
The role of inflammation in cerebral aneurysms

Abstract
The natural history of unruptured intracranial aneurysms (IAs) is poorly understood. At present, risk factors for aneurysm rupture are limited to demographics and rudimentary anatomic features of the aneurysm. The first sign of aneurysm destabilization and rupture may be subarachnoid hemorrhage, a potentially devastating brain injury with high morbidity and mortality. An emerging body of literature suggests a complex inflammatory cascade likely promotes aneurysm wall remodeling and progressive ballooning of the arterial wall, ultimately terminating in aneurysm rupture. These events likely begin with hemodynamic, flow-related endothelial injury; the injured endothelium stimulates inflammation, including the recruitment and transmigration of inflammatory cells, particularly macrophages. Various proteases are secreted by the inflammatory infiltrate, resulting in degradation of the extracellular matrix and the structural changes unique to IAs. Detailed understanding of these inflammatory processes may result in (1) early identification of patients at high risk for aneurysm rupture, perhaps via arterial wall imaging, and (2) targeted, noninvasive therapies to treat or even prevent cerebral aneurysms.
Keywords
Introduction
Subarachnoid hemorrhage due to intracranial aneurysm (IA) rupture is a devastating disease. Initial mortality may be as high as 40-50%, and of those who survive, one-third to one-half are left with permanent neurologic deficits.[1] When an unruptured aneurysm is discovered in a patient, current therapeutic options to prevent aneurysm rupture include invasive endovascular occlusion versus surgical therapy, or close radiologic follow-up with intervention when the risk of rupture is deemed high enough. That being said, risk stratification of patients with an unruptured IA is based on a limited understanding of natural history, size appears to contribute to aneurysm destabilization,[2] but there are likely other, poorly understood factors at play. And as demonstrated in the International Study of Unruptured Intracranial Aneurysms (ISUIA), invasive endovascular or surgical treatments are associated with an overall 1-year morbidity/mortality of 10%.[3]
In order to appropriately tailor treatment decisions, further understanding of the pathophysiology behind aneurysm growth and rupture is needed. In recent years, a growing body of literature has identified inflammation as a key player in the pathogenesis of intracranial aneurysms (IAs), from aneurysmogenesis and vascular remodeling to aneurysm destabilization and rupture. Here, we will review the pathology of IAs along with the literature supporting a role for inflammation in this pathology; we will also examine potential inflammatory targets for noninvasive treatment of IAs.
Structural characteristics of intracranial aneurysms
Intracranial aneurysms are believed to be acquired vascular lesions; they are exceedingly rare in children and their incidence increases with age.[4,5] As IAs are preferentially located at bifurcations and sharp curves, hemodynamics (e.g. various shear stressors) are believed to trigger aneurysmogenesis. From a structural perspective, compared to extracranial vessels, intracranial vessels have less elastic fiber in the tunica media and adventitia, less smooth muscle in the media, and a thinner adventitia.[6] At vessel bifurcations, the apical portion of the intracranial vessel lacks smooth muscle cells (SMCs), a gap referred to as the “medial raphe”.[7] During the initiation of aneurysms, the luminal surface of the vessel becomes irregular and often damaged, even denuded, a probable consequence of disturbed hemodynamic stress.[8]
Although shear stressors likely trigger the initial injury, further degradation and disorganization of the vascular wall leading to the aneurysmal growth is likely the result of an inflammatory cascade.[9-11] In general, the vessel wall is transformed into a disorganized array, with fragmentation/loss of the internal elastic lamina, myointimal hyperplasia, and disorganization of muscle fiber structure.[12-14] SMCs transition from a contractile phenotype to a pro-remodeling, pro-inflammatory synthetic phenotype, and finally to a dedifferentiated phenotype prior to aneurysm rupture.[15] Though the initial vascular injury was from high shear stress, the cavity of the aneurysm is subjected to low, atheroprone-like shear stress, the type conducive to inflammatory cell adhesion and infiltration.[16] In large aneurysms (e.g. those prone to rupture), there are often advanced atherosclerotic changes, phenotypically modified SMCs, lipid-laden macrophages, and lymphocytes.[17]
Inflammatory mediators of aneurysm wall remodeling
The histological findings in the walls of IAs, those of degeneration and pathologic vascular remodeling, are similar to the findings evident in inflammatory atherosclerotic lesions. Summarized here and depicted in Figure 1 are the mediators of inflammation likely to play a role in IA pathogenesis.
