
Total tumor RNA pulsed dendritic cells plus adoptive transfer of ex-vivo enriched autologous T-lymphocytes in the treatment of children with primary brain tumors


Abstract
The therapeutic approach of adoptive lymphocyte transfer (ALT) using lymphocytes primed and expanded ex-vivo by exposure to total tumor RNA (ttRNA) containing dendritic cells (DCs) and administered after lymphodepletive host conditioning in patients with refractory melanoma with brain metastases has shown excellent objective responses indicating that the central nervous system (CNS) is not an immune privileged site and further paved the way for utilization of a similar approach in other cancers. We have shown that the use of ALT + ttRNA DCs following either myeloablative or non-myeloablative host conditioning is feasible and safe and appears to prolong survival in a proportion of children with recurrent medulloblastoma who had failed standard cytotoxic therapy. Further refinements in this promising approach are needed to improve outcomes and extend this treatment to a broad range of CNS malignancies.
Keywords
Introduction
Primary pediatric central nervous system tumors
Pediatric brain tumors are the commonest solid tumors in children and unfortunately the cause for the most cancer related mortality in this age group[1]. Standard treatment for these patients at diagnosis includes surgery, chemotherapy, and/or irradiation leading to cures around 70% overall but with a high cost in terms of permanent neuro-cognitive and hormonal deficits[2]. The prognosis for those with recurrent disease remains dismal and novel approaches are urgently needed for refractory tumors. With the recent spurt in enthusiasm in the field of cancer immunotherapy, there is an increasing interest to utilizing this approach to treat children with recurrent central nervous system (CNS) tumors. The focus of this review is our experience in using adoptive lymphocyte transfer (ALT) with total tumor derived mRNA loaded dendritic cells (DCs) following myeloablative (MA) conditioning chemotherapy in pediatric brain tumors. This work is a form of adoptive cellular therapy (ACT) pioneered in large part by Dr. Rosenberg at the National Cancer Institute that resulted in durable remissions in patients with metastatic melanoma[3] and heralded a new approach in the field of adoptive cellular immunotherapy that could be extended to other cancers.
The CNS is not an immune privileged site
The CNS and by extension brain tumors have been thought to be protected from the immune system[4,5]. Several lines of evidence lent credence to this theory including an intact blood brain barrier (BBB), lack of prototypical lymphatic structures, a general lack of antigen presenting cells (APCs) within the brain tissue, low to absent expression of major histocompatibility (MHC) class I molecules, constitutive expression of immunosuppressive cytokines including transforming growth factor-β and IL-10, and the slow rejection allogenic tissue implanted in the brain as compared to other sites[5]. However, there is also ample evidence to the contrary including the occurrence of paraneoplastic syndromes in which a spontaneous immune response to tumors causes immune damage to the central and peripheral nervous system structures[6], objective immune responses in brain metastases in patients with recurrent metastatic melanomas treated with adoptive T-cell therapy[3], and immune infiltrates seen in primary CNS tumors including malignant gliomas that are a few examples of how the CNS cannot be considered an immune privileged site[5]. While the BBB is relatively impermeable in the normal state, tumors generally disrupt the BBB, and tumor release of inflammatory cytokines can further induce migration of immune cells into the brain[5]. Released tumor antigens can either be engulfed by APCs and migrate through the cerebrospinal fluid and exit from the cribriform plate to the nasal mucosa or be transported via the interstitial fluid and drain along the capillary walls of blood vessels to reach cervical nodes to sensitize the immune system. Microglia (the CNS resident macrophages) can also play a role through innate or adaptive immunity mechanisms[5].
Immune system and cancer
The immune system and the responses thereof have been traditionally divided into innate and adaptive immunity with a considerable interaction and cross talk between the two systems[7]. Innate immunity is mediated by phagocytes (DCs, macrophages, neutrophils), natural killer (NK) cells, mast cells, eosinophils, and basophils[8]. Innate immunity is mediated through receptors that are pre-determined in the germline by approximately 100 genes and are called pattern recognition receptors targeting a specific set of ligands grouped as pathogen or damage associated molecular patterns[8]. Innate responses are rapid and occur within a matter of hours following a pathogenic threat.
The key players in the acquired form of immunity include the APCs, T-helper (CD4), T-suppressor cells, and cytotoxic CD8+ cells)[9]. Acquired immune responses depend on T and B cell receptor diversity generated somatically during lymphocyte development that are not genetically pre-determined towards a specific antigen and arise on exposure to a pathogen or foreign antigen by random site specific recombination in the immunoglobulin (B-cells) or T-cell receptor (TCR) genes and clonal expansion of such lymphocytes on further antigen exposure[7,9]. Degeneracy (in contrast to specificity) is a typical characteristic of antigen recognition in adaptive immunity, which refers to the cellular response from a single receptor that interacts with several ligands (antigens in this case) that are structurally different[7]. Acquired immune responses on an average takes 7-10 days to initiate and peak following antigen encounter.
