi R, Aoki T, Nishimura M, Miyamoto S. Imidapril inhibits cerebral aneurysm formation in an angiotensin-converting enzyme-independent and matrix metalloproteinase-9-dependent manner. Neurosurgery 2012;70:722-30.
55. Kimura N, Shimizu H, Eldawoody H, Nakayama T, Saito A, Tominaga T, Takahashi A. Effect of olmesartan and pravastatin on experimental cerebral aneurysms in rats. Brain Res 2010;1322:144-52.
56. Tada Y, Kitazato KT, Tamura T, Yagi K, Shimada K, Kinouchi T, Satomi J, Nagahiro S. Role of mineralocorticoid receptor on experimental cerebral aneurysms in rats. Hypertension 2009;54:552-7.
57. Makino H, Tada Y, Wada K, Liang EI, Chang M, Mobashery S, Kanematsu Y, Kurihara C, Palova E, Kanematsu M, Kitazato K, Hashimoto T. Pharmacological stabilization of intracranial aneurysms in mice: a feasibility study. Stroke 2012;43:2450-6.
58. Yagi K, Tada Y, Kitazato KT, Tamura T, Satomi J, Nagahiro S. Ibudilast inhibits cerebral aneurysms by down-regulating inflammation-related molecules in the vascular wall of rats. Neurosurgery 2010;66:551-9.
59. Blum A, Shamburek R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation and thrombogenesis. Atherosclerosis 2009;203:325-30.
60. Shovman O, Levy Y, Gilburd B, Shoenfeld Y. Antiinflammatory and immunomodulatory properties of statins. Immunol Res 2002;25:271-85.
61. Bellosta S, Bernini F, Ferri N, Quarato P, Canavesi M, Arnaboldi L, Fumagalli R, Paoletti R, Corsini A. Direct vascular effects of HMG-CoA reductase inhibitors. Atherosclerosis 1998;137 Suppl:S101-9.
62. Matsumori A, Nunokawa Y, Sasayama S. Nifedipine inhibits activation of transcription factor NF-kappaB. Life Sci 2000;67:2655-61.
63. Yamagishi S, Inagaki Y, Abe R, Kikuchi S, Sasaki N, Takeuchi M. Nifedipine inhibits apoptotic cell death of cultured endothelial cells induced by tumor necrosis factor-alpha. Drugs Exp Clin Res 2003;29:141-5.
64. Yamagishi S, Inagaki Y, Kikuchi S. Nifedipine inhibits tumor necrosis factor-alpha-induced monocyte chemoattractant protein-1 overexpression by blocking NADPH oxidase-mediated reactive oxygen species generation. Drugs Exp Clin Res 2003;29:147-52.
65. Krishnaswamy G, Ajitawi O, Chi DS. The human mast cell: an overview. Methods Mol Biol 2006;315:13-34.
66. Liu J, Divoux A, Sun J, Zhang J, Clément K, Glickman JN, Sukhova GK, Wolters PJ, Du J, Gorgun CZ, Doria A, Libby P, Blumberg RS, Kahn BB, Hotamisligil GS, Shi GP. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med 2009;15:940-5.
67. Sun J, Sukhova GK, Wolters PJ, Yang M, Kitamoto S, Libby P, MacFarlane LA, Mallen-St Clair J, Shi GP. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med 2007;13:719-24.
68. Sun J, Sukhova GK, Yang M, Wolters PJ, MacFarlane LA, Libby P, Sun C, Zhang Y, Liu J, Ennis TL, Knispel R, Xiong W, Thompson RW, Baxter BT, Shi GP. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J Clin Invest 2007;117:3359-68.
69. Sun J, Zhang J, Lindholt JS, Sukhova GK, Liu J, He A, Abrink M, Pejler G, Stevens RL, Thompson RW, Ennis TL, Gurish MF, Libby P, Shi GP. Critical role of mast cell chymase in mouse abdominal aortic aneurysm formation. Circulation 2009;120:973-82.
70. Xu JM, Shi GP. Emerging role of mast cells and macrophages in cardiovascular and metabolic diseases. Endocr Rev 2012;33:71-108.
71. Yoshimura Y, Murakami Y, Saitoh M, Yokoi T, Aoki T, Miura K, Ueshima H, Nozaki K. Statin use and risk of cerebral aneurysm rupture: a hospital-based case-control study in Japan. J Stroke Cerebrovasc Dis 2014;23:343-8.
72. Kurth T, Kase CS, Berger K, Schaeffner ES, Buring JE, Gaziano JM. Smoking and the risk of hemorrhagic stroke in men. Stroke 2003;34:1151-5.
73. Sandvei MS, Romundstad PR, Müller TB, Vatten L, Vik A. Risk factors for aneurysmal subarachnoid hemorrhage in a prospective population study: the HUNT study in Norway. Stroke 2009;40:1958-62.
74. Hasan DM, Mahaney KB, Brown RD Jr, Meissner I, Piepgras DG, Huston J, Capuano AW, Torner JC. Aspirin as a promising agent for decreasing incidence of cerebral aneurysm rupture. Stroke 2011;42:3156-62.
75. Gross BA, Rosalind Lai PM, Frerichs KU, Du R. Aspirin and aneurysmal subarachnoid hemorrhage. World Neurosurg 2014;82:1127-30.
76. Garbe E, Kreisel SH, Behr S. Risk of subarachnoid hemorrhage and early case fatality associated with outpatient antithrombotic drug use. Stroke 2013;44:2422-6.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Aoki T. Inflammation mediates the pathogenesis of cerebral aneurysm and becomes therapeutic target. Neurosciences 2015;2:86-92. http://dx.doi.org/10.4103/2347-8659.154381
AMA Style
Aoki T. Inflammation mediates the pathogenesis of cerebral aneurysm and becomes therapeutic target. Neuroimmunology and Neuroinflammation. 2015; 2: 86-92. http://dx.doi.org/10.4103/2347-8659.154381
Chicago/Turabian Style
Aoki, Tomohiro. 2015. "Inflammation mediates the pathogenesis of cerebral aneurysm and becomes therapeutic target" Neuroimmunology and Neuroinflammation. 2: 86-92. http://dx.doi.org/10.4103/2347-8659.154381
ACS Style
Aoki, T. Inflammation mediates the pathogenesis of cerebral aneurysm and becomes therapeutic target. Neurosciences. 2015, 2, 86-92. http://dx.doi.org/10.4103/2347-8659.154381
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.