
It takes two: potential therapies and insights involving microglia and macrophages in glioblastoma

Abstract
Microglia and macrophages, two myeloid cell lineages with different origins, make up the majority of immune cells present in glioblastoma (GBM). However, much of the literature does not distinguish between microglia and macrophages, despite a growing body of evidence that demonstrates key structural and functional differences between the cell types. Furthermore, the current M1/M2 paradigm used to sub-classify microglia and macrophages has proven to be incomplete at best, with the growing amount of in vivo and genomic data incompatible with this dichotomy. Finally, a number of studies have already established that in the setting of the GBM tumor microenvironment, both microglia and macrophages are complicit in tumor progression. This review highlights the differences between microglia and macrophages, particularly in the context of GBM, and discusses at length several potential therapeutic strategies made possible by understanding specific pro-tumor and anti-tumor pathways in these myeloid populations. Ultimately, investigating the differences between microglia and macrophages offers insight into the progression of GBM, its marked resistance to current immunotherapy regimens, and future directions for new treatment modalities.
Keywords
Introduction
The advent of immunotherapy as a viable cancer treatment option has resulted in the rapid emergence of new therapeutic strategies, with immune checkpoint inhibitors (ICI) serving as the cornerstone for mounting anti-tumor immune responses against several types of cancers like non-small-cell lung cancer, bladder cancer, and advanced-stage melanoma[1-5]. Intrinsic to ICI-based therapies, particularly those blocking cytotoxic T lymphocyte antigen 4 and programmed cell death protein 1 (PD-1), is the recruitment of CD8+ T cells from tumor infiltrating lymphocytes (TILs) that are normally immunosuppressed in the tumor microenvironment (TME)[6]. However, certain cancers have remained resistant to current immunotherapeutic strategies and are considered “cold tumors”; the recent phase III CheckMate 143 trial involving nivolumab, an anti-PD-1 drug, failed to meet its primary endpoint of improved overall survival in patients with glioblastoma (GBM)[7,8].
GBM is the most aggressive intrinsic brain tumor, with median overall survival ranging from 12 to 15 months in patients who receive current standard of care[9]. While ICI has shown some promise in preclinical GBM models - particularly in combination with radiation therapy - emerging studies support the idea that GBM is a cold tumor, meaning that it shows more resistance to anti-PD-1 when compared to other hot tumors like melanoma that respond to ICI therapies; in regards to this difference in response, GBM appears to have a (1) T cell population with high expression of exhaustion markers such as lymphocyte-activation gene 3 (LAG3/CD223) and T-cell immunoglobulin mucin 3 (TIM3); (2) relatively few TILs; and (3) a high volume of myeloid cells (i.e., microglia and macrophages) that make up about 30%-40% of the tumor cell population[10,11].
As such, there is interest in exploring additional candidates for immune cell reactivation beyond lymphocytes, particularly within the myeloid population[12]. Along this line, several studies in the last decade have revealed immunosuppressive and pro-tumor characteristics in microglia and macrophages within the TME, resulting in a growing interest in viewing these myeloid cells as potential therapeutic targets[12-14]. It should be noted that while granulocytic or monocytic myeloid-derived suppressor cells (MDSCs) are also considered to be a part of the myeloid compartment, there is limited data regarding specific markers that easily distinguish MDSCs from monocytes and will therefore not be addressed further as a distinct population from microglia and macrophages at this time[15].
As such, the purpose of this review is to distinguish the structural and functional differences of microglia and macrophages in the context of the TME of GBM, expand upon the roles of microglia and macrophages in GBM progression and invasion, and discuss current and potential treatment strategies involving these two cell populations.
Macrophages and microglia: similar but distinct populations
Historically, microglia and macrophages have generally been considered interchangeable in the TME, with the former functionally described as the macrophages of the central nervous system (CNS). While both cell types have shared immunologic functions, including phagocytosis of microorganisms and cell debris with subsequent antigen presentation to lymphocytes[16], the advent of genome-wide microarray analyses and detection of specific cellular markers have phenotypically distinguished these cell populations [Figure 1][17,18].
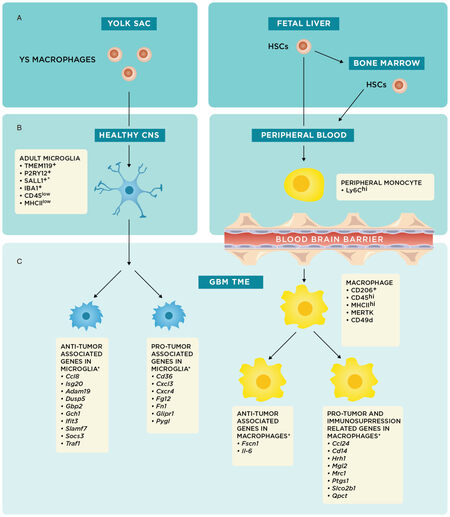
Figure 1. *These markers and/or genes have been described in detail mostly in preclinical mouse models as described in this review. Origins and specific cellular markers and genes of microglia and macrophages. (A) Schematic summarizing the embryonic origins of microglia and macrophages from the yolk sac (YS) and fetal liver, respectively; (B) YS macrophages migrate to the central nervous system (CNS) early in embryonic development and remain in the brain as tissue resident macrophages, or microglia. Fetal liver and bone marrow hematopoietic stem cells (HSCs) mature into monocytes and enter the peripheral blood[13]; and (C) during states of CNS inflammation, particularly in the context of glioblastoma (GBM) tumor microenvironment (TME), microglia and macrophages experience shifts in phenotype; the underlying genetic changes are schematically represented here to show pro-tumor or anti-tumor associated genes[12]. In reality, there are most likely microglial and macrophage populations that have concurrent expression of anti-tumor and pro-tumor genes
Moreover, embryological studies like those from Janossy et al.[19] and Ginhoux et al.[20] have shown that microglia and macrophages come from distinct embryological origins [Figure 1A][13]. Microglia, which are endemic to the CNS, come from yolk sac progenitors and migrate to the brain early in development[19]. The prevailing thought is that microglial populations are enduring and maintain their numbers primarily through local self-renewal[13,20]. Recent fluorescent fate-mapping studies from Tay et al.[21] support this model and also suggest that this self-renewal process is stochastic in the normal steady-state, independent of bone marrow (BM) input, and dependent upon brain geography and inflammatory status.