APCs
Amongst several APCs, DCs are the key APC in humans[10-12]. The human DC arises from a common bone marrow CD14+ or CD14- myeloid progenitor that also gives rise to the monocyte and is uniquely dependent on Flt3-ligand for proliferation[13,14]. Antigens presenting cells typically present endogenous or exogenous antigens on the cell surface in the context of MHC complex class I (for endogenous proteins) or class II (for exogenous proteins) molecules[12]. The proteasomes within each cell converts proteins into small peptide fragments (15-20 amino acids in length) which are then loaded on to MHC complexes and transported to the cell surface. The peptide fragments are bound within the major clefts of the MHC molecules and are recognized by the T-lymphocytes (CD8+ T cell for MHC class I and CD4+ T cell for MHC class II) via its clonotypically unique TCR[15].
Cross priming of T-lymphocytes
The mere encounter of a cytotoxic T-cell and a cognate foreign antigen is not enough for antigen recognition and expansion. Additional signals that arise out of receptor-ligand interaction between the T-lymphocytes and APCs are necessary for an active immune response to occur and is called cross priming of T-cells. DCs are called “immature” when they have not encountered antigen yet but have a higher capacity for phagocytosis[16]. Immature DCs are not capable of causing T-lymphocyte expansion but can produce immune tolerance when presenting self-antigens by causing T cell deletion or induction of regulatory T-lymphocytes[11,16,17]. Maturation of DCs occur in the lymph nodes and within areas of lymphocyte predominance thus increasing the chances of lymphocyte encounter and cross priming. Productive encounter of the TCR of CD8+ or CD4+ lymphocytes with p-MHC-I or p-MHC-II complexes respectively on the mature DCs (first signal) requires the ligation of lymphocyte receptor CD28 of the CD8+ lymphocytes with its ligand (CD86) on DCs (the second signal or co-stimulation)[18-20], as CD-28 deficient mice frequently have deficient T-cell responses[21]. The interaction of CD40 on CD4+ lymphocytes with the CD40-L on the mature DCs is required for proper priming of the CTLs via CD28 and CD86[17]. Around the same time as the CD28-CD86 interaction, upregulation of the inhibitory receptor, cytotoxic T-lymphocyte antigen-4 (CTLA4), occurs and engages with CD80 (B7-2) (with a higher affinity compared to CD28) on DCs resulting in dampening of the T-cell response to prevent excessive immune reaction to antigen stimulus[22].
CD8+ lymphocyte subsets and immunologic memory
The CD8+ cytotoxic T-cells are essential for killing viral, protozoal, intracellular bacteria organisms and has a key role in preventing tumor growth and eradicating established tumors[23]. It also plays a significant part in mediating effectiveness of standard cytotoxic therapies for cancer[24,25]. In the context of eliminating microorganisms and tumor control, its cytocidal effects are mediated via (1) perforin and granzymes through induction of caspases (identified by the expression of CD107a on degranulating cells); (2) the Fas/Fas Ligand; (3) cytotoxicity aimed at tumor stromal cells including tumor vasculature; (4) secretion of tumor necrosis factor-α (TNFα) and interferon-γ (IFN-γ) that in turn induce tumor cell senescence; and (5) an anti-angiogenic effect by targeting tumor associated macrophages and secretion of IFN-γ (a known inhibitor of tumor angiogenesis)[23]. Encounter of naïve CD8+ T cells with p-MHC I complex on DCs results in a series of events grouped into three phases; (1) a multi-log clonal expansion of CTLs with capabilities of peripheral tissue homing, release of effector cytokine, and consequent cytotoxicity; (2) a contraction or “death” phase when there is rapid apoptosis of antigen-specific T-cells; and (3) development of long-lived antigen-specific cells that represent “memory” cells which reside in the peripheral lymph nodes (central memory cells) or can home in into the peripheral tissues where infection/tumor exist (effector memory cells)[26,27]. The attributes of these memory cells encapsulate the hallmarks of immunologic memory; increased precursor frequency compared to naïve lymphocytes, capacity for antigen-independent renewal in response to cytokines (IL-7, IL-15, and IL-21), procurement of effector functions and clonal expansion upon re-challenge providing long-term protection of the host from future pathogens or tumor recurrences[26]. Immunophenotypes of the different CD8+ T cells are listed in Figure 1[27]. Naïve CD8+ T cells express C-C chemokine receptor type 7 (CCR7) and CD62L to allow homing to lymphoid tissues[26,27]. Central memory CD8+ lymphocytes are antigen-experienced cells that express CCR7 and CD62L allowing them to easily extravasate through venules to populate T-cell zones within regional lymph nodes and provide protection against systemic antigen re-challenge[26,27]. These cells also have the ability to migrate to secondary lymph nodes by virtue of these two markers and secrete IL-2. Effector memory cells on the other hand, lose expression of these two molecules allowing them to migrate to peripheral tissues and protect against a peripheral challenge.