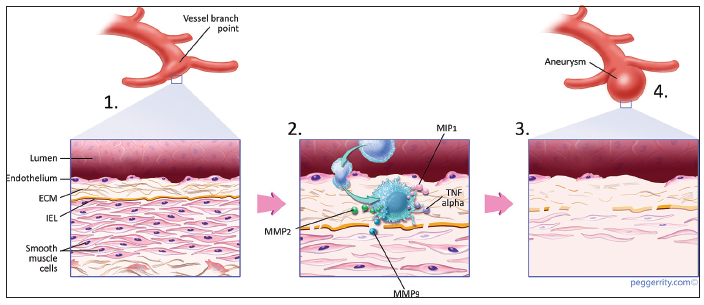
Figure 1. (1) Flow-related endothelial injury; (2) triggers an inflammatory response whereby cells (macrophages) infiltrate the arterial wall and secrete pro-inflammatory cytokines and metalloproteinases; (3) the mounting inflammatory response results in proteolytic destruction of the extracellular matrix and smooth muscle cell phenotypic modulation; (4) macroscopically, the arterial wall is remodeled into an aneurysm wall with progressive aneurysmal ballooning
Endothelial dysfunction
Flow-mediated endothelial dysfunction is likely pivotal in aneurysm formation.[18] Several mechanosensors, such as ion channels, integrins, cell adhesion molecules, G-protein-coupled receptors, have been identified at the apical and basal surfaces of the endothelium;[14] these sensors can identify variations in wall shear stress and adapt lumen diameter accordingly. High shear stress can result in activation of inflammatory mediators, such as the master regulator of inflammation, nuclear factor-kappaB (NF-κB).[19,20] Mechanical stressors can denude the endothelium, triggering the expression of chemoattractants, pro-inflammatory cytokines, and cell adhesion molecules at the surface of endothelial cells.[21] Absent from normal control arteries, monocyte chemoattractant protein-1 (MCP-1) and interleukin-8 (IL-8) are expressed in human and experimental IAs[22] and vascular cell adhesion molecule-1 (VCAM-1) is expressed in the walls of human and rat model IAs.[23]
Macrophages and other inflammatory infiltrates
Numerous studies have demonstrated the presence of inflammatory cell infiltrates, particularly macrophages, in IAs.[24] In one study, inflammatory infiltrates were present in half of all unruptured aneurysms (10/20) versus 100% of all ruptured aneurysms (40/40).[25] And in a study by Frösen et al.[26] whereby 42 ruptured IAs were histologically compared with 24 unruptured IAs, infiltration of the aneurysm wall by macrophages correlated strongly with aneurysm rupture. Macrophages are thought to be a key mediator of IA vascular remodeling as they release matrix metalloproteinases (MMP) such as MMP-9 and MMP-2.[27,28] In one study by Kanematsu et al.,[29] macrophage-depleted mice had a substantially lower risk of IA development compared with control mice (10% vs. 60%).