However, during states of CNS inflammation, BM-derived macrophages can be drawn into the CNS from the periphery by the same chemokines that increase permeability of the blood-brain-barrier[22]. As a result, while microglia will always be present in brain tissue regardless of inflammatory status, peripheral macrophages should only be present in significant numbers during periods of inflammation[22].
While embryological studies clearly reveal that these two populations have distinct origins, the phenotypic differences between microglia and macrophages have often been overlooked. During recruitment of peripheral monocytes under inflammatory conditions in both the neonatal and adult brain, Ling[23] showed that peripheral monocytes have the potential to histologically differentiate into microglia-appearing cells within the CNS. This finding perpetuated the notion that circulating blood monocytes could act as microglial progenitors that replenished microglial populations[24]. It should be stressed that because these original findings were based on histological morphology, the conclusions drawn on their functional differences were limited; moreover, using histologic structural characteristics on microscopy to differentiate microglia and macrophages is unreliable since both cell types have morphologies that are plastic and inconsistent[25,26]. These older studies also could not take advantage of the results from more recent bulk-RNA sequencing studies, which have since elucidated specific cellular markers for microglia, such as CD45low, major histocompatibility complex II (MHCII)low, transmembrane protein 119, P2Y purinoceptor 12, IBA1, and Sal-like protein 1 [Figure 1B][27-29]. Similarly, macrophages have their own specific cellular markers, including CD45hi, MHCIIhi, CD49d, CD206, and MER receptor tyrosine kinase (MERTK) [Figure 1C][30-35].
Beyond cellular markers, microglia have characteristics that are functionally distinct from macrophages. While microglia are considered the resident immune cells of the CNS and perform roles similar to macrophages including phagocytosis and antigen presentation, they are also thought to have additional roles in homeostasis such as secretion of neurotrophic factors that are essential for both normal maintenance and response to pathological conditions[36]. As a key component to normal parenchymal surveillance, microglia are mobile within their own distinct territories and completely scan the brain parenchyma several times a day[37]. While scanning, microglia are sensitive to ATP, potassium, and purinoceptor inhibitors, and as such can detect neuronal cell death or other pathological features with high acuity[38,39]. Upon activation, they convert to an amoeboid phenotype and act similarly to macrophages with a high metabolic rate, rapidly migrating to the source lesion and secreting IL-6, IL-1β, and TNFα before phagocytosing as needed[40].
Of note, while tissue-specific macrophages can be found in other organs outside of the CNS, microglia are special in part due to the brain’s privileged status behind the blood-brain barrier (BBB); after embryonic migration, they remain and exert their effect only in their original tissue with minimal interaction with other systems[41]. This is in contrast to other tissue-specific macrophages, like Langerhans cells, which are epidermal-specific macrophages that have the capacity to migrate to peripheral lymph nodes upon activation[19], or intestinal macrophages, which act locally but rely less upon self-renewal and more upon recruitment of circulating macrophages to maintain their numbers[42].
In the context of this review however, the strongest rationale for viewing microglia and macrophages as distinct populations emanates from their functional differences in the context of the TME, specifically in that of GBM. In the CNS, mild or moderate inflammation leads to the protective function of microglia as outlined above and features minimal interaction with peripheral macrophages. However, more intense acute injury or chronic inflammatory states - as experienced in GBM - can lead to neurotoxic and tumor-promoting activation of microglia, recruitment of peripheral macrophages, and subsequent immunosuppression[41]. More specifically, chemokines released in the TME attract peripherally derived macrophages, which then migrate into the brain through the BBB and express anti-inflammatory cytokines that attenuate the recruitment and aggregation of pro-inflammatory leukocytes (e.g., additional microglia or neutrophils)[35]. The complexity of the GBM TME with this consequent anti-inflammatory attenuation ultimately contributes to pathology and promotes gliomagenesis[43].
M1/M2: an outdated paradigm
Within the GBM TME, microglia and macrophages have classically been subdivided into M1 and M2 phenotypes to characterize them as either having anti-tumor or tumor-promoting (pro-tumor) properties, respectively. The M1/M2 dichotomy was first discussed by Mills et al.[44] as a way to distinguish the phenotypic predilections of macrophages from the perspective of Th1 and Th2 lineages in CD4+ T cells; they proposed that M1 refer to macrophages with Th1 backgrounds that tended to produce inflammatory induced nitric-oxide species (iNOS), while M2 would refer to Th2 derived macrophages that produced more cell-division stimulating polyamines, like ornithine. In short, the original M1/M2 terminology sought to extrapolate the same phenotypic dichotomy assigned to T-helper cells (Th1/Th2) to macrophages[44]. However, later research elucidated further phenotypic pathways for macrophages related to other cytokines and factors (e.g., IL-10, TGF-B) that made the extrapolation from the initial binary Th1/Th2 characterization less robust[45]. As a result, Mantovani et al.[46] proposed the conversion of the M1/M2 dichotomy into a continuum, with M1 and M2 representing two opposite poles of immune function.
In this vein, M1 macrophages, or classically activated macrophages, are typically noted as inducing prototypic inflammatory (pro-inflammatory) responses, while M2 macrophages, or alternatively activated macrophages, are those with antagonism of normal inflammatory (anti-inflammatory) responses[47,48]. More specifically, M2 has been further divided into subtypes; M2a correlates to Th2 responses, type II inflammation, and pathogen elimination. M2b correlates to Th2 activation and immunoregulation. Finally, M2c correlates to immunoregulation, matrix deposition, and tissue remodeling[46]. Ultimately, the popularity of using the M1/M2 paradigm in studies came from its simplicity; by mirroring the nomenclature used for the Th1/Th2 phenotypes, M1/M2 provided a simple and easy to conceptualize model to understand immunosuppressed myeloid populations[46].
