
Role of the complement cascade in cerebral aneurysm formation, growth, and rupture

Abstract
Rupture of intracranial aneurysms is the most common cause of nontraumatic subarachnoid hemorrhage, but the intricate neuroinflammatory processes which contribute to aneurysm pathophysiology are not well-understood. Mounting evidence has implicated the complement cascade in the progression of aneurysms from their formation to rupture. In this article, we identify and review studies that have sought to determine the role of the complement system in the aneurysm pathogenesis. The studies were generally conducted by immunhistological analyses on aneurysm tissue collected intraoperatively, and multiple components of the complement cascade and its modulators were identified in specific regions of the aneurysm wall. The results of the studies suggest that the complement cascade is locally upregulated and disinhibited in the perianeurysmal environment, and that it contributes to chronic as well as acute immunological damage to the aneurysm wall. In the future, understanding the mechanisms at work in complement-mediated damage is necessary to leading the development of novel therapies.
Keywords
Introduction
Saccular or “berry” aneurysms, which are characterized by an outpouching from one side of an affected artery, have a prevalence of 3.2% in the general population[1,2] and account for 90% of intracranial aneurysms.[3,4] Ruptured saccular aneurysms are responsible for 85% of cases of nontraumatic subarachnoid hemorrhage (SAH),[5] which carries a high case-fatality rate of 27-44%[6] and often leaves survivors with significant functional and cognitive deficits.[2,7] Clinically, aneurysms have mainly been characterized by their location and morphological features (size, shape, etc.),[8] but less attention has been paid to the underlying immune processes, which contribute to their formation, growth, or rupture. Understanding these mechanisms is important to facilitate the development of novel diagnostic and therapeutic strategies, and histopathological findings may contribute to conventional clinical and radiological factors.[9,10] Recently, there has been growing evidence that the complement cascade, a major effector arm of the innate immune system,[11] plays a role in the pathophysiology of intracranial aneurysms, and more broadly, in cerebrovascular conditions. In this article, we review the complement system as it relates to the pathogenesis of intracranial saccular aneurysms. A thorough review of the literature was conducted on PubMed, MEDLINE, EMBASE, and Cochrane library databases using the search terms: “complement”, “aneurysm”, “SAH”, “hemorrhagic stroke”, “neuroinflammation”, and “saccular” in varying combinations. Only original research articles that, at least in part, investigated the role of the complement cascade in intracranial aneurysms were selected.
Histopathological mechanisms of saccular aneurysm formation, growth, and rupture
The progression of saccular aneurysms from formation to rupture involves a complex interplay of hemodynamic forces and changes in aneurysm morphology (luminal factors), vessel wall integrity (extra-luminal factors), immunological pathways, and patient-specific factors.[8,12-14] Clinical management and risk stratification has generally relied on luminal characteristics (i.e. size, location), and patient-specific risk factors, mainly smoking, hypertension, heavy alcohol consumption, positive family history,[15-18] but this does not differentiate among aneurysms that have similar morphological features yet heterogeneous natural histories and pathological features.
Strong hemodynamic forces are thought to play a role in initiating aneurysm formation as a result of high shear stress on vessel walls, which is greatest at the bifurcations of cerebral arteries, where aneurysms occur most often.[19] Shear stress has been shown to lead to loss and damage of endothelial cells, loss of the internal elastic lamina (IEL), migration of vascular smooth muscle cells (SMCs), and induction of intracellular pathways in endothelial cells that induce pro-inflammatory cytokine release.[8,20-24]
Endothelial injury is considered to be a necessary first step in the aneurysm pathogenesis.[25,26] In response to increased wall stress, endothelial cells undergo not only morphological, but also functional changes, in which they upregulate production of pro-inflammatory signals (cytokines, interleukins such as interleukin-1β [IL-1β], leukocyte chemoattractants) as well as matrix metalloproteinases (MMPs),[27] resulting in recruitment of inflammatory cells and MMP-mediated enzymatic remodeling of the vessel wall. In addition, disruption of the IEL, which may initially be in the form of shear stress-induced tears, is a characteristic histopathological feature of saccular aneurysms.[8,24] As the IEL is damaged, the hemodynamic environment becomes turbulent within the lumen of the nascent aneurysm, leading to further endothelial damage and eventually de-endothelialization within the luminal surface of the aneurysm.[22,28] This hemodynamic environment, in concert with the exposed subendothelial matrix, promotes formation of intraluminal thrombi which consist of layers of platelets, red blood cells, lipid-laden macrophages, and other leukocytes leading to increased oxidative stress, inflammation, and further cell death.[14,29-30] The subsequent inflammatory response within the damaged vessel wall consists of leukocytic infiltration with macrophages, T-cells, and mast cells, and is found in both ruptured and unruptured aneurysms.[8,15,21,31-33] This inflammatory response results in progressive disorganization and loss of SMC and extracellular matrix (ECM) from the media and open and stretched collagen in the adventitia.[8]
Hemodynamic and environmental factors both contribute to phenotypic modulation and eventually damage of and death to SMCs.[25] Initially, SMCs adapt to hemodynamic stress by migrating from the tunica media into the intima to form myointimal hyperplasia. However, in response to local signals, they also become a secretory, pro-inflammatory cell type characterized by upregulation of necrosis factor-κB signaling, increased production of MMP, IL-1β, tumor necrosis factor-α, and initiation of pathways leading to SMC apoptosis.[34-40] As SMCs are largely responsible for the production of the ECM,[41] their apoptosis results in decreased synthesis of the new matrix, which exacerbates the effects of the overabundant MMPs and further weakens the vessel wall. MMPs are also produced by macrophages,[42,43] which play a large role in neuroinflammation, and in intracranial aneurysms there is downregulation of tissue inhibitors of MMPs[42,44] and up-regulation of proteolytic cathepsins.[45]
In effect, the key processes believed to be responsible for weakening of the vessel wall, and potential subsequent rupture, include enzymatic degradation of the ECM, progressive loss of SMCs, and also the decreased synthesis of new collagen fibers.[34] The result is a chronic state of remodeling and inflammation that ultimately results in critical weakening of the vessel wall that is progressively less capable of withstanding hemodynamic stress, thus leading to growth and potentially rupture of the aneurysm.[25] Inflammation causing enzymatic remodeling of the vascular ECM is a shared pathological mechanism also seen in several other vascular diseases including atherosclerosis, abdominal aortic aneurysms, and arteritides.[46-48] Atherosclerosis is itself a common feature in saccular intracranial aneurysms and as in abdominal aortic aneurysms, is associated with aneurysmal progression and likelihood of rupture.[25] The contribution of atherosclerosis and patient risk factors, including hypertension and smoking are similar to its contribution to abdominal aortic aneurysms.[46]
Although the acute mechanism of aneurysm rupture has yet to be fully elucidated, there are several key factors that play a role. Clinically, a major predictor of rupture is increased diameter > 7 mm, above which the 5-year risk of rupture increases from 2.6% for 12 mm to 40% for 25 mm.[49] In addition, using patient-specific computation flow dynamics analyses, it has been shown that small size of the impingement region within the aneurysm, unsteady flow dynamics, and concentrated inflow jet are all associated with rupture of the aneurysm.[14,50,51] However, repeated studies have shown that a higher degree of inflammatory infiltration is associated with aneurysm wall degradation and subsequent higher risk of rupture.[13,31,52,53] Owing to the highly effective and quickly-amplified nature of the activated complement cascade, the acute pathogenesis of aneurysm rupture is of particular interest. Understanding the role of complement in this mechanism as well as the chronic processes responsible for aneurysm development is invaluable for future clinical endeavors.