Extracellular matrix remodeling
An essential feature of IAs is fragmentation of the internal elastic lamina (IEL) and thinning of the arterial media. These changes alter the mechanical properties of the aneurysm wall; in response to further shear stress, the destabilized arterial wall may progressively balloon. MMPs are proteolytic enzymes secreted by activated macrophages and by phenotypically modified SMCs. MMPs are capable of degrading the principal structural components in the artery wall, collagen, and elastin, and are, therefore, likely responsible for the structural changes in the internal elastic lamina and media in IAs.[30,31]
Thrombus formation
Normally, an intact endothelial wall protects the luminal surface from thrombosis and platelet aggregation, in part via the expression of CD39, nitric oxide, and prostacyclin.[32] In contrast, damaged or denuded endothelial cells may instead activate thrombosis and platelet aggregation pathways. Altered hemodynamic flow within the IA cavity may also promote thrombus formation.[33] Neutrophils and macrophages are recruited to the site of endothelial injury and thrombosis. These cells often release proteolytic enzymes, MMPs, cathepsin G, and elastase, to try and promote fibrinolysis and thrombus degradation. Instead, these proteolytic enzymes may further degrade the IA wall.[34] SMCs and myofibroblasts may invade the thrombus, incorporating the thrombus into the IA wall itself.[35]
Complement cascade
The complement cascade has also been studied as a contributor to the pathogenesis of IAs. Activation of complement leads to robust and efficient proteolytic cascades, typically terminating in opsonization and lysis of pathogens as well as in the generation of the classic inflammatory response through the production of potent pro-inflammatory molecules. Immunostaining of IA walls both in humans and animal models have identified complement components, particularly C3 and C9.[36] Two studies using microarray analysis have demonstrated variable expression of complement-related genes in IAs as compared with control arterial tissue.[37,38] And in another study comparing ruptured with unruptured aneurysms, the expression of the complement cascade end product (the membrane attack complex) was greater in ruptured samples and correlated significantly with aneurysm wall degeneration and inflammatory cell infiltration.[39] It continues to be unclear, though, how complement activation results in IA rupture, further studies are needed to define the exact pathways linking the two.
Imaging of arterial wall inflammation
Noninvasive imaging of vascular inflammation within the aneurysm wall may in the future help differentiate stable IAs from destabilized IAs at greater risk for rupture. For instance, protocols have been developed to visualize arterial wall inflammation in patients with intracranial atherosclerosis. Preliminary studies of atherosclerotic plaques suggest vulnerable plaques prone to rupture have arterial wall imaging profiles separate from stable, asymptomatic plaques.[40-42] Regarding IAs, DeLeo et al.[43] published a pilot study whereby active inflammation was imaged in vivo in a rabbit model of common carotid artery aneurysms. This group utilized a myeloperoxidase-specific paramagnetic contrast agent in conjunction with magnetic resonance imaging (MRI). Several years ago, Hasan et al.[44] reported on the use of ferumoxytol-enhanced MRI to image macrophages within aneurysm walls in 11 patients with unruptured IAs. Ferumoxytol is an iron oxide nanoparticle theoretically macrophage-selective as it is cleared by reticuloendothelial system macrophages. Interestingly, early ferumoxytol-associated imaging changes (24 h postinfusion) were identified in five patients, and several of these patients had “symptomatic” IAs (progressive headache; rapid aneurysmal enlargement; aneurysm rupture). Further studies with larger sample sizes are needed to confirm whether ferumoxytol-associated imaging changes correlate with greater IA rupture risk.
Potential anti-inflammatory pharmacologic targets
Advances in our understanding of the inflammatory cascade leading to aneurysm destabilization and rupture may result in the designing of novel therapies individualized to specific patients. Preliminary data in animal models of IA suggest therapies targeting the inflammatory response may have efficacy in the future treatment of IA. For instance, in their rat model of IA, Aoki et al.[45,46] demonstrated reduction of IA wall inflammation and cessation of aneurysm progression via various statin agents. The expression of MCP-1, VCAM-1, IL-1β, inducible nitric oxide synthase, and MMP-9 were all reduced in statin-treated rats, likely via inhibition of NF-κB. However, other studies have demonstrated dose-dependent effects of statins on IAs, including aneurysm growth and/or rupture with high doses of statins.[47] A case-control study by Marbacher et al.[48] did not find a reduction in the incidence of IAs in patients with a history of statin use. Additional prospective studies are needed to clarify the role statins may play in patients with IAs. Other promising therapeutics include edavarone, a synthetic free radical scavenger, and nifedipine, a calcium channel antagonist.[45,49] In an experimental model of IA, nifedipine inhibited DNA binding of NF-κB, preventing progression of IA wall degeneration and limiting IA size.