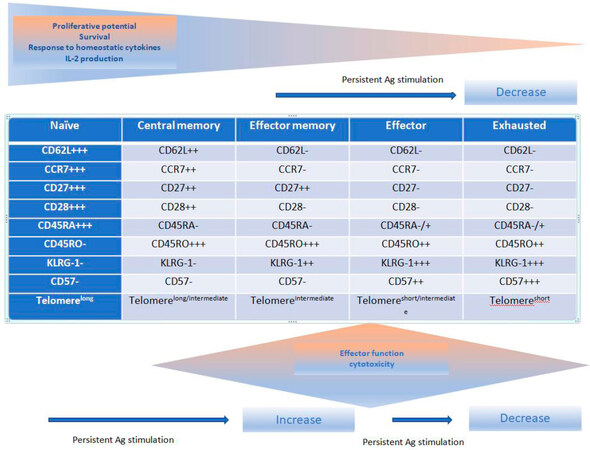
Figure 1. Phenotypic and function changes in CD8+ T cells following antigen (Ag) stimulation[27]
TCRs
The TCR is a membrane bound receptor consisting of two polypeptide heterodimers (αβ or γδ) connected by a disulfide bond and anchored to the membrane via a protein complex called CD3[28]. In the α chain, the constant region is followed by the J region whereas in the β chain there is an intervening D region between the C and J regions. The complementarity determining region 3 (CDR3) (peptide region of 15 amino acids) consists of the VJ junction in the α chain and the VDJ junctions in the β chain[28]. The CDR3 region confers the area of antigen contact and specificity for the TCR and is created by random joining of the V (54 regions), D (2 regions), and J (13 regions) genes. Further specificity is provided by random insertion and deletion of nucleotides in the V-D, V-J, and D-J junctions of the CDR3 domain during somatic recombination[28]. A similar process occurs with the α chain without the D region. The TCR α and β genes are located on chromosome 14 and 7 respectively.
Adoptive T cell therapies with total tumor RNA DC vaccines in children with primary CNS tumors
Our lab has expertise in the development of ex vivo “educated” adoptive T-cell transfer along with total tumor RNA (ttRNA) DC vaccines for the treatment of brain tumors in the context of either MA or non-myeloablative (NMA) conditioning regimens.
Neoantigen load and immune landscape of common pediatric brain tumors
In the last few years, advances in next generation sequencing in pediatric brain tumors has yielded a wealth of knowledge on the molecular landscape of various tumors including low grade gliomas, medulloblastomas, malignant gliomas (including diffuse pontine gliomas), ependymomas, and atypical teratoid rhabdoid tumors[29]. However, the overarching theme from these studies is that while actionable mutations do exist in all these tumors, the mutational load is significantly much lower than carcinogen-induced adult malignancies (non-small lung cancer or melanomas) except in the case of mismatch repair deficiency (MMR) induced malignant gliomas in children[30,31]. Unrepaired DNA damage in these tumors due to the somatic or germline mutations of MSH1, MSH2, MSH6, or PMS-2, POLE, or POLD1 genes results in accumulation of an inordinate number of additional non-synonymous mutations resulting in a high neoantigen load and increased immunogenicity against these tumors due to lack of induction of central tolerance[30-32].
Recent studies of the immune landscape of pediatric brain tumors demonstrates a tumor micro-environment that is variable across different tumor types. In an immune assay of 91 gliomas [glioblastoma multiforme (GBM) 68 and pilocytic astrocytoma 23] by immunohistochemistry (IHC), the CD8+ lymphocytes, CD56+ NK cells, and CD68+ macrophages were significantly higher in grade IV infiltrative glioma as compared to non-infiltrative grade I pilocytic astrocytoma[33]. Similarly, in a prospective randomized therapeutic trial in pediatric high grade gliomas using a backbone of bevacizumab (AvastinTM. Genentech corporation, San Francisco, CA) in patients receiving radiotherapy plus temozolomide (TemodarTM, Merck Co., Kenilworth, NJ), CD8+ infiltration (both perivascular and intratumoral) was highest in both MMR deficient malignant gliomas (four cases with somatic POLE or POLD1 mutations; median mutation count of 4848, range 2197-5332) and anaplastic pleomorphic xanthoastrocytomas (with BRAF alterations)[32]. In this same study, RNA-sequencing data in a subset of samples revealed CD8 T cell effector/T cell signature that correlated with CD8+ T cell infiltration by IHC. This signature was particularly prominent in tumors with mitogen activated protein kinase pathway activation (including BRAFv600e, NF-1, and FGFR1 mutations and NTRK2 translocation). However, the histone mutant mid-line tumors (carry H3F3A mutations) had notable absence of immune infiltrates[32]. An IHC analysis of the antigen processing machinery (APM) in astrocytic tumors (4 each of grade I-IV gliomas) revealed down regulation of the APM proteins LMP-2, TAP1, and β2 microglobulin without change in the surface human leukocyte antigen (HLA)-class I protein expression[34]. In 26 medulloblastoma samples, Vermeulen et al.