Function of the complement system
The complement system, a network of approximately 30 plasma and membrane-associated proteins, is a major mediator of innate immunity, functioning in cell lysis (e.g. lysis of microbes, virus-infected cells, tumor cells), inflammation, cell signaling, chemotaxis, opsonization, and vascular effects.[54-57] In addition, complement facilitates the adaptive immune response by functioning in antigen presentation, immunologic memory, and costimulation of B-cells via antigen receptors. The presentation of “nonself ” or damaged cells leads to a cascade of events that result in the destruction of the microbes or targeted cells and subsequent inflammation. The cascade is catalyzed by complement components (many of which are proteases) that circulate in inactive forms (zymogens) until they are activated by several mechanisms.[10] Excessive complement activation, however, damages healthy tissue, and is implicated in a variety of central nervous system conditions (SAH, intracerebral hemorrhage, ischemic stroke, ischemia-reperfusion injury, and multiple sclerosis[58-60]) as well as myocardial infarctions and asthma.[61-64] In SAH in particular, complement activation has been associated with poorer functional outcomes and even vasospasm.[60,65-67]
Dysregulation of any of the above processes, deficiencies in the complement proteins, and activation by various molecules can lead to a pathological over-or under-activation of the complement system. These complement disorders [Table 1] include paroxysmal nocturnal hemoglobulinuria (PNH), hereditary angioedema, and atypical hemolytic uremic syndrome.
Complement pathway disorders
| Classic pathway | Membrane attack complex | Alternative pathway | Control proteins | Others |
|---|---|---|---|---|
| C1q deficiency | C5-9 deficiency | Factor B deficiency | Factor I deficiency | Serosal protease deficiency |
| C1r/C1s deficiency | Factor D deficiency | Factor H deficiency | Mannose binding lectin deficiency | |
| C4 deficiency | C4 binding protein deficiency | |||
| C2 deficiency | C1 inhibitor protein deficiency | |||
| C3 deficiency | Complement receptor 1-3 deficiency | |||
| Scleroderma | Paroxysmal noctural hemoglobinuria | |||
| Immunoglobulin A nephropathy | Leukocyte adhesion deficiency syndrome | |||
| Henoch-Schonlein purpura | Hereditary angioedema | |||
| Membranous nephropathy | Age-related macular degeneration | |||
| Systemic lupus erythematosus |
In PNH, for example, decreased expression of the complement regulators CD55 and CD59 allows for complement-mediated lysis of red blood cells in a predominantly intravascular hemolysis.[10,68] Anticomplement therapy already exists to treat many of these conditions that directly result from complement dysregulation. These include a complement component-1 (C1)-inhibitor concentrate (which inactivates C1r and C1s and mannose-binding lectin (MBL)-associated protein 2 (MASP 2) and is approved to treat hereditary angioedema[69,70]), as well as eculizumab (a monoclonal antibody approved for PNH and atypical hemolytic uremic syndrome[71]). Anticomplement therapy may also attenuate the damage from ischemia-reperfusion injury.[72-74]
Classical, lectin, and alternative pathways
There are three recognized pathways of complement system activation: the classical, lectin, and alternative pathways [Figure 1]. The common point of each pathway is the formation of a C3 convertase, which activates C3 by cleaving it into C3b and C3a.[75] C3 activation serves as a nidus for amplification of the complement response. All three pathways eventually form C5 convertases that cleave C5 into C5a and C5b, after which the C5b fragment initiates assembly of C6, C7, C8, and C9 into the membrane attack complex (MAC; also known as the terminal complement cascade, or C5b-9) which lyses the cell by forming a pore in the lipid bilayer.[57]
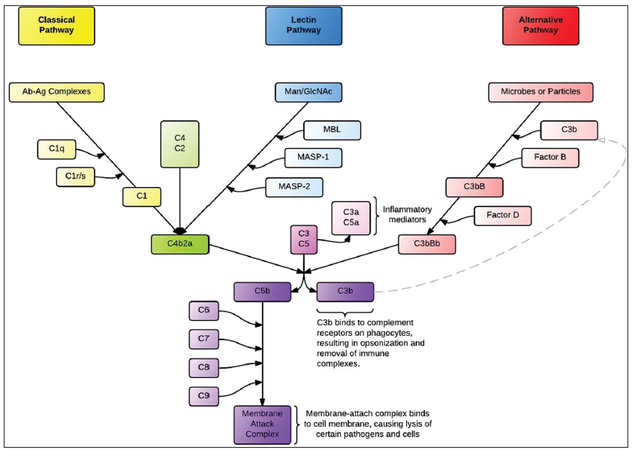
Figure 1. The three pathways of the complement cascade
The classical pathway is primarily activated by antigen-antibody complexes. After binding to an antigen, the Fc region of the antibody (typically IgM or IgG) undergoes a conformational change that allows it to bind to the C1q subunit of C1, a multimer that also contains C1r and C1s subunits. The C1s subunit then cleaves C4 and C2, and then two of the products, C4b and C2a, associate to form the C3-convertase, C4bC2a. C4bC2a also serves as the C3 convertase in the lectin (mannose-binding) pathway, a complement pathway triggered when MBL or Ficolin bind to terminal mannose groups on the surfaces of microbes. This binding allows MBL to associate with circulating MASPs, which then cleave C4 and C2 to form the C4bC2a C3 convertase.[76,77] In both the classical and lectin pathways, C3b may then bind to C4b2a to form the C5 convertase, C4b2a3b. This is different from the C5 convertase in the alternative pathway, where C2 and C4 do not play a major role.