However, while the M1/M2 framework was designed to be a simplified operational concept that provided foundational language to a rapidly growing field[48], it has since been used erroneously in much of the literature as a solid classification scheme for macrophages, and to an increasingly greater extent, microglia. From a generalized view, using M1/M2 as a classification system is problematic for a variety of reasons. First, the vast majority of data supporting the system comes from in vitro studies that have not been reliably recapitulated in vivo[49]. These concepts rarely translate to systemic models, as in vitro systems have limited engagement with larger systemic variables beyond characteristics of cell maturation, adhesion, and cytokine production[50]. More specifically, macrophages in vitro versus macrophages in vivo have been documented to have different morphologies, functions, and expression of specific cellular markers[49].
Beyond experimental inconsistencies, the M1/M2 phenotypes are also outdated in the sense that their original formulation predated the significant new body of genomics research that has emerged in the last 15 years. Genome-wide microarray analysis of both glioma-associated microglia and macrophages in GL261 murine gliomas by Szulzewsky et al.[17] have shown that both cell types have expression profiles that do not fit within any previously documented M1/M2 classification scheme[51]. Indeed, these myeloid populations only had partial overlap with previously documented M1/M2 phenotypes, with 59.6% of genes that were significantly upregulated (261/438 analyzed) not characterized as either M1 or M2; this indicates that there is far more complexity than the M1/M2 label can provide, at least from a genomics standpoint[17]. Of note, some of the genes identified outside of the classic M1/M2 phenotype were associated with angiogenesis (Vegfa, Hgf), suppression of immunity (Arg1, Tgfb3), and tumor invasion (Mmp2, Mmp14, Ctgf) in mouse models[17].
In the context of this review, the M1/M2 classification is further problematic when applied to microglia. The original nomenclature for the M1/M2 classification came from studying macrophages and a significant portion of the current literature has merely transposed this M1/M2 nomenclature to microglia without respect to the differences between these two myeloid populations[45]. As discussed previously, however, it was also only recently that we have come to recognize the numerous differences between microglia and macrophages in terms of structure, function, and expression of tumor-related pathways[17,18]. As a result, while the M1/M2 nomenclature for macrophages has issues with oversimplification, the use of said classification scheme for microglia may be simply inaccurate. Importantly, a variety of genome-wide expression studies of microglia in a variety of disease state models, including generalized inflammatory states[18], amyotrophic lateral sclerosis (ALS)[49], autoimmune encephalomyelitis[52], Alzheimer’s disease[53], traumatic brain injury[54], and GBM[55] all showed no clear evidence of true M1/M2 differentiation among microglia and instead mostly showed that they simultaneously express both M1 and M2 phenotypic markers.
Furthermore, it is difficult to classify microglia along the M1/M2 continuum, especially in the context of GBM, since activated microglia in the TME have several more functions that do not fit into the classic functional categories associated with M1/M2 in macrophages. For example, while the M2 phenotype for macrophages has largely been regarded as being immunosuppressed - specifically in the context of increased trophic polyamines - the M2 subtype in microglia may actually have a more active pro-tumor role that supports gliomagenesis and invasion[22,46,47,56-60].
As a result, we recognize the equivocal and limited nature of using M1/M2 as a classification scheme and will be utilizing a more flexible paradigm to organize our discussion of microglia and macrophages, specifically within the context of the TME in GBM. To address these differences as well as move away from the M1/M2 nomenclature, we focus on specific markers and pathways present on microglia or macrophages and designate these molecular targets as having either anti-tumor or pro-tumor/immunosuppressive characteristics [Figure 2].
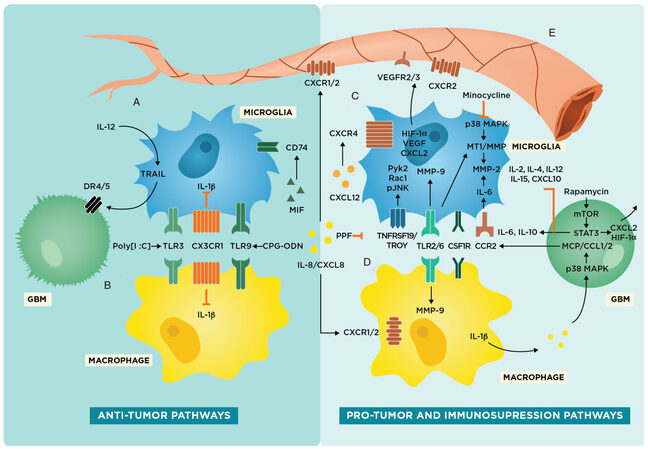
Figure 2. Glioma-associated microglia and macrophage pathways in the tumor microenvironment including potential therapeutic targets. Schematic representing anti-tumor and pro-tumor pathways in microglia. (A) Intrinsic anti-tumor pathways present in microglia involving TRAIL, IL-1β inhibition, and CD74 upregulation; (B) Intrinsic anti-tumor pathways present in macrophages involving IL-1β inhibition. toll-like receptor 3 (TLR3), CX3CR1, and TLR9 are both present on both microglia and macrophages; (C) pro-tumor or tumor-progressive pathways in microglia that are associated with increased gliomagenesis and invasion; (D) pro-tumor and immunosuppressing pathways present in macrophages that result in decreased immune response against glioblastoma (GBM); and (E) tumor angiogenesis and vasculogenesis pathways involving microglia and macrophages
Therapeutic strategies involving glioma-associated microglia and macrophages in the GBM TME
Several preclinical and clinical studies have already examined the efficacy of targeting glioma-associated microglia and macrophages for anti-tumor therapy [Table 1]. In general, there are two main strategies for treatment: inhibition of tumor-promoting microglia and macrophages [Figure 2C and D] or upregulation of anti-tumor receptors and cytokines in microglia and macrophages [Figure 2A and B][61,62]. It should be noted that while there are some receptors and resultant cascades that are present in both microglia and macrophages, there are also a variety of signaling pathways unique to either microglia or macrophages, which lends credence to the idea that these cell types have distinct roles in the TME.