[35] found CD3+ T cell intratumoral and/or perivascular in distribution. The number of CD3+ tumor infiltrating lymphocytes (TILs) was at a median of 23.5 per 2 mm3 of tumor tissue. The phenotype was predominantly CD8+ T cells (52%) followed by CD4+ (35%) and CD4+CD25+ Fox P3 (2.5%) regulatory T cells. The number of TILs was not different between histologic or molecular subtypes of this tumor. There was also low CTL activation evidenced by only a small percentage of T-cells expressing granzyme B (3.9%; maximum 35%). In addition, decreased cell activation was attributed to a complete lack of expression of MHC class I on tumor cells (HLA-A and B) and CD1 d. Down regulation of MHC class I complex in medulloblastoma has similarly been reported in other studies[36,37]. One study of 10 primary medulloblastoma samples observed that while down regulation of class I molecules and associated proteins of APM machinery was found in these samples, HLA class I restricted tumor antigen specific CTLs that were generated by stimulation with DCs containing tumor mRNA, were able to effectively lyse medulloblastoma cell lines in a HLA-restricted manner, suggesting that down regulation or absence of MHC class I molecules or APM did not impact on tumor recognition by CTLs[37]. In addition, tumor cells expressed serpins (granzyme inhibitors) including serpin B1 and serpin B4 as additional means of immune evasion. The immune environment in ependymomas has been shown to determine patient prognosis between the two recently described molecular groups (Group A and B)[38,39]; in the unfavorable group A tumors, the gene expression pattern has more cell infiltration and immune gene signatures that indicate an immunosuppressive environment; in group B tumors there exists more of an immune stimulating response possibly predicting for a better prognosis in patients with these tumors both at initial diagnosis and following recurrence[40]. Furthermore, recurrent tumors from group A had higher expression of genes related to inflammation and immunoregulatory function and recurrent group B tumors had antiviral and adaptive immune gene signatures[40]. In keeping with these gene signatures, group B recurrent tumors had higher infiltration of CD4+ and CD8+ T cells. When group A and group B tumors from diagnosis were evaluated for secretion of immune cytokines following stimulation, group B tumors secreted higher amounts of TNF-α (2.7 fold), IFN-γ (5.3 fold), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (5.1 fold)[40]. In contrast to ependymomas, the tumor microenvironment of diffuse pontine glioma, a midline tumor with a dismal prognosis, is predominantly populated by CD11b+ macrophages which contain scant CD3+ T-lymphocytes; moreover, in contrast to malignant glioma, diffuse intrinsic pontine glioma ex-vivo cell cultures (patient-derived) release significantly less cytokines[41].
TtRNA DC vaccines in pediatric brain tumors
DC vaccines used in cancer are of four main categories and include peptide vaccines, cellular vaccines (tumor or immune cells), viral vector vaccines, and nucleic acid (DNA and RNA) vaccines[42,43]. Our lab has shown the use of RNA-pulsed DCs to be a versatile platform for activating tumor-specific T cells in vitro and in vivo in several murine and human systems. Several clinical trials have been conducted demonstrating the feasibility and safety of tumor lysate or RNA-pulsed DCs in human patients[44-46]. While specific tumor-associated antigens such as carcinoembryonic antigen, telomerase reverse transcriptase, melanoma antigens, epidermal growth factor receptor variant III (EGFRvIII), and human cytomegalovirus (CMV) phosphoprotein 65 (pp65) have all been successfully utilized in trials as either peptide vaccines or RNA-encoded antigens in DCs[46-48], studies have demonstrated that the majority of endogenous anti-tumor immune responses in patients with malignancy are against unidentified, patient-specific antigens[49]. While use of ttRNA pulsed DCs to expand tumor-specific lymphocytes allows for these patient-specific antigens to be targeted, sufficient tumor tissue for clinical-scale vaccination is not always readily available. Our laboratory has utilized amplification of ttRNA with reverse-transcriptase primed polymerase chain reaction to generate cDNA library templates encoding for the antigenic content of tumor cells from as few as 500 starting tumor cells. Through inclusion of a T7 RNA polymerase binding site in the 5’ primer used for amplification, ttRNA can be readily generated through in vitro transcription after cDNA amplification. Using such techniques, we have been able to generate enough RNA for clinical DC vaccine preparations from colorectal tumors, renal carcinoma, and pediatric and adult brain tumor specimens using excess tumor material harvested during surgical resection[50-52].