The alternative pathway is triggered by carbohydrates or proteins found on self and nonself surfaces.[10] In the plasma, C3 is spontaneously hydrolyzed (but not cleaved) at a low rate to form iC3, which is amplified in the environment near pathogens due to these carbohydrates or proteins. iC3 binds to Factor B, which Factor D then cleaves into to Bb and Ba, producing the fluid-phase C3 convertase (iC3Bb). The fluid-phase C3 convertase cleaves C3 to C3a and C3b, creating sufficient C3b to bind with Bb and form C3bBb, which, when stabilized by Properdin on a microbial surface, acts as the principal C3 convertase of the alternative complement pathway. The “alternative” C3 convertase C3bBb may then associate with an additional C3b to form the C5 convertase, C3bBbC3b.[75] The C5 convertases from any of the three pathways may then facilitate the assembly of the MAC. In addition, several of the cleavage byproducts, chiefly, C3a and C5a, known as anaphylatoxins, initiate a cascade of signals which induces leukocyte chemotaxis,[57] the effects of which include degranulation and resulting smooth muscle contraction and increased vascular permeability.[55,56] Cleavage products of C3 also cause a parallel pathway of direct cerebral injury via iron toxicity.[78,79]
A link exists between the complement and coagulation cascades, as kallikrein and thrombin may also cleave C3 and C5.[80,81] In addition, C-reactive protein (CRP) may eitheractivatecomplementbyrecruitingC1qtothesurface of damaged cells or regulate the cascade by recruiting complement inhibitors.[82,83] Cholesterol-containing lipids and enzymatically-modified or oxidized LDLs in atherosclerosis can activate complement via C1,[84-87] and these also play a role in the pathogenesis of coronary atherosclerotic plaques.[88]
Modulators of the complement system
The complement system is highly regulated by activators and inhibitors so as to confine the destructive mechanisms of the complement system to nonhost surfaces while protecting the healthy tissue, and alteration of these modulators plays a role in a variety of disease processes.[57] As noted the alternative pathway C3 convertase (C3bBb) is stabilized on the microbial cell surface by Properdin,[89] which is released by a variety of leukocytes,[90-92] some of which, as mentioned, are present in aneurysm walls. In addition to microbial surfaces, Properdin can also bind to apoptotic and necrotic cells and facilitate a complement response.[89] Regulators of complement activation inhibit the complement system at two predominant steps: either at the level of convertases, or in the assembly of the MAC.[57,93] Factor I acts at the convertase level by cleaving C3b into its inactive form, iC3b, which is unable to form an active C3 convertase with Bb. This process achieves specificity to protecting host cells from complement activation because it requires the host membrane-bound cofactors including complement receptor-1 (CR-1) and membrane-cofactor protein (MCP).[57,75,94-96] CR-1 in particular is a potent inhibitor of the classical and alternative pathways, and also plays a role in clearing immune complexes and antigen presentation to B-cells.[10,57] Other inhibitors at the convertase level include decay accelerating factor (DAF), which inhibits assembly of C3 convertases, and Factor H, which serves as a cofactor for factor I and DAF. Inhibitors of assembly of the MAC include CD59, vitronectin, and S-protein.[10,95,97]
Role of complement in aneurysms
There has been a growing interest in the role that complement plays in the pathogenesis of cerebral aneurysms, which has paralleled similar work done with aortic aneurysms.[47,98,99] Much of the esteemed work has been done by the Neurosurgery Research Group at the Helsinki University Central Hospital, Helsinki, Finland, and has taken the research model of previous studies which have examined ruptured versus unruptured aneurysms.[32] The studies have largely been done by standard immunohistological localization of complement in aneurysm tissue samples taken intraoperatively immediately after aneurysm clipping.
The first study to demonstrate complement deposition in aneurysm walls was by Chyatte et al.,[48] who compared 25 aneurysm samples taken during microsurgical repair to 11 control samples from basilar arteries taken at autopsy. Compared to the basilar samples, they found significantly more deposition of immunoreactive C9 and C3c (a breakdown product of C3), and both C3c and C9 were deposited throughout the aneurysm wall, often diffusely. They also found increased presence of immunoglobulins (IgG and IgM) and leukocytes (CD68 macrophages and T-lymphocytes). Due to the deposition of earlier (C3c) as well as terminal complement products (C9), the authors concluded that complement activation, in concert with other inflammatory mediators, played a role in the pathogenesis of aneurysms.