Recently, aspirin has emerged as a candidate for noninvasive pharmacotherapy in patients with unruptured IAs. Depending on the dose, aspirin can inhibit several inflammatory mediators via its irreversible inhibition of cyclooxygenase-2. Among patients enrolled in ISUIA, those with a history of aspirin use 3 times weekly, or greater had a lower risk of cerebral aneurysm rupture and subarachnoid hemorrhage compared with those who never used aspirin.[50] These findings were reproduced in a retrospective study of 747 patients with IAs by Gross et al.[51] subarachnoid hemorrhage occurred in 28% of patients with a history of aspirin use versus 40% of patients without a history of aspirin use. In the previous section, we described an imaging protocol (ferumoxytol-enhanced MRI) whereby macrophages in IA walls can be visualized. In one ferumoxytol-enhanced MRI study, patients were imaged pre- and post-daily aspirin therapy (for several months). Ferumoxytol signal within the IA walls was decreased in these patients postaspirin therapy, suggesting IA inflammation may have decreased.[52]
Conclusion
Inflammation has emerged as a probable key mediator of both aneurysmogenesis and aneurysmal destabilization and rupture. The inflammatory cascade is likely interrelated with mechanical flow-induced vascular dysfunction. Further studies will, hopefully, further define these pathways, aid in our prediction of the natural history of an IA in a patient-specific manner, and identify novel pharmacologic targets to prevent aneurysm growth and rupture.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Bederson JB, Awad IA, Wiebers DO, Piepgras D, Haley EC Jr, Brott T, Hademenos G, Chyatte D, Rosenwasser R, Caroselli C. Recommendations for the management of patients with unruptured intracranial aneurysms: a Statement for healthcare professionals from the Stroke Council of the American Heart Association. Stroke 2000;31:2742-50.
2. Rinkel GJ, Djibuti M, Algra A, van Gijn J. Prevalence and risk of rupture of intracranial aneurysms: a systematic review. Stroke 1998;29:251-6.
3. Wiebers DO, Whisnant JP, Huston J 3rd, Meissner I, Brown RD Jr, Piepgras DG, Forbes GS, Thielen K, Nichols D, O'Fallon WM, Peacock J, Jaeger L, Kassell NF, Kongable-Beckman GL, Torner JC; International Study of Unruptured Intracranial Aneurysms Investigators. Unruptured intracranial aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet 2003;362:103-10.
4. Heiskanen O, Vilkki J. Intracranial arterial aneurysms in children and adolescents. Acta Neurochir (Wien) 1981;59:55-63.
6. Wilkinson IM. The vertebral artery. Extracranial and intracranial structure. Arch Neurol 1972;27:392-6.
7. Finlay HM, Whittaker P, Canham PB. Collagen organization in the branching region of human brain arteries. Stroke 1998;29:1595-601.
8. Meng H, Metaxa E, Gao L, Liaw N, Natarajan SK, Swartz DD, Siddiqui AH, Kolega J, Mocco J. Progressive aneurysm development following hemodynamic insult. J Neurosurg 2011;114:1095-103.
9. Aoki T, Nishimura M. The development and the use of experimental animal models to study the underlying mechanisms of CA formation. J Biomed Biotechnol 2011;2011:535921.
10. Tulamo R, Frösen J, Hernesniemi J, Niemelä M. Inflammatory changes in the aneurysm wall: a review. J Neurointerv Surg 2010;2:120-30.
11. Hashimoto T, Meng H, Young WL. Intracranial aneurysms: links among inflammation, hemodynamics and vascular remodeling. Neurol Res 2006;28:372-80.
12. Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, Jaaskelainen J. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke 2004;35:2287-93.
13. Rajesh BJ, Sandhyamani S, Bhattacharya RN. Clinico-pathological study of cerebral aneurysms. Neurol India 2004;52:82-6.
14. Schlote W, Gaus C. Histologic aspects from ruptured and nonruptured aneurysms. Neurol Res 1994;16:59-62.
15. Chalouhi N, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Koch WJ, Dumont AS. Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab 2012;32:1659-76.
16. Turjman AS, Turjman F, Edelman ER. Role of fluid dynamics and inflammation in intracranial aneurysm formation. Circulation 2014;129:373-82.
17. Kosierkiewicz TA, Factor SM, Dickson DW. Immunocytochemical studies of atherosclerotic lesions of cerebral berry aneurysms. J Neuropathol Exp Neurol 1994;53:399-406.
18. Tamura T, Jamous MA, Kitazato KT, Yagi K, Tada Y, Uno M, Nagahiro S. Endothelial damage due to impaired nitric oxide bioavailability triggers cerebral aneurysm formation in female rats. J Hypertens 2009;27:1284-92.