While expansion of tumor cells using in vitro culture is feasible, primary brain tumor cells are often difficult to propagate and gene expression microarray analysis has demonstrated that most tumor specific genes expressed in vivo are not recapitulated within in vitro propagated tumor cells. Furthermore, we have demonstrated in murine intracranial glioma models that a significant shift in brain tumor gene expression is induced in response to host anti-tumor immunity[53]. This strongly suggests that the antigenic content of tumor cells propagated in vitro will be significantly different than in vivo propagated tumors, and thus the relevance of in vitro propagated tumors as an antigenic source for immunotherapy is questionable. This observation prompted us to investigate the capacity to amplify the RNA content of tumor cells isolated directly from surgically resected malignant glioma specimens to utilize an antigenic source more representative of the antigens expressed within patients’ tumor cells in vivo. Based on current clinical protocols utilizing ttRNA pulsed DCs[54], it is possible to produce up to 750 μg of amplified tumor mRNA per patient. We have successfully amplified tumor mRNA to clinical scale (over 1mg) from as few as 500 astrocytoma cells from resected human glioma specimens from adult and pediatric brain tumors. Enrichment of tumor antigens can be done using subtractive hybridization of excess pooled normal brain RNA from tumor RNA prior to amplification and in vitro RNA synthesis and verification of enrichment of tumor-associated genes by comparative real-time PCR.
Once enough ttRNA is obtained, it can then be introduced via electroporation (300 V for 500 μsecs) into immature DCs derived from patient derived peripheral blood monocytes obtained via a peripheral blood mononuclear cell (PBMC) collection via apheresis. Differentiation of monocytes into DCs is achieved in in vitro cultures under the influence of cytokines including GM-CSF and IL-4 for 7 days. The brief electrical current used during electroporation is enough to create a reversible breach in the cell membrane for the ttRNA to rapidly enter the cytoplasm before degradation of the RNA. Electroporated ttRNA results in better translation to tumor proteins in the cytoplasm of DCs and induction of tumor immunity in the host[42]. Although such immature ttRNA containing DCs (ttRNA DCs) can be administered as vaccine[55], induction of immune responses is vastly inferior compared to mature ttRNA DCs; maturity can be achieved by combination of cytokines including IL-1β, TNF-α, IFN-γ, and IL-6. Once matured, ttRNA DCs can be frozen in aliquots of 1 × 107 cells for clinical use.
Dose and route of administration of ttRNA DC vaccine and immunoadjuvants
While studies in adult cancers have shown that efficacy of monocyte derived ttRNA DC vaccines might be dose dependent, larger dose vaccines do not necessarily produce proportional T cell responses but might rather dampen them by inducing immune tolerance[56]. In fact, pre-clinical studies have shown that as few as 85 mature DCs can produce adequate T-cell responses and ideally should be less than 5 × 106 cells per dose[56,57]. Several routes of administration have been tried including intravenous, intradermal, and intranodal, injections[56]. Most vaccine cells end up in kidneys, lung, and spleen when given intravenously (as assessed by radiolabeling studies) and intranodal administration does not result in major distribution of DCs to other regional lymph nodes[56]. The skin is an ideal site for vaccination due to its rich plexus of blood vessels and lymphatics that coalesce and drain into the regional inguinal lymph nodes [also called vaccine site draining lymph nodes (VDLN)]. Although only 4% of an administered intradermal dose of vaccine reaches the VDLN (the rest either degrading locally or engulfed by macrophages)[57], the number of mature DCs that migrate is still within the realm of less than 5 × 106 cells when a total of a 10 million (1 × 107) cells are given with each vaccine dose. DC function and migration can be improved with immunoadjuvants[46,47]. The ex vivo maturation of immature DCs (which lack the capacity to migrate well) with a cytokine cocktail including IL-1β, TNF-α, IFN-γ, and IL-6, results in increased expression of the CCR7 and activation of the CCR7/C-C chemokine ligand type 21 (CCL21) mediated axis of migration of the DCs to the draining lymph nodes. In addition, co-stimulatory molecules expression is improved resulting in better cross-presentation and lymphocyte expansion in the regional nodes. Our laboratory has also shown that pre-conditioning of the one vaccine site with a recall antigen such as tetanus-diphtheria toxoid followed by bilateral administration of DCs pulsed with CMV pp65 RNA in patients with malignant glioma (which express pp65 in over 90% of tested samples) results in improved DC migration and improved survival as compared to control patients who receive only the pp65 DC vaccine[46]. A parallel experiment in mice confirmed these findings in a manner dependent on the increased systemic release of the chemokine CCL3[46].