In a 2006 study, Tulamo et al.[100] investigated the role of the MAC in aneurysm rupture by comparing samples from 26 unruptured to 32 ruptured samples. Using a monoclonal mouse antibody to stain the C5b-9 complex (MAC), the authors found that the immunostaining for the MAC was approximately twice as dense in ruptured versus unruptured samples (median: 39% vs. 20%, P < 0.001, respectively). A greater concentration of MAC was also significantly associated with structural pathology of the aneurysmal wall including wall degeneration, de-endothelialization, and degenerative change of the outer wall, as well as infiltrates of CD163+ macrophages and T-lymphocytes. Together, these results suggested a role for activated complement in saccular aneurysm degeneration and rupture.
Using a similar experimental model as their first study, Tulamo et al.[84] then sought to determine, which complement pathway was involved in aneurysm rupture by staining for complement components specific to the alternative and classical pathways, as well as their potential activators. It was found that components of the classical pathway, C1q and C4b/iC4b, as well as the MAC and C3b/iC3b and C3d, were present in significantly greater concentrations and were more widely distributed in ruptured versus unruptured aneurysms, and specifically the staining tended to localize along the ECM in a band-like pattern in the outer aneurysm wall. In a smaller, separate sample of unruptured aneurysms, the authors reported heavy immunostaining for CRP, MAC, oxidized LDL, and IgG, which was increased in concentration from the lumen-to-adventitia direction. Of note, however, although the authors report that tissue form human tonsils was used as a positive control (presumably due to the dense immune elements), they did not have separate, nonaneurysmal tissue that served as a negative control. Tulamo et al.[84] concluded that these findings most likely represented activation of the classical pathway, due to the presence of its potential activators (including IgG, oxidized LDL, and CRP) and C1q deposition, which is specific to the classical pathway. The alternative pathway was less likely to play a role because there was little staining of the specific marker, Properdin. They suggested these immunoglobulins and complement components leak out and accumulated in the aneurysmal wall due to endothelial dysfunction and impaired clearance mechanisms.
A third similar study by Tulamo et al.[101] found that deposition of the MAC was greater in the outer wall than in the lumen of ruptured compared with unruptured aneurysms. This was associated with increased deposition of the complement inhibitors, including the Factor H polymorphic variant Y402H (associated with age-related macular degeneration[102]), C4b binding protein, and protectin (CD59, a MAC inhibitor); however, the outer wall lacked inhibitors, especially protectin. Other inhibitors, such as MCP and DAF, were only sparsely expressed by adventitial mural cells. The authors suggested that the increased MAC activity in the outer wall may be the result of decreased complement inhibitors in that region, and that the outer wall’s decreased ability to inhibit the complement cascade may facilitate eventual rupture. However, judging from the finding of their second study, in which IgG was more densely deposited towards the outer aneurysm wall, it is unclear whether this graded IgG concentration could be the primary factor in the greater MAC activity in the outer wall.
Studies on genetic expression profiles have also identified the role of complement-related genes in aneurysmal tissue. In a small study,[103] which compared aneurysm samples to control superficial temporal artery tissue, there was upregulated expression of three genes for C1q, the deposition of which was found in the aforementioned study by Tulamo et al.,[84] as well as those for complement Factor D, Factor H, Factor B, and C3a. The authors pointed out that these alterations in the expression profile of these genes represent a change in equilibrium of the complement system in the perianeurysmal environment. There has also been an animal study by Aoki et al.,[104] in which the investigators used a DNA microarray to compare intimal and medial gene expression in cerebral aneurysms versus normal cerebral arteries. It was found that in the media there was upregulation of Factor H and C4 expression, although downregulation of C3 and C6. By contrast, C3 and C6 were upregulated in the intima. The authors argue that their differing results from the initial study by Tulamo et al.,[100] may be explained by different regulation of complement mRNA expression between the endothelial cells and the SMCs in cerebral aneurysms.
Although the genetic and histopathological studies mentioned were no doubt pioneering, there are certain points of discussion. Among the studies assessing the role of the complement in aneurysms, the experimental design often differed significantly. In general, the study sizes were small, although this is what would be expected given the limits of intraoperative sample collection. In addition, one study was an animal model.[104] Whereas some of the studies compared aneurysmal tissue to control samples (such as the superficial temporal artery),[48,103,104] the three studies by Tulamo et al.[84,100,101] only compared ruptured to unruptured aneurysms. In effect, although several studies provide compelling evidence that there is more complement deposition and activation in aneurysms than in healthy cerebrovascular tissue, those by Tulamo et al.[84,100,101] generally cannot make this comparison since these studies lacked controls from nonaneurysmal vascular tissue.
In general, the method of specimen collection involved taking a sample of aneurysm tissue, intraoperatively, that was distal to the placement of a surgical clip and then running immunhistological analyses. There is a possibility, therefore, that the pathology of the sample and/or its immunological characteristics may have changed between the time that the sample was collected and analyzed. This time period varied widely among and within studies, including one of the studies in which samples varied from 4 h to 47 days.[101] However, although this is a consideration for judging the quality of the immunologically analyzed specimens, they were generally flash-frozen in liquid nitrogen so as to minimize degradation and preserve their immunohistological staining characteristics. In addition, the aneurysm samples represent patients requiring surgical, but not endovascular intervention, which imparts a selection bias to patients with certain clinical and aneurysmal morphological characteristics. The results of the Tulamo studies from Finland may also have limitations in their external validity because Finland represents a relatively homogenous population, and the prevalence of intracranial aneurysms is twice as high as other comparable countries.[1]
A larger issue with interpreting the results of the analyses, in which authors have attributed the aneurysm pathogenesis to complement activation, are confounding causes of the observed immunohistological properties of aneurysm tissue. In several of the human studies there were statistically significant differences in the proportions of patients with characteristics known to affect aneurysm pathology (including smoking history, hypertension, and family history of aneurysms) as well as differences in the gross aneurysm morphology. Furthermore, it is unclear whether the increased complement activation in ruptured compared with unruptured samples (as was reported in the studies by Tulamo et al.[84,100,101]) contributed to the acute mechanism of rupture, or whether the resulting rupture and SAH hemorrhage occurred for other reasons (i.e. hemodynamic factors, etc.) and then initiated a secondary complement and inflammatory response as a result of the injury. Indeed, as mentioned, there is evidence that the complement system is upregulated in patients with SAH.[60,65,66] In response to this concern, Tulamo et al.[84,100,101] have argued that the increased density of macrophage infiltration in ruptured versus unruptured aneurysms argues for a more chronic inflammatory process, as dense accumulation of macrophages typically occurs over days to weeks following an acute injury. In addition, they point out that although less concentrated, complement deposits were found in unruptured aneurysms. Their argument is also supported by the numerous animal and human studies that have shown inflammatory infiltrate in aneurysms that have not ruptured.[22,32,36,37] Tulamo et al.[84,100,101] suggest that the hemorrhagic insult and physical factors may have contributed to complement activation but that more studies will be needed.