19. Gimbrone MA Jr, Topper JN, Nagel T, Anderson KR, Garcia-Carde-a G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci 2000;902:230-9.
20. Wang Z, Kolega J, Hoi Y, Gao L, Swartz DD, Levy EI, Mocco J, Meng H. Molecular alterations associated with aneurysmal remodeling are localized in the high hemodynamic stress region of a created carotid bifurcation. Neurosurgery 2009;65:169-77.
21. Kilic T, Sohrabifar M, Kurtkaya O, Yildirim O, Elmaci I, Günel M, Pamir MN. Expression of structural proteins and angiogenic factors in normal arterial and unruptured and ruptured aneurysm walls. Neurosurgery 2005;57:997-1007.
22. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Egashira K, Hashimoto N. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke 2009;40:942-51.
23. Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery 1999;45:1137-46.
24. Crompton MR. Mechanism of growth and rupture in cerebral berry aneurysms. Br Med J 1966;1:1138-42.
25. Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke 1999;30:1396-401.
26. Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, Jaaskelainen J. Growth factor receptor expression and remodeling of saccular cerebral artery aneurysm walls: implications for biological therapy preventing rupture. Neurosurgery 2006;58:534-41.
27. Jamous MA, Nagahiro S, Kitazato KT, Tamura T, Aziz HA, Shono M, Satoh K. Endothelial injury and inflammatory response induced by hemodynamic changes preceding intracranial aneurysm formation: experimental study in rats. J Neurosurg 2007;107:405-11.
28. Boyle JJ. Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture. Curr Vasc Pharmacol 2005;3:63-8.
29. Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N, Lawton MT, Young WL, Liang EI, Nuki Y, Hashimoto T. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke 2011;42:173-8.
30. Caird J, Napoli C, Taggart C, Farrell M, Bouchier-Hayes D. Matrix metalloproteinases 2 and 9 in human atherosclerotic and non-atherosclerotic cerebral aneurysms. Eur J Neurol 2006;13:1098-105.
31. Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke 2007;38:162-9.
32. Watson SP. Platelet activation by extracellular matrix proteins in haemostasis and thrombosis. Curr Pharm Des 2009;15:1358-72.
33. Wohner N. Role of cellular elements in thrombus formation and dissolution. Cardiovasc Hematol Agents Med Chem 2008;6:224-8.
34. Fontaine V, Touat Z, Mtairag el M, Vranckx R, Louedec L, Houard X, Andreassian B, Sebbag U, Palombi T, Jacob MP, Meilhac O, Michel JB. Role of leukocyte elastase in preventing cellular re-colonization of the mural thrombus. Am J Pathol 2004;164:2077-87.
35. Lee D, Yuki I, Murayama Y, Chiang A, Nishimura I, Vinters HV, Wang CJ, Nien YL, Ishil A, Wu BM, Vinuela F. Thrombus organization and healing in the swine experimental aneurysm model. Part I. A histological and molecular analysis. J Neurosurg 2007;107:94-108.
36. Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery 1999;45:1137-46.
37. Krischek B, K asuya H, Tajima A, Akagawa H, Sasaki T, Yoneyama T, Ujiie H, Kubo O, Bonin M, Takakura K, Hori T, Inoue I. Network-based gene expression analysis of intracranial aneurysm tissue reveals role of antigen presenting cells. Neuroscience 2008;154:1398-407.
38. Shi C, Awad IA, Jafari N, Lin S, Du P, Hage ZA, Shenkar R, Getch CC, Bredel M, Batjer HH, Bendok BR. Genomics of human intracranial aneurysm wall. Stroke 2009;40:1252-61.
39. Tulamo R, Frösen J, Junnikkala S, Paetau A, Pitkäniemi J, Kangasniemi M, Niemela M, Jaaskelainen J, Jokitalo E, Karatas A, Hernesniemi J, Meri S. Complement activation associates with saccular cerebral artery aneurysm wall degeneration and rupture. Neurosurgery 2006;59:1069-76.