Summary of molecular targets for myeloid interactions with glioblastoma (GBM) Table summarizing receptors and ligands on microglia, macrophages, and glioma cells that are present in the context of GBM tumor microenvironment, along with their interactions with each other in preclinical and clinical investigations
| Molecular targets involved with tumor progression in GBM | Normal function | Aberrant function in tumors | Preclinical studies | |
|---|---|---|---|---|
| Chemokine (C-X-C motif) ligand 2 (CXCL2)- from microglia, macrophages, gliomas | Chemokine (C-X-C motif) receptor 2 (CXCR2)- on endothelial cells | Chemoattractant for neutrophils | Angiogenesis | Brandenburg 2015[97] - CXCL2 was upregulated and stronger than VEGF in vitro -blocking CXCL2-CXCR2 resulted in diminished glioma sizes - in vivo deletion of microglia and macrophages decreased vessel density by 50% |
| CX3CL1- from neurons | CX3CR1- on microglia and macrophages | Chemokine mediation of immune response | Deletion on macrophages promotes gliomagenesis; accumulation of inflammatory myocytes | Feng 2015[79] - Deletion of Cx3cr1 from microglia and macrophages leads to increased tumor incidence and shorter survival times |
| Chemokine (C-C motif) ligand 2 (CCL2)- from microglia and GBM | Chemokine (C-C motif) receptor 2 (CCR2)- on microglia | Chemoattractant for microglia | Recruitment of microglia to tumor site; IL-6 positive feedback cycle for inflammation | Carvalho da Fonseca[62], Zhang 2012[80] - CCL2 was found to be produced by GBM - in vitro glioma lines displayed increased invasion of extracellular collagen matrices when co-cultured with CCR2 expressing microglia |
| CXCL12, from TME | CXCR4- on microglia | Proinflammatory chemokine signaling | Vasculogenesis in radiation-resistant gliomas | Tabouret 2015[98] - CXCR4 blockade in GBM implants lead to decreased VEGf and Hif1a expression |
| HiF1α- from microglia + GBM | Angiogenesis | Angiogenesis | Tabouret 2015[98], Brandenberg 2015[97] - Decreased Hif1a expression lead to less angiogenesis and smaller tumor size | |
| VEGF- from microglia, macrophages | VEGFR-2/3- on endothelial cells | Angiogenesis | Angiogenesis | Tabouret 2015[98], Brandenberg 2015[97] - Decreased VEGF expression lead to less angiogenesis and smaller tumor size |
| CpG DNA | Toll-like receptor 9 (TLR9) on microglia and macrophages | Innate Immune Response | Gliomagenesis | Carpentier 2010[67] - Phase II clinical trial investigating CpG as a therapeutic did not find a significant improvement in progression-free survival, though a few long-term survivors suggest potential benefit |
| CXCL8/IL8 from GBM, microglia, macrophages | CXCR1, CXCR2, on endothelial cells, macrophages, and microglia | Neutrophil chemotaxis, angiogenesis | Gliomagenesis, chemoresistance, invasion, angiogenesis | Waugh 2008[73], Brandenburg 2015[97] - Blocking CXCR2 resulted in considerably diminished glioma sizes |
| Membrane type 1 metalloprotease (MT1-MMP) | Pro-MMP2 to MMP2 | Extracellular matrix breakdown | Invasion | Markovic 2011[83] - Oral minocycline administration greatly reduced glioma growth in orthotopically implanted mice - MT1-MMP was decreased in treated mice and highly upregulated in untreated mice |
| Colony stimulating factor 1 (CSF1) | Colony stimulating factor 1 receptor (CSF1R) - on macrophages and microglia | Production, differentiation, and function of macrophages and microglia | Gliomagenesis and immunosuppression | Pyonteck 2013[76] - The brain-penetrant CSF1R inhibitor BLZ945 resulted in increased expression of anti-tumor responses in glioma associated macrophages and resulted in decreased intracranial growth of patient-derived glioma xenografts in mice Yan 2017[77] - Combination therapy with CSF1R inhibitor PLX3397 and tyrosine kinase inhibitors dovitinib and vatalanib resulted in increased survival in mice glioma models |
Activation of the intrinsic anti-tumor properties of microglia and macrophages
First, while much of the scientific literature corroborates a story of microglia and macrophages having mostly a pro-tumor or immunosuppressive role in the GBM TME, there is some evidence that microglia and macrophages have intrinsic anti-tumor properties as well. An in vitro study by Hwang et al.[63] demonstrated that microglia conditioned culture medium (MCM) promotes apoptosis of glioma cells, with additional cytotoxic effect when exposing microglial cells to lipopolysaccharides (LPS) or IFNγ. Moreover, this effect was glioma-specific, without unwanted astrocyte cytotoxicity. Proteonomic analysis of the MCM revealed LPS- and IFNγ-related proteins along with markedly elevated expression of cathepsin proteases - particularly cathepsin B. When cathepsin B was suppressed, glioma-apoptosis was no longer observed, indicating this protein’s importance in microglial anti-tumor function[63].