Conclusion
There is mounting evidence that the complement cascade plays a role in the chronic as well as acute inflammation that has been associated with aneurysm formation, growth, and rupture. In effect, the physicochemical stress that occurs in intracranial arteries may lead to exposure and deposition of complement-activating factors in the aneurysm wall, which later predispose it to rupture. Specifically, assembly of MACs in the outer aneurysmal wall may be involved in the acute mechanism of rupture. Future, multicenter studies will need to compare a large number of aneurysmal samples to appropriate controls so that the aneurysm pathophysiology may be better understood. Furthermore, the complement cascade may provide a target for future therapies in the treatment of aneurysms and SAH.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Vlak MH, Algra A, Brandenburg R, Rinkel GJ. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: a systematic review and meta-analysis. Lancet Neurol 2011;10:626-36.
2. Rinkel GJ, Algra A. Long-term outcomes of patients with aneurysmal subarachnoid haemorrhage. Lancet Neurol 2011;10:349-56.
3. Brown RD Jr, Broderick JP. Unruptured intracranial aneurysms: epidemiology, natural history, management options, and familial screening. Lancet Neurol 2014;13:393-404.
6. Nieuwkamp DJ, Setz LE, Algra A, Linn FH, de Rooij NK, Rinkel GJ. Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta-analysis. Lancet Neurol 2009;8:635-42.
7. Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 2010;41:e519-36.
8. Krings T, Mandell DM, Kiehl TR, Geibprasert S, Tymianski M, Alvarez H, Terbrugge KG, Hans FJ. Intracranial aneurysms: from vessel wall pathology to therapeutic approach. Nat Rev Neurol 2011;7:547-59.
9. Holling M, Jeibmann A, Gerss J, Fischer BR, Wassmann H, Paulus W, Hasselblatt M, Albert FK. Prognostic value of histopathological findings in aneurysmal subarachnoid hemorrhage. J Neurosurg 2009;110:487-91.
10. Ruigrok YM, Rinkel GJ. From GWAS to the clinic: risk factors for intracranial aneurysms. Genome Med 2010;2:61.
12. Chalouhi N, Ali MS, Starke RM, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Koch WJ, Dumont AS. Cigarette smoke and inflammation: role in cerebral aneurysm formation and rupture. Mediators Inflamm 2012;2012:271582.
13. Sforza DM, Putman CM, Cebral JR. Hemodynamics of cerebral aneurysms. Annu Rev Fluid Mech 2009;41:91-107.
14. Frösen J, Tulamo R, Paetau A, Laaksamo E, Korja M, Laakso A, Niemelä M, Hernesniemi J. Saccular intracranial aneurysm: pathology and mechanisms. Acta Neuropathol 2012;123:773-86.
15. Juvela S, Hillbom M, Numminen H, Koskinen P. Cigarette smoking and alcohol consumption as risk factors for aneurysmal subarachnoid hemorrhage. Stroke 1993;24:639-46.
16. Vlak MH, Rinkel GJ, Greebe P, Greving JP, Algra A. Lifetime risks for aneurysmal subarachnoid haemorrhage: multivariable risk stratification. J Neurol Neurosurg Psychiatry 2013;84:619-23.
17. Feigin VL, Rinkel GJ, Lawes CM, Algra A, Bennett DA, van Gijn J, Anderson CS. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005;36:2773-80.
18. Broderick JP, Brown RD Jr, Sauerbeck L, Hornung R, Huston J 3rd, Woo D, Anderson C, Rouleau G, Kleindorfer D, Flaherty ML, Meissner I, Foroud T, Moomaw EC, Connolly ES; FIA Study Investigators. Greater rupture risk for familial as compared to sporadic unruptured intracranial aneurysms. Stroke 2009;40:1952-7.
19. Bonneville F, Sourour N, Biondi A. Intracranial aneurysms: an overview. Neuroimaging Clin N Am 2006;16:371-82, vii.
20. Turjman AS, Turjman F, Edelman ER. Role of fluid dynamics and inflammation in intracranial aneurysm formation. Circulation 2014;129:373-82.
21. Jamous MA, Nagahiro S, Kitazato KT, Tamura T, Aziz HA, Shono M, Satoh K. Endothelial injury and inflammatory response induced by hemodynamic changes preceding intracranial aneurysm formation: experimental study in rats. J Neurosurg 2007;107:405-11.
22. Meng H, Wang Z, Hoi Y, Gao L, Metaxa E, Swartz DD, Kolega J. Complex hemodynamics at the apex of an arterial bifurcation induces vascular remodeling resembling cerebral aneurysm initiation. Stroke 2007;38:1924-31.
23. Sho E, Sho M, Singh TM, Nanjo H, Komatsu M, Xu C, Masuda H, Zarins CK. Arterial enlargement in response to high flow requires early expression of matrix metalloproteinases to degrade extracellular matrix. Exp Mol Pathol 2002;73:142-53.
24. Greenhill NS, Stehbens WE. Scanning electron microscopic investigation of the afferent arteries of experimental femoral arteriovenous fistulae in rabbits. Pathology 1987;19:22-7.