GM-CSF, a potent stimulant of the bone marrow granulocyte-monocyte progenitors, is secreted by endothelial cells, fibroblasts, and lymphocytes[58]. The cytokine can cause DC maturation on its own including expression of co-stimulatory molecules, induce CCR7 and migration towards CCL9, IL6 and TNF-α secretion and hence lymphocyte stimulation and expansion. In the context of immunotherapy, GM-CSF can be given either parenterally at a dose of 250 μg/m2/day for 5-7 days or pre-embedded in the vaccine using a significantly lower dose (75-150 μg per vaccine) to minimize possible systemic adverse reactions with parenteral administration as well as minimize mobilization of immunosuppressive myeloid-derived suppressor cells (MDSCs) from the bone marrow with higher doses[58,59]. Cellular vaccines can also be genetically modified to secrete GM-CSF in the local milieu[60]. It should be noted that GM-CSF hypersensitivity due to autoantibody formation has been reported to occur when given with a vaccine[61]. At our institution ttRNA-DC vials contain 5.7 × 106 cells in 1mL. The final ttRNA-DC product is a patient-specific single-dose syringe containing a total dose of 1 × 107 cells formulated in 400 μL of preservative free saline. The administered volume is 0.4 mL embedded with 150 μg of GM-CSF. Half the dose is administered intradermally into each thigh about 5 cm below inguinal ligament. Patients in our clinical trials receive at least three biweekly vaccines with the first dose administered along with ALT infusion.
Ex-vivo T cell expansion
The discovery in 1976 of IL-2, a T-cell growth factor, in the supernatant of phytohemagglutinin-activated human lymphocytes[62], facilitated ex-vivo expansion of autologous T-cells without loss of effector function as well as the parenteral use of this cytokine to induce T-cell proliferation in the host[3]. Using this approach, impressive responses, albeit transient, were seen with bulky tumors in patients with lymphomas and metastatic melanomas. In a search of reactive T-cells and cognate tumor antigens, it was found that certain tumors, especially melanomas, harbored reactive lymphocytes in the tumor stroma that were of both CD4+ and CD8+[63]. These cells provided a rich source of possible TILs that if harvested, expanded ex-vivo, and reinfused could provide better therapeutic benefit[63]. Hence, TILs obtained from a resected melanoma and injected into the same host resulted in objective tumor regressions[63]. However, T-cells administered following such ex-vivo TILs expansion did not survive more than a few days due to immunosuppressive factors related to the tumor[63]. This obstacle was circumvented by using host NMA lymphodepletive conditioning using cyclophosphamide and fludarabine immediately prior to TIL transfer that led to increased longevity of the infused T cells and more oligoclonal expansion directed at specific antigenic epitopes (see later under “lymphodepletion with chemotherapy”)[64]. While TIL expansion and transfer can theoretically be possible in many human tumors with T-cell infiltration, it appears to be particularly suitable in melanomas due to high mutation burden and immunogenicity, characteristics of a carcinogen - induced tumor.
While it seems attractive to employ utilize this strategy in children with brain tumors, the lack of a prominent lymphocyte infiltrate in such tumors makes this an unsuitable approach in this patient population. However, the approach in our lab involves using ttRNA DCs to “educate” and expand naïve autologous lymphocytes ex vivo and reinfuse these lymphocytes (ttRNA-xALT) into the host after lymphodepletive conditioning using either MA or NMA chemotherapy followed by hematopoietic stem cell (HSC) rescue and sustained the expansion of these “educated” lymphocytes by regular administration of ttRNA DCs. We have demonstrated the capacity to induce anti-tumor lymphocytes in vitro and in vivo using tumor RNA-pulsed DCs from a variety of human and murine tumors, including malignant brain tumors[50,52,55,65-73]. We have explored the capacity to enhance the yield of antigen-specific T cells ex-vivo through a rapid expansion protocol (REP) developed at the National Cancer Institute for expansion of melanoma-reactive tumor-infiltrating lymphocytes[74]. The REP employs irradiated allogeneic PBMCs as feeder cells, low-dose anti-CD3 monoclonal antibodies (OKT3), and IL-2 for the rapid expansion of activated lymphocytes in culture[74]. This process has the capacity to expand lymphocytes stimulated by RNA-pulsed DCs 200-500 fold and is possible to achieve greater than 1 × 1010 T-cells with an input of 1 × 108 activated lymphocytes. The addition of IL-21 and IL-7 during co-culture of T cells with RNA-pulsed DCs prior to REP leads to a greater generation of central memory antigen-reactive T cells which have been shown to be superior in anti-tumor efficacy.
Lymphodepletion with chemotherapy augments immune responses
After episodes of lymphopenia, host recovery ensues before regaining normal lymphocyte counts[75]. After profound lymphopenia[75,76], there may be an increase in cytokines (i.e., IL-7, IL-15) which can induce lymphocyte differentiation into effector memory T cells imbued with memory recall against target antigens[77]. However, under these conditions, competition for homeostatic cytokines remains a potent barrier for lymphocyte proliferation[75], which may allow antigen-specific B- or T-cells (generated through vaccination) to predominate during recovery from lymphodepletive therapy; data to support this has been shown in murine model[78,79] and in humans[80]. The predominance of these cell types may augment anti-tumor response[78,79,81], but may theoretically also precipitate autoimmunity[82,83].