A similar study by Kees et al.[64] examined toll-like receptor 3 (TLR3) and its agonist, polyinosinic-polycytidylic acid [poly(I:C)], on both microglia and macrophages. The activation of TLR3 on these myeloid cells resulted in the secretion of glioma specific toxic soluble factors in co-culture with GBM cells [Figure 2A and B]. Of note, a similar phenomenon involving poly(I:C)-induced TLR3 activation in dendritic cells demonstrated by Garzon-Muvdi et al.[65] showed anti-tumor effect. Likewise, TLR9 has also been implicated in anti-tumoral pathways. Several pre-clinical trials have shown that local treatment with oligodeoxynucleotides containing CpG motifs (CpG-ODN) have strong immunostimulatory effects and activate TLR9 in both microglia and macrophages[66]; in a murine glioma in vivo study by Carpentier et al.[67], the use of CpG-ODN resulted in decreased tumor size without toxicity to brain parenchyma [Figure 2A and B]. Unfortunately, follow-up studies in humans including a phase II trial did not show significant progression free survival or radiological response in patients treated with CpG-ODN[68,69].
More promisingly, another study demonstrated the importance of IL-12 in the modulation of microglial anti-tumor activity in mouse models. Using recombinant adenovirus-carrying IL-12 (rAAV2/IL-12), Chiu et al.[70] demonstrated that IL-12 resulted in increased activation of microglia as demonstrated by increased expression of ED1 and tumor necrosis factor-related apoptosis-inducing ligand; in vitro, IL-12 exposure also resulted in microglial-mediated apoptosis of GBM cells through DR4/5 binding[70,71]. In a follow-up study, they observed a similar effect in vivo, with murine GBM models treated with IL-12 exhibiting greater infiltration of activated microglial cells within the tumor mass. Additionally, IL-12 treated mice had significantly reduced tumor volume and increased survival compared to non-treated tumor control groups [Figure 2A][70].
Finally, Zeiner et al.[72] found that GBM has high expression of macrophage migration inhibitory factor, which can bind to the receptor CD74 on glioma-associated microglia. Interestingly, they found a positive correlation between CD74-positive glioma-associated microglia and patient survival, indicating anti-tumoral characterization of this marker. This positive prognostic factor offers a potential area of exploration into pathways involving CD74 to further elucidate candidate receptors or cytokines for encouraging microglial recruitment for anti-GBM response.
Inhibition of pro-tumor functions of microglia and macrophages
There are also several strategies that aim to inhibit pro-tumor or reactivate immunosuppressive pathways in microglia and macrophages. Interleukin 8 [IL-8 or chemokine (C-X-C motif) ligand 8, CXCL8] has been implicated in several tumorigenic pathways, most pronouncedly via its binding CXCR1/2 on endothelial cells and macrophages; this has been associated with tumor growth and chemoresistance, increased invasion, and tumor angiogenesis[73,74]. Furthermore, increased presence of IL-8 has been found in the TME of GBM along with upregulation of its receptors in macrophages and endothelial cells [Figure 2D and E][73]. A follow-up study by Infanger et al.[75] demonstrated similar findings, with IL-8 linked to maintenance and growth of GBM cancer stem-like cells. In the same investigation, they found that CXCR2 silencing reversed the tumor-promoting effects of endothelial cells in vivo, demonstrating the potential therapeutic benefit of inhibiting IL-8 signaling for anti-tumor response.
Further work in understanding the TME and its impact on glioma-associated macrophages include studies on BLZ945 and PLX3397: inhibitors of colony stimulating factor-1 receptor (CSF1R). Contrary to their original hypothesis that CSF1R inhibition would lead to tumor inhibition through global depletion of tumor-associated microglia and macrophages, they actually achieved tumor inhibition in xenograft mouse models through enhanced survival and promotion of tumor-associated macrophages that demonstrated antitumor properties[76,77]. Specifically, when Pyonteck et al.[76] used BLZ945 to inhibit CSF1R in mice, they reported shifts in the gene signatures of macrophage populations away from pro-tumor/immunosuppressive phenotypes with consequent inhibition of GBM progression, in vivo. However, continued use of CSF1R inhibitors resulted in acquired resistance to further CSF1R inhibition in GBM mouse models[78]. Therefore, a follow-up study by Yan et al.[77], focused on using a combinatorial approach with PLX3397 (another potent CSF1R inhibitor) and the tyrosine kinase inhibitors dovitinib or vatalanib; this combination therapy demonstrated significant and lasting reduction in tumor volume compared to PLX3397 alone, indicating that these anti-tumor macrophages rendered glioma cells more sensitive to treatment.
Interestingly, while exposure to PLX3397 preserved macrophage density and resulted in a phenotypic shift, non-glioma associated stromal microglia were almost fully depleted. Yet, while this data suggests the preservation and redirection of tumor-associated macrophages to an anti-tumor phenotype coupled with depletion of stromal microglia in surrounding tissues, neither study adequately characterized the true ratio of microglia to macrophages in the surviving tumor-associated milieu, nor were they able to adequately attribute the ratio of anti-tumor cells to that of peripheral macrophages[77,78]. As a result, while their results suggest enhancement of anti-tumor tumor-associated macrophages alone, further characterization of both populations of cells in the context of CSF1R inhibition is necessary to accept that assertion without doubt. Regardless, the CSF1R pathway potentially indicates a promising therapeutic avenue for targeting macrophages in the GBM TME[77].
Additionally, Cx3cr1 knock-out (KO) experiments by Feng et al.[79], gave further insight into the complexity of tumor adaptive pathways involving both microglia and macrophages. The ligand for CX3C chemokine receptor 1 (CX3CR1), CX3CL1 (fractalkine), is an important chemokine-signaling protein in the healthy CNS that mediates inflammatory response of both microglia and macrophages, including properties of adhesion and migration [Table 1]. When deleted, Cx3cr1 KO mice experienced increased gliomagenesis and greater tumor burden [Figure 2A and B]. Interestingly though, there was no effect on microglial migration in peri-tumoral areas and instead resulted in a significant increase in macrophage recruitment from the periphery and subsequent infiltration. Of key importance is that deletion of Cx3cr1 in mice saw an increase in IL-1β production from both microglia and macrophages, implicating this receptor in the suppression of IL-1β production[79].