25. Chalouhi N, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Koch WJ, Dumont AS. Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab 2012;32:1659-76.
26. Killer-Oberpfalzer M, Aichholzer M, Weis S, Richling B, Jones R, Virmani R, Cruise GM. Histological analysis of clipped human intracranial aneurysms and parent arteries with short-term follow-up. Cardiovasc Pathol 2012;21:299-306.
27. Aoki T, Nishimura M, Matsuoka T, Yamamoto K, Furuyashiki T, Kataoka H, Kitaoka S, Ishibashi R, Ishibazawa A, Miyamoto S, Morishita R, Ando J, Hashimoto N, Nozaki K, Narumiya S. PGE(2) -EP(2) signalling in endothelium is activated by haemodynamic stress and induces cerebral aneurysm through an amplifying loop via NF-kB. Br J Pharmacol 2011;163:1237-49.
28. Meng H, Tutino VM, Xiang J, Siddiqui A. High WSS or low WSS? Complex interactions of hemodynamics with intracranial aneurysm initiation, growth, and rupture: toward a unifying hypothesis. AJNR Am J Neuroradiol 2014;35:1254-62.
29. Laaksamo E, Tulamo R, Liiman A, Baumann M, Friedlander RM, Hernesniemi J, Kangasniemi M, Niemelä M, Laakso A, Frösen J. Oxidative stress is associated with cell death, wall degradation, and increased risk of rupture of the intracranial aneurysm wall. Neurosurgery 2013;72:109-17.
30. Kosierkiewicz TA, Factor SM, Dickson DW. Immunocytochemical studies of atherosclerotic lesions of cerebral berry aneurysms. J Neuropathol Exp Neurol 1994;53:399-406.
31. Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, Jääskeläinen J. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke 2004;35:2287-93.
32. Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke 1999;30:1396-401.
34. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Morishita R, Hashimoto N. Reduced collagen biosynthesis is the hallmark of cerebral aneurysm: contribution of interleukin-1beta and nuclear factor-kappaB. Arterioscler Thromb Vasc Biol 2009;29:1080-6.
35. Ali MS, Starke RM, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Owens GK, Koch WJ, Greig NH, Dumont AS. TNF-α induces phenotypic modulation in cerebral vascular smooth muscle cells: implications for cerebral aneurysm pathology. J Cereb Blood Flow Metab 2013;33:1564-73.
36. Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke 2007;38:162-9.
37. Aoki T, Kataoka H, Shimamura M, Nakagami H, Wakayama K, Moriwaki T, Ishibashi R, Nozaki K, Morishita R, Hashimoto N. NF-kappaB is a key mediator of cerebral aneurysm formation. Circulation 2007;116:2830-40.
38. Ishibashi R, Aoki T, Nishimura M, Hashimoto N, Miyamoto S. Contribution of mast cells to cerebral aneurysm formation. Curr Neurovasc Res 2010;7:113-24.
39. Pera J, Korostynski M, Krzyszkowski T, Czopek J, Slowik A, Dziedzic T, Piechota M, Stachura K, Moskala M, Przewlocki R, Szczudlik A. Gene expression profiles in human ruptured and unruptured intracranial aneurysms: what is the role of inflammation? Stroke 2010;41:224-31.
40. Kadir vel R, Ding YH, Dai D, Zakaria H, Rober tson AM, Danielson MA, Lewis DA, Cloft HJ, Kallmes DF. The influence of hemodynamic forces on biomarkers in the walls of elastase-induced aneurysms in rabbits. Neuroradiology 2007;49:1041-53.
41. Metz RP, Patterson JL, Wilson E. Vascular smooth muscle cells: isolation, culture, and characterization. Methods Mol Biol 2012;843:169-76.
42. Aoki T, Kataoka H, Moriwaki T, Nozaki K, Hashimoto N. Role of TIMP-1 and TIMP-2 in the progression of cerebral aneurysms. Stroke 2007;38:2337-45.
43. Aoki T, Fukuda M, Nishimura M, Nozaki K, Narumiya S. Critical role of TNF-alpha-TNFR1 signaling in intracranial aneurysm formation. Acta Neuropathol Commun 2014;2:34.
44. Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta 2010;1803:55-71.
45. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Cathepsin B, K, and S are expressed in cerebral aneurysms and promote the progression of cerebral aneurysms. Stroke 2008;39:2603-10.
46. Nordon IM, Hinchliffe RJ, Holt PJ, Loftus IM, Thompson MM. Review of current theories for abdominal aortic aneurysm pathogenesis. Vascular 2009;17:253-63.
47. Capella JF, Paik DC, Yin NX, Gervasoni JE, Tilson MD. Complement activation and subclassification of tissue immunoglobulin G in the abdominal aortic aneurysm. J Surg Res 1996;65:31-3.
48. Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery 1999;45:1137-46.
50. Cebral JR, Castro MA, Burgess JE, Pergolizzi RS, Sheridan MJ, Putman CM. Characterization of cerebral aneurysms for assessing risk of rupture by using patient-specific computational hemodynamics models. AJNR Am J Neuroradiol 2005;26:2550-9.
51. Cebral JR, Mut F, Weir J, Putman C. Quantitative characterization of the hemodynamic environment in ruptured and unruptured brain aneurysms. AJNR Am J Neuroradiol 2011;32:145-51.
52. Rinkel GJ, Djibuti M, Algra A, van Gijn J. Prevalence and risk of rupture of intracranial aneurysms: a systematic review. Stroke 1998;29:251-6.
53. Starke RM, Chalouhi N, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Wada K, Shimada K, Hasan DM, Greig NH, Owens GK, Dumont AS. Critical role of TNF-α in cerebral aneurysm formation and progression to rupture. J Neuroinflammation 2014;11:77.
54. Bacle F, Haeffner-Cavaillon N, L aude M, Couturier C, Kazatchkine MD. Induction of IL-1 release through stimulation of the C3b/C4b complement receptor type one (CR1, CD35) on human monocytes. J Immunol 1990;144:147-52.