Rosenberg et al.[84] used NMA lymphodepletion to sustain and augment these antigen specific memory T cell subsets against refractory malignancies, and achieved impressive responses in patients[84-88] despite autoimmunity[64,86]. Under these NMA contexts, adoptively transferred T cells proliferate intensely following lymphopenia and comprise the bulk of a T cell repertoire in a treated host, which persists for months after cell transfer[64,89]. Moreover, disease regression strongly associates with the amount and persistence of these antigen-specific T cells[84-88,90]. This anti-tumor effect can also be significantly enhanced by MA conditioning regimens coupled with autologous HSC support. Interestingly, HSCs were found to confer an enhancing effect on the in vivo expansion and persistence of tumor-specific lymphocytes transferred simultaneously with HSCs independent of the effects of lymphopenia[91]. Pursuant to these findings, Rosenberg and colleagues proceeded to evaluate MA conditioning regimens coupled with peripheral blood stem cell (PBSC) rescue in patients with malignant melanoma receiving ACT. ALT was administered in the peri-transplant period within 24 h of PBSC infusion and was feasible and safe in humans[92]. As demonstrated in murine studies, this conditioning regimen enhanced anti-tumor responses in patients with refractory metastatic disease, resulting in increased objective clinical responses from 30%-50% of patients with NMA regimens to over 70% in patients receiving MA conditioning coupled with PBSC infusion.
The mechanisms by which lymphodepletion leads to an enhancement of immune responses in humans are not well elucidated but elegant murine studies have implicated the following important processes: (1) increased in production of homeostatic cytokines such as IL-7 and IL-15 that drive lymphocyte proliferation[93]; (2) decreased competition with adoptively transferred tumor-specific lymphocytes through removal of “cytokine sinks” consisting of host lymphocytes and NK cells that decrease the bioavailability of growth factors[94]; (3) removal of CD4+CD25+Fox P3+ tregs that attenuate anti-tumor immunity[95]; (4) increased toll-like receptor agonistic signals and inflammatory cytokines through release of gut microbial antigens such as endotoxin during damage to gut endothelium by MA therapy[96]; and (5) direct enhancing effects of HSC transplant on the in vivo expansion and function of adoptively transferred lymphocytes[97].
HSCs augment immune responses during ttRNA-DC + ttRNA-xALT therapy
MA chemotherapy with HSC rescue is frequently used in children with primary brain tumors including medulloblastoma, other central primitive neuro-ectodermal tumors (PNETs), and in malignant glioma both at diagnosis as well as at recurrence[98]. The HSC rescue serves to repopulate the bone marrow and recovery following myeloablation and additionally helps in the reconstitution of the host immune system. Since MA conditioning with HSC rescue is used in our adoptive T-cell therapy protocols, we wanted to explore potential immune-modulatory effects of HSC in addition to its role in recovery from lethal bone marrow damage. It has been previously shown that HSCs can promote the expansion and function of CD8+ T-cells by secreting homeostatic cytokines IL-7 and IL-15[91]. In a pre-clinical highly invasive chemotherapy and radiotherapy resistant orthotopic glioma mouse model (KR158B glioma), administration of MA conditioning + HSC + x-ALT + ttRNA DC (× 3 vaccines) produced significantly improved survival and cures in 30% of animals as compared to tumor bearing controls (no treatment, MA conditioning alone, MA + HSC + x-ALT only, or ttRNA DC only)[99]. The x-ALT cells were syngeneic splenocytes harvested from tumor antigen-primed mice and expanded ex-vivo with ttRNA DCs in the presence of IL-2. The HSCs were found to migrate into the tumor and attract activated T-cells into the tumor. Correlative studies found that tumor elaborating C-X-C motif chemokine 12 (CXCL12) attracted the HSCs into the tumor by expressing the cognate receptor C-X-C motif chemokine receptor 4 (CXCR4). T-cells (both CD4+ and CD8+) were attracted into the tumor milieu following secretion of CCL3 by the HSCs[99]. In addition, maintenance DC vaccines were crucial in maintaining this immune response[99]. In probing the role of HSCs further in inducing immune responses, it was found that in addition to attracting effector T-cells into the tumor microenvironment, HSC infusion precipitated production of activated CD86+CD11c+MHC class II+ cells consistent with a DC phenotype in this tumor milieu and replacement of host MDSCs[100]. This was attributed to the differentiation of the HSCs into DC under the influence of T-cell secreted IFN-γ[100].