The importance of IL-1β lies in the IL-1β/CCL2/IL-6 interaction between microglia and glioma cells [Figure 2A-C]. Specifically, IL-1β released from both microglia and macrophages activates the p38 mitogen-activated protein kinase (MAPK) pathway in glioma cells, which in turn results in increased expression of CCL2-the agonist for CCR2 on microglia [Table 1][80]. This results in an increase in microglial production of IL-6 and eventually MMP-2, which facilitates tumor migration, invasion, and gliomagenesis [Figure 2C][81,82]. The CX3CR1/IL-1β/CCL2 pathway continues to be an area of active interest, particularly in regard to reduction of IL-6 pro-tumor signaling and inhibition of MMP-based pathways in microglia [Figure 2C and D][80].
Similarly, studies have examined the p38 MAPK pathway in microglia and its potential for anti-tumor therapy. Minocycline, a tetracycline that inhibits the p38 MAPK pathway, appears to counteract the pro-tumor phenotype of microglia and reduce tumor growth in vitro and in vivo by inhibiting downstream microglial MT1-MMP expression in mouse models. Decrease in MT1-MMP expression is in turn associated with decreased MMP-2 activity, which follows a similar treatment schema as mentioned above in the IL-1β/CCL2/IL-6 pathway [Figure 2C][83,84].
Also stimulating secretion of CCL2, IL-6, IL-1, and NO is TNFα, which is readily produced by glioma-associated microglia[79,85]. TNF receptor 1 (TNFR1) activation leads to the degradation of IκBa, an inhibitor of NFκB signaling. This degradation leads to a positive feedback loop with p65/p50 nuclear translocation and subsequent transcriptional activation of TNFα[86,87]. Meanwhile, NFκB activation also activates pro-migratory genes that contribute to tumor invasiveness involving several pro-tumor chemokines and MMP pathways[88]. As such, there is ongoing interest in targeting TNFR1 and the related NFκB pathway in microglia[86].
Moreover, GBM cells induce TLR2/6 activation in both macrophages and microglia via the myeloid differentiation primary response 88/TLR8 signaling pathway, which in turn leads to an increase in metalloproteinases like MMP-9 that facilitate tumor invasion and angiogenesis [Figure 2C and D][89]. TLR2 on microglia is also directly involved with promoting tumor invasion with downstream production of MT1-MMP[90]. Studies done with murine GL261 glioma cells injected into TLR2 KO mice resulted in smaller tumor burden and reduced MT1-MMP levels in glioma-associated microglia[90].
Contributing further empiric credence to the idea that macrophages and microglia are distinct populations in the TME, Jacobs et al.[91] found that propentofylline, a methylxanthine, directly acts only on microglia-and not on macrophages-through tumor necrosis factor receptor superfamily, member 19 (TNFRSF19)/TROY inhibition. In a rat glioma model, they demonstrated that TROY is upregulated in infiltrating microglia, with downstream expression of pro-tumor genes Pyk2, Rac1, and pJNK. The potential effectiveness of targeting TROY was shown through a series of siRNA experiments that resulted in inhibition of microglial migration towards glioma cells and, as a result, decreased pro-tumor activity [Figure 2C].
Another important pathway involves signal transducer and activator of transcription 3 (STAT3), which is upregulated in both GBM and glioma-associated microglia and is associated with GBM pathogenesis, progression, and immune evasion[92-94]. A study by Lisi et al.[95] in 2014 examined the use of rapamycin to inhibit mTOR in glioma; the result was the reversal of pro-tumor functions in microglia with glioma-specific cytotoxic behavior. Normally, mTOR leads to an increase in STAT3 expression, which in turn results in increased production of pro-tumor cytokines IL-6, IL-10, CXCL2, and HIF-1α [Figure 2C-E]. With mTOR inhibition, iNOS production was increased in glioma-associated microglia along with concomitant decrease in IL-10 gene expression[94,95]. Moreover, several preclinical studies have demonstrated that siRNA inhibition of STAT3 in glioma cells leads to microglial activation and tumor growth inhibition in murine models, with increases in IL-2, IL-4, IL-12, IL-15, and CXCL10, along with upregulation of CD80 and CD86 on myeloid cells[94,96].
Finally, the production of HIF-1α, vascular endothelial growth factor (VEGF), and CXCL2 in myeloid and glioma cells have long since been known to have roles in tumor angiogenesis [Figure 2E][97]. Specific to microglia however is CXCL12 and its ligand CXCR4, which have been implicated in radiation resistance and increased tumor vasculogenesis. Tabouret et al.[98] demonstrate that with tumor recurrence, there is a switch in expression profile from VEGFR3-HIF-1α to CXCL12-CXCR4 predominance in glioma-associated microglia. As such, microglia may have roles in propagating additional mechanisms of immune resistance in tumor recurrence, providing another rationale for studying and targeting this population to optimize anti-tumor strategies [Figure 2E].
Conclusion
In this review, we discussed the importance of the roles that microglia and macrophages play in GBM. These two cell types have been shown to be complicit in contributing to an immunosuppressed and/or tumor-progressive milieu; however, more data need to be collected on the interactions between microglia and macrophages within these populations in the TME. This review also highlights the importance of semantically distinguishing between microglia and macrophages. As there are certain cancer-specific interactions with either microglia or macrophages, we recommend clearly delineating between the two in order to avoid complicating future experimental designs and discussions. Furthermore, this review provides a note of caution in strictly following the M1/M2 phenotype for macrophages and microglia, as they are complex and have several differences with each other that make this transposed classification scheme largely unfounded. However, it is this same complexity we appreciate for the potential exploration of new pathways; we look forward to further studying these populations and pathways to work towards a clearer understanding of immunotherapy for GBM.
Declarations
Authors’ contributionsConceived of the presented idea: Choi J, Lim M
Underwent literature review and synthesized a draft: Choi J
Looked over and edited draft: Lim M, Mai N, Jackson C, Belcaid Z
Developed the figures and tables and organized the final draft: Choi J, Mai N
Contributed ideas throughout the process and approved the final draft: Lim M
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin Ther 2015;37:764-82.
2. Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol 2017;14:463-82.
3. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16:275-87.
4. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54.
5. Lipson EJ, Forde PM, Hammers HJ, Emens LA, Taube JM, et al. Antagonists of PD-1 and PD-L1 in cancer treatment. Semin Oncol 2015;42:587-600.
6. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol 2018;15:422-42.
7. Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, et al. European association for neuro-oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol 2017;18:e315-29.
8. Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro Oncol 2017;19:iii21.
9. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987-96.
10. Li B, Severson E, Pignon JC, Zhao H, Li T, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol 2016;17:174.
11. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res 2018;24:4175-86.
12. Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol 2018;9:1004.
13. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 2018;18:225-42.
14. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, et al. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res 2013; doi: 10.1158/1078-0432.CCR-12-3314.
15. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol 2018;19:108-19.
16. Mallat M, Marín-Teva JL, Chéret C. Phagocytosis in the developing CNS: more than clearing the corpses. Curr Opin Neurobiol 2005;15:101-7.
17. Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One 2015; doi: 10.1371/journal.pone.0116644.
18. Satoh J. Gene expression profiles of M1 and M2 microglia characterized by comparative analysis of public datasets. Clin Exp Neuroimmunol 2018;9:124-38.
19. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010;330:841-5.
20. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 2007;10:1538-43.
21. Tay TL, Mai D, Dautzenberg J, Fernández-Klett F, Lin G, et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci 2017;20:793-803.
22. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 2016;19:20-7.
23. Ling EA. Some aspects of amoeboid microglia in the corpus callosum and neighbouring regions of neonatal rats. J Anat 1976;121:29-45.
24. Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci 2013;7:45.
25. Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol 2017;79:619-43.
26. Sobaniec-Lotowska ME. A transmission electron microscopic study of microglia/macrophages in the hippocampal cortex and neocortex following chronic exposure to valproate. Int J Exp Pathol 2005;86:91-6.
27. Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016;36:39-49.
28. Lou N, Takano T, Pei Y, Xavier AL, Goldman SA, et al. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci U S A 2016;113:1074-9.
29. Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol 2016;17:1397-406.
30. Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol 2017;18:385-92.
31. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014;159:1327-40.
32. Goldmann T, Wieghofer P, Jordão MJ, Prutek F, Hagemeyer N, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 2016;17:797-805.
33. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol 2017;18:234.
34. Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017; doi: 10.1126/science.aai8478.
35. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep 2016;17:2445-59.
36. Paolicelli RC, Ferretti MT. Function and dysfunction of microglia during brain development: consequences for synapses and neural circuits. Front Synaptic Neurosci 2017;9:9.
37. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 2007;10:1387-94.
38. Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, et al. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci 2001;21:1975-82.
39. Koizumi S, Fujishita K, Inoue K. Regulation of cell-to-cell communication mediated by astrocytic ATP in the CNS. Purinergic Signal 2005;1:211-7.
40. Cornejo F, von Bernhardi R. Age-dependent changes in the activation and regulation of microglia. Adv Exp Med Biol 2016;949:205-26.
41. London A, Cohen M, Schwartz M. Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci 2013;7:34.
42. Mowat AM, Bain CC. Mucosal macrophages in intestinal homeostasis and inflammation. J Innate Immun 2011;3:550-64.
43. Shechter R, London A, Varol C, Raposo C, Cusimano M, et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med 2009; doi: 10.1371/journal.pmed.1000113.
44. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol 2000;164:6166-73.
45. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 2014;6:13.
46. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 2004;25:677-86.
47. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 2016;173:649-65.
49. Sousa C, Biber K, Michelucci A. Cellular and molecular characterization of microglia: a unique immune cell population. Front Immunol 2017;8:198.
50. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol 2013;14:986-95.
51. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016; doi: 10.1172/jci.insight.85841.
52. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 2014;211:1533-49.
53. Efthymiou AG, Goate AM. Late onset Alzheimer's disease genetics implicates microglial pathways in disease risk. Mol Neurodegener 2017;12:43.
54. Morganti JM, Riparip LK, Rosi S. Call off the dog(ma): M1/M2 polarization is concurrent following traumatic brain injury. PLoS One 2016; doi: 10.1371/journal.pone.0148001.
55. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol 2017;18:234.
56. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol 2014;5:514.
57. Wu SY, Watabe K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front Biosci (Landmark Ed) 2017;22:1805-29.
58. Annovazzi L, Mellai M, Bovio E, Mazzetti S, Pollo B, et al. Microglia immunophenotyping in gliomas. Oncol Lett 2018;15:998-1006.
59. Laudati E, Currò D, Navarra P, Lisi L. Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem Int 2017;108:100-8.
60. Roesch S, Rapp C, Dettling S, Herold-Mende C. When immune cells turn bad-tumor-associated microglia/macrophages in glioma. Int J Mol Sci 2018; doi: 10.3390/ijms19020436.
61. Li W, Graeber MB. The molecular profile of microglia under the influence of glioma. Neuro Oncol 2012;14:958-78.
62. da Fonseca AC, Amaral R, Garcia C, Geraldo LH, Matias D, et al. Microglia in cancer: for good or for bad? Adv Exp Med Biol 2016;949:245-61.
63. Hwang SY, Yoo BC, Jung JW, Oh ES, Hwang JS, et al. Induction of glioma apoptosis by microglia-secreted molecules: the role of nitric oxide and cathepsin B. Biochim Biophys Acta 2009;1793:1656-68.
64. Kees T, Lohr J, Noack J, Mora R, Gdynia G, et al. Microglia isolated from patients with glioma gain antitumor activities on poly (I:C) stimulation. Neuro Oncol 2012;14:64-78.