40. Swartz RH, Bhuta SS, Farb RI, Agid R, Willinsky RA, Terbrugge KG, Butany J, Wasserman BA, Johnstone DM, Silver FL, Mikulis DJ. Intracranial arterial wall imaging using high-resolution 3-tesla contrast-enhanced MRI. Neurology 2009;72:627-34.
41. Rudd JH, Warburton EA, Fryer TD, Jones HA, Clark JC, Antoun N, Johnstrom P, Davenport AP, Kirkpatrick PJ, Arch BN, Pickard JD, Weissberg PL. Imaging atherosclerotic plaque inflammation with [18F]-fluorodeoxyglucose positron emission tomography. Circulation 2002;105:2708-11.
42. Tawakol A, Migrino RQ, Bashian GG, Bedri S, Vermylen D, Cury RC, Yates D, LaMuraglia GM, Furie K, Houser S, Gewirtz H, Muller JE, Brady TJ, Fischman AJ. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J Am Coll Cardiol 2006;48:1818-24.
43. DeLeo MJ 3rd, Gounis MJ, Hong B, Ford JC, Wakhloo AK, Bogdanov AA, Jr. Carotid artery brain aneurysm model: in vivo molecular enzyme-specific MR imaging of active inflammation in a pilot study. Radiology 2009;252:696-703.
44. Hasan DM, Mahaney KB, Magnotta VA, Kung DK, Lawton MT, Hashimoto T, Winn HR, Saloner D, Martin A, Gahramanov S, Dosa E, Neuwelt E, Young WL. Macrophage imaging within human cerebral aneurysms wall using ferumoxytol-enhanced MRI: a pilot study. Arterioscler Thromb Vasc Biol 2012;32:1032-8.
45. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Simvastatin suppresses the progression of experimentally induced cerebral aneurysms in rats. Stroke 2008;39:1276-85.
46. Aoki T, Kataoka H, Ishibashi R, Nakagami H, Nozaki K, Morishita R, Hashimoto N. Pitavastatin suppresses formation and progression of cerebral aneurysms through inhibition of the nuclear factor kappaB pathway. Neurosurgery 2009;64:357-65.
47. Tada Y, Kitazato KT, Yagi K, Shimada K, Matsushita N, Kinouchi T, Kanematsu Y, Satomi J, Kageji T, Nagahiro S. Statins promote the growth of experimentally induced cerebral aneurysms in estrogen-deficient rats. Stroke 2011;42:2286-93.
48. Marbacher S, Schläppi JA, Fung C, Hüsler J, Beck J, Raabe A. Do statins reduce the risk of aneurysm development? A case-control study. J Neurosurg 2012;116:638-42.
49. Aoki T, Nishimura M, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Reactive oxygen species modulate growth of cerebral aneurysms: a study using the free radical scavenger edaravone and p47phox(-/-) mice. Lab Invest 2009;89:730-41.
50. Hasan DM, Mahaney KB, Brown RD Jr, Meissner I, Piepgras DG, Huston J, Capuano AW, Torner JC. Aspirin as a promising agent for decreasing incidence of cerebral aneurysm rupture. Stroke 2011;42:3156-62.
51. Gross BA, Rosalind Lai PM, Frerichs KU, Du R. Aspirin and aneurysmal subarachnoid hemorrhage. World Neurosurg 2014;82:1127-30.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Turkmani AH, Edwards NJ, Chen PR. The role of inflammation in cerebral aneurysms. Neurosciences 2015;2:102-6. http://dx.doi.org/10.4103/2347-8659.153982
AMA Style
Turkmani AH, Edwards NJ, Chen PR. The role of inflammation in cerebral aneurysms. Neuroimmunology and Neuroinflammation. 2015; 2: 102-6. http://dx.doi.org/10.4103/2347-8659.153982
Chicago/Turabian Style
Turkmani, Ali H., Nancy J. Edwards, Peng R. Chen. 2015. "The role of inflammation in cerebral aneurysms" Neuroimmunology and Neuroinflammation. 2: 102-6. http://dx.doi.org/10.4103/2347-8659.153982
ACS Style
Turkmani, AH.; Edwards NJ.; Chen PR. The role of inflammation in cerebral aneurysms. Neurosciences. 2015, 2, 102-6. http://dx.doi.org/10.4103/2347-8659.153982
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 11 clicks
Cite This Article 11 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.