55. Haas PJ, van Strijp J. Anaphylatoxins: their role in bacterial infection and inflammation. Immunol Res 2007;37:161-75.
56. Wetsel RA. Structure, function and cellular expression of complement anaphylatoxin receptors. Curr Opin Immunol 1995;7:48-53.
57. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res 2010;20:34-50.
58. Ducruet AF, Zacharia BE, Hickman ZL, Grobelny BT, Yeh ML, Sosunov SA, Connolly ES, Jr. The complement cascade as a therapeutic target in intracerebral hemorrhage. Exp Neurol 2009;219:398-403.
59. Gasque P, Singhrao SK, Neal JW, Wang P, Sayah S, Fontaine M, Morgan BP. The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and nonmyeloid cells in inflamed human central nervous system: analysis in multiple sclerosis and bacterial meningitis. J Immunol 1998;160:3543-54.
60. Mack WJ, Ducruet AF, Hickman ZL, Garrett MC, Albert EJ, Kellner CP, Mocco J, Connolly ES, Jr. Early plasma complement C3a levels correlate with functional outcome after aneurysmal subarachnoid hemorrhage. Neurosurgery 2007;61:255-60.
61. Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O'Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009;119:480-6.
64. Qu H, Ricklin D, Lambris JD. Recent developments in low molecular weight complement inhibitors. Mol Immunol 2009;47:185-95.
65. Zanier ER, Zangari R, Munthe-Fog L, Hein E, Zoerle T, Conte V, Orsini F, Tettamanti M, Stocchetti N, Garred P, De Simoni MG. Ficolin-3-mediated lectin complement pathway activation in patients with subarachnoid hemorrhage. Neurology 2014;82:126-34.
66. Ostergaard JR, Kristensen BO, Svehag SE, Teisner B, Miletic T. Immune complexes and complement activation following rupture of intracranial saccular aneurysms. J Neurosurg 1987;66:891-7.
67. Lindsberg PJ, Ohman J, Lehto T, Karjalainen-Lindsberg ML, Paetau A, Wuorimaa T, Carpén O, Kaste M, Meri S. Complement activation in the central nervous system following blood-brain barrier damage in man. Ann Neurol 1996;40:587-96.
68. Rosti V. The molecular basis of paroxysmal nocturnal hemoglobinuria. Haematologica 2000;85:82-7.
69. Davis AE 3rd, Mejia P, Lu F. Biological activities of C1 inhibitor. Mol Immunol 2008;45:4057-63.
70. Bowen T, Cicardi M, Farkas H, Bork K, Longhurst HJ, Zuraw B, Aygoeren-Pürsün E, Craig T, Binkley K, Hebert J, Ritchie B, Bouillet L, Betschel S, Cogar D, Dean J, Devaraj R, Hamed A, Kamra P, Keith PK, Lacuesta G, Leith E, Lyons H, Mace S, Mako B, Neurath D, Poon MC, Rivard GE, Schellenberg R, Rowan D, Rowe A, Stark D, Sur S, Tsai E, Warrington R, Waserman S, Ameratunga R, Bernstein J, Björkander J, Brosz K, Brosz J, Bygum A, Caballero T, Frank M, Fust G, Harmat G, Kanani A, Kreuz W, Levi M, Li H, Martinez-Saguer I, Moldovan D, Nagy I, Nielsen EW, Nordenfelt P, Reshef A, Rusicke E, Smith-Foltz S, Späth P, Varga L, Xiang ZY. 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol 2010;6:24.
71. Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, Röth A, Szer J, Elebute MO, Nakamura R, Browne P, Risitano AM, Hill A, Schrezenmeier H, Fu CL, Maciejewski J, Rollins SA, Mojcik CF, Rother RP, Luzzatto L. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med 2006;355:1233-43.
72. Yang J, Ahn HN, Chang M, Narasimhan P, Chan PH, Song YS. Complement component 3 inhibition by an antioxidant is neuroprotective after cerebral ischemia and reperfusion in mice. J Neurochem 2013;124:523-35.
73. Heydenreich N, Nolte MW, Göb E, Langhauser F, Hofmeister M, Kraft P, Albert-Weissenberger C, Brede M, Varallyay C, Göbel K, Meuth SG, Nieswandt B, Dickneite G, Stoll G, Kleinschnitz C. C1-inhibitor protects from brain ischemia-reperfusion injury by combined antiinflammatory and antithrombotic mechanisms. Stroke 2012;43:2457-67.
74. Kim GH, Mocco J, Hahn DK, Kellner CP, Komotar RJ, Ducruet AF, Mack WJ, Connolly ES, Jr. Protective effect of C5a receptor inhibition after murine reperfused stroke. Neurosurgery 2008;63:122-5.
75. Parham P. Elements of the immune system and their roles in defens. In: The Immune System. 3rd ed. New York: Garland Science; 2009. pp. 2-27.
76. Sørensen R, Thiel S, Jensenius JC. Mannan-binding-lectin-associated serine proteases, characteristics and disease associations. Springer Semin Immunopathol 2005;27:299-319.
77. Wallis R. Interactions between mannose-binding lectin and MASPs during complement activation by the lectin pathway. Immunobiology 2007;212:289-99.
78. Wang X, Mori T, Sumii T, Lo EH. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke 2002;33:1882-8.
79. Katsu M, Niizuma K, Yoshioka H, Okami N, Sakata H, Chan PH. Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. J Cereb Blood Flow Metab 2010;30:1939-50.
80. Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol 2007;28:184-92.
81. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 2006;12:682-7.
82. Mihlan M, Blom AM, Kupreishvili K, Lauer N, Stelzner K, Bergström F, Niessen HW, Zipfel PF. Monomeric C-reactive protein modulates classic complement activation on necrotic cells. FASEB J 2011;25:4198-210.