65. Garzon-Muvdi T, Theodros D, Luksik AS, Maxwell R, Kim E, et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 2018;9:20681-97.
66. Weeratna RD, Makinen SR, McCluskie MJ, Davis HL. TLR agonists as vaccine adjuvants: comparison of CpG ODN and resiquimod (R-848). Vaccine 2005;23:5263-70.
67. Carpentier A, Metellus P, Ursu R, Zohar S, Lafitte F, et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol 2010;12:401-8.
68. Meng Y, Carpentier AF, Chen L, Boisserie G, Simon JM, et al. Successful combination of local CpG-ODN and radiotherapy in malignant glioma. Int J Cancer 2005;116:992-7.
69. Ursu R, Carpentier A, Metellus P, Lubrano V, Laigle-Donadey F, et al. Intracerebral injection of CpG oligonucleotide for patients with de novo glioblastoma-A phase II multicentric, randomised study. Eur J Cancer 2017;73:30-7.
70. Chiu TL, Wang MJ, Su CC. The treatment of glioblastoma multiforme through activation of microglia and TRAIL induced by rAAV2-mediated IL-12 in a syngeneic rat model. J Biomed Sci 2012;19:45.
71. Chiu TL, Peng CW, Wang MJ. Enhanced anti-glioblastoma activity of microglia by AAV2-mediated IL-12 through TRAIL and phagocytosis in vitro. Oncol Rep 2011;25:1373-80.
72. Zeiner PS, Preusse C, Blank AE, Zachskorn C, Baumgarten P, et al. MIF receptor CD74 is restricted to microglia/macrophages, associated with a M1-polarized immune milieu and prolonged patient survival in gliomas. Brain Pathol 2015;25:491-504.
74. Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol 2005;7:122-33.
75. Infanger DW, Cho Y, Lopez BS, Mohanan S, Liu SC, et al. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res 2013;73:7079-89.
76. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013;19:1264-72.
77. Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017;36:6049-58.
78. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016; doi: 10.1126/science.aad3018.
79. Feng X, Szulzewsky F, Yerevanian A, Chen Z, Heinzmann D, et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015;6:15077-94.
80. Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, et al. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012;33:312-9.
81. Li R, Li G, Deng L, Liu Q, Dai J, et al. IL-6 augments the invasiveness of U87MG human glioblastoma multiforme cells via up-regulation of MMP-2 and fascin-1. Oncol Rep 2010;23:1553-9.
82. Wang H, Lathia JD, Wu Q, Wang J, Li Z, et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 2009;27:2393-404.
83. Markovic DS, Vinnakota K, van Rooijen N, Kiwit J, Synowitz M, et al. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain Behav Immun 2011;25:624-8.
84. Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and IkappaBalpha degradation in a stimulus-specific manner in microglia. J Neurochem 2006;96:314-23.
85. Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol 2008;83:1136-44.
86. Tchoghandjian A, Jennewein C, Eckhardt I, Rajalingam K, Fulda S. Identification of non-canonical NF-κB signaling as a critical mediator of Smac mimetic-stimulated migration and invasion of glioblastoma cells. Cell Death Dis 2013; doi: 10.1038/cddis.2013.70.
87. Taetzsch T, Levesque S, McGraw C, Brookins S, Luqa R, et al. Redox regulation of NF-κB p50 and M1 polarization in microglia. Glia 2015;63:423-40.
88. Wu Y, Zhou BP. TNF-alpha/NF-kappaB/snail pathway in cancer cell migration and invasion. Br J Cancer 2010;102:639-44.
89. Hu F, Ku MC, Markovic D, a Dzaye OD, Lehnardt S, et al. Glioma-associated microglial MMP9 expression is upregulated by TLR2 signaling and sensitive to minocycline. Int J Cancer 2014;135:2569-78.
90. Vinnakota K, Hu F, Ku MC, Georgieva PB, Szulzewsky F, et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro Oncol 2013;15:1457-68.
91. Jacobs VL, Liu Y, De Leo JA. Propentofylline targets TROY, a novel microglial signaling pathway. PLoS One 2012; doi: 10.1371/journal.pone.0037955.
92. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 2007;7:41-51.
93. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 1999;10:39-49.
94. Zhang L, Alizadeh D, Van Handel M, Kortylewski M, Yu H, et al. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia 2009;57:1458-67.
95. Lisi L, Laudati E, Navarra P, Dello Russo C. The mTOR kinase inhibitors polarize glioma-activated microglia to express a M1 phenotype. J Neuroinflammation 2014;11:125.
96. Hussain SF, Kong LY, Jordan J, Conrad C, Madden T, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res 2007;67:9630-6.
97. Brandenburg S, Müller A, Turkowski K, Radev YT, Rot S, et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol 2016;131:365-78.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Choi J, Mai N, Jackson C, Belcaid Z, Lim M. It takes two: potential therapies and insights involving microglia and macrophages in glioblastoma. Neurosciences 2018;5:42. http://dx.doi.org/10.20517/2347-8659.2018.47
AMA Style
Choi J, Mai N, Jackson C, Belcaid Z, Lim M. It takes two: potential therapies and insights involving microglia and macrophages in glioblastoma. Neuroimmunology and Neuroinflammation. 2018; 5: 42. http://dx.doi.org/10.20517/2347-8659.2018.47
Chicago/Turabian Style
Choi, John, Nicholas Mai, Christopher Jackson, Zineb Belcaid, Michael Lim. 2018. "It takes two: potential therapies and insights involving microglia and macrophages in glioblastoma" Neuroimmunology and Neuroinflammation. 5: 42. http://dx.doi.org/10.20517/2347-8659.2018.47
ACS Style
Choi, J.; Mai N.; Jackson C.; Belcaid Z.; Lim M. It takes two: potential therapies and insights involving microglia and macrophages in glioblastoma. Neurosciences. 2018, 5, 42. http://dx.doi.org/10.20517/2347-8659.2018.47
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.