83. Ji SR, Wu Y, Potempa LA, Liang YH, Zhao J. Effect of modified C-reactive protein on complement activation: a possible complement regulatory role of modified or monomeric C-reactive protein in atherosclerotic lesions. Arterioscler Thromb Vasc Biol 2006;26:935-41.
84. Tulamo R, Frösen J, Junnikkala S, Paetau A, Kangasniemi M, Peláez J, Hernesniemi J, Niemelä M, Meri S. Complement system becomes activated by the classical pathway in intracranial aneurysm walls. Lab Invest 2010;90:168-79.
86. Sjöberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol 2009;30:83-90.
87. Biró A, Thielens NM, Cervenák L, Prohászka Z, Füst G, Arlaud GJ. Modified low density lipoproteins differentially bind and activate the C1 complex of complement. Mol Immunol 2007;44:1169-77.
88. Meuwissen M, van der Wal AC, Niessen HW, Koch KT, de Winter RJ, van der Loos CM, Rittersma SZ, Chamuleau SA, Tijssen JG, Becker AE, Piek JJ. Colocalisation of intraplaque C reactive protein, complement, oxidised low density lipoprotein, and macrophages in stable and unstable angina and acute myocardial infarction. J Clin Pathol 2006;59:196-201.
89. Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Annu Rev Immunol 2010;28:131-55.
90. Wirthmueller U, Dewald B, Thelen M, Schäfer MK, Stover C, Whaley K, North J, Eggleton P, Reid KB, Schwaeble WJ. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J Immunol 1997;158:4444-51.
91. Schwaeble W, Huemer HP, Möst J, Dierich MP, Ströbel M, Claus C, Reid KB, Ziegler-Heitbrock HW. Expression of properdin in human monocytes. Eur J Biochem 1994;219:759-64.
92. Schwaeble W, Dippold WG, Schäfer MK, Pohla H, Jonas D, Luttig B, Weihe E, Huemer HP, Dierich MP, Reid KB. Properdin, a positive regulator of complement activation, is expressed in human T cell lines and peripheral blood T cells. J Immunol 1993;151:2521-8.
93. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol 2009;9:729-40.
94. Blackmore TK, Hellwage J, Sadlon TA, Higgs N, Zipfel PF, Ward HM, Gordon DL. Identification of the second heparin-binding domain in human complement factor H. J Immunol 1998;160:3342-8.
95. Turnberg D, Botto M. The regulation of the complement system: insights from genetically-engineered mice. Mol Immunol 2003;40:145-53.
96. Krych-Goldberg M, Atkinson JP. Structure-function relationships of complement receptor type 1. Immunol Rev 2001;180:112-22.
97. Huang Y, Qiao F, Abagyan R, Hazard S, Tomlinson S. Defining the CD59-C9 binding interaction. J Biol Chem 2006;281:27398-404.
98. Pagano MB, Zhou HF, Ennis TL, Wu X, Lambris JD, Atkinson JP, Thompson RW, Hourcade DE, Pham CT. Complement-dependent neutrophil recruitment is critical for the development of elastase-induced abdominal aortic aneurysm. Circulation 2009;119:1805-13.
99. Bradley DT, Badger SA, Bown MJ, Sayers RD, Hughes AE. Coding polymorphisms in the genes of the alternative complement pathway and abdominal aortic aneurysm. Int J Immunogenet 2011;38:243-8.
100. Tulamo R, Frösen J, Junnikkala S, Paetau A, Pitkäniemi J, Kangasniemi M, Niemelä M, Jääskeläinen J, Jokitalo E, Karatas A, Hernesniemi J, Meri S. Complement activation associates with saccular cerebral artery aneurysm wall degeneration and rupture. Neurosurgery 2006;59:1069-76.
101. Tulamo R, Frösen J, Paetau A, Seitsonen S, Hernesniemi J, Niemelä M, Järvelä I, Meri S. Lack of complement inhibitors in the outer intracranial artery aneurysm wall associates with complement terminal pathway activation. Am J Pathol 2010;177:3224-32.
102. Thakkinstian A, Han P, McEvoy M, Smith W, Hoh J, Magnusson K, Zhang K, Attia J. Systematic review and meta-analysis of the association between complement factor H Y402H polymorphisms and age-related macular degeneration. Hum Mol Genet 2006;15:2784-90.
103. Shi C, Awad IA, Jafari N, Lin S, Du P, Hage ZA, Shenkar R, Getch CC, Bredel M, Batjer HH, Bendok BR. Genomics of human intracranial aneurysm wall. Stroke 2009;40:1252-61.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Taylor BES, Appelboom G, Zilinyi R, Goodman A, Chapel D, LoPresti M, Jr. ESC. Role of the complement cascade in cerebral aneurysm formation, growth, and rupture. Neurosciences 2015;2:93-101. http://dx.doi.org/10.4103/2347-8659.154888
AMA Style
Taylor BES, Appelboom G, Zilinyi R, Goodman A, Chapel D, LoPresti M, Jr. ESC. Role of the complement cascade in cerebral aneurysm formation, growth, and rupture. Neuroimmunology and Neuroinflammation. 2015; 2: 93-101. http://dx.doi.org/10.4103/2347-8659.154888
Chicago/Turabian Style
Taylor, Blake E. S., Geoff Appelboom, Robert Zilinyi, Ariana Goodman, David Chapel, Melissa LoPresti, Edward Sander Connolly Jr.. 2015. "Role of the complement cascade in cerebral aneurysm formation, growth, and rupture" Neuroimmunology and Neuroinflammation. 2: 93-101. http://dx.doi.org/10.4103/2347-8659.154888
ACS Style
Taylor, BES.; Appelboom G.; Zilinyi R.; Goodman A.; Chapel D.; LoPresti M.; Jr. ESC. Role of the complement cascade in cerebral aneurysm formation, growth, and rupture. Neurosciences. 2015, 2, 93-101. http://dx.doi.org/10.4103/2347-8659.154888
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 23
likes
Like This Article 23
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.