Pre-clinical and clinical studies of ttRNA DC vaccine -/+ x-ALT in primary CNS tumors
In our laboratories and those of others, systemic immunization using DCs co-cultured with uncharacterized tumor homogenate[44,101], whole tumor RNA[55], unidentified peptides eluted from tumor cells by gentle acid washing[102], or a distinct peptide encompassing the tumor-specific EGFRvIII mutation[103] have been shown to induce humoral and cell-mediated systemic immune responses and to prolong the survival of mice with intracranial brain tumors. We have used a strain of mice (VMDk) that is susceptible to experimental autoimmune encephalitis to demonstrate the safety and efficacy of ttRNA pulsed DCs in mediating potent antitumor immune responses and regression of established tumor that has prolonged survival in treated animals without causing inflammatory reactions in the CNS[65,66]. Our group has previously demonstrated the efficacy of adoptive cell therapy employing tumor-specific T cells generated from bone marrow-derived DCs pulsed with ttRNA against intracranial glioma[99].
Phase 1 study in pediatric patients with recurrent medulloblastoma and primitive neuroectodermal tumors (reMATCH trial, NCT01326104)
Using a strategy similar to adoptive T-cell therapy pioneered by Dr. Rosenberg at the National Cancer Institute in patients with metastatic melanoma, we recently completed a phase I study of ttRNA - pulsed DC vaccine + ttRNA-xALT following MA or NMA conditioning in 10 patients with recurrent PNET and GBM (medulloblastoma 8, pineoblastoma 1, and GBM 1)[104]. All patients had tumor resection/biopsy to confirm recurrence and obtain tissue for vaccine preparation. PBMCs were collected following surgical recovery for DC preparation and T-cell expansion. Patients then received either NMA conditioning with cyclophosphamide + fludarabine (n = 9) or MA conditioning with carboplatin + thiotepa + etoposide with PBSC support (n = 1) followed by xALT in one of two dose levels; 3 × 106 cells/kg (n = 3) and 3 × 107 cells/kg (n = 7). All patients received at least 3 doses of ttRNA DCs once every 2 weeks at a 1 × 107/kg per dose. The median number of vaccines given was 3 (range, 3-9). Of 8 evaluable patients for dose-limiting toxicity by receiving ttRNA x-ALT and at least one dose of ttRNA DC, there were no dose limiting toxicities associated with ttRNA DCs + ALT. Toxicities that were possibly attributable to immunotherapy included grade I rash (n = 1) and a transient grade III elevation of serum alkaline phosphatase (n = 1) 3 months after the 3rd dose of ttRNA DC. Median time to progression in 9 patients from 1st ttRNA DC + x-ALT administration was 5 months (range, 2-24) and median survival 13 months (range, 2-46+ months). In a recurrent tumor with a dismal prognosis, 5 patients survived for > 20 months following first dose of ttRNA DC + x-ALT. Three of 10 patients are currently alive; 2 patients who relapsed 12 and 24 months respectively following immunotherapy but currently alive following additional salvage therapies at 45+ and 46+ months. One additional patient with Gorlin’s syndrome and recurrent medulloblastoma is currently alive 42+ months following immunotherapy (received a total of 9 doses of ttRNA DCs) and never received radiotherapy either at diagnosis or at relapse. Measurement of inflammatory cytokines (IFN-γ, TNF, IL-6, IL-8, and IL-17A) was elevated in general following MA or NMA chemotherapy. Lymphocyte recovery occurred in all patients at variable intervals. Using next-generation sequencing (NGS) we evaluated TCR-vβ clones in the PBMC samples from all patients at baseline and at regular intervals during and post immunotherapy. Clonal diversity during recovery was higher in patients with prolonged survival (> 20 months; n = 5). Clonal hyper-expansion and persistence appeared to be higher in patients with prolonged survival and reached significance at day 7 following x-ALT administration. One patient who received MA conditioning prior to x-ALT and 3 ttRNA DCs and is a long-term survivor at 46+ months, experienced massive and selective expansion of 4 tumor-reactive TCR Vβ clones in the peripheral blood up to four months (16 weeks) post-treatment (Flores et al.[99], 2018, submitted for publication). T-cells from one of these clones was tested for anti-tumor function against patient’s ttRNA DCs. IFN-γ secretion was measured to indicate recognition of cognate tumor antigen and found to be elevated compared to control ovalbumin-RNA containing DCs. This data suggests that expansion of productive frequency of TCR Vβ family is potentially predictive of T cell clonal expansion within the larger family. Analysis of TCR Vβ family expansion in peripheral blood of treated patients could be predictive of response to adoptive immunotherapy. We have subsequently enrolled 23 subjects (screened 34) in an ongoing multi-institutional phase II study in which our institution serves as the central GMP manufacturing facility for autologous cellular products.
Ongoing phase I studies in newly diagnosed malignant glioma and diffuse pontine glioma
We have also initiated two additional upfront phase I clinical trials in children with newly diagnosed malignant glioma (ACTION trial, NCT03334305) and diffuse brain stem glioma (BRAVO trial, NCT03396575) using the ttRNA DC vaccine + x-ALT platform with some modifications from the reMATCH trials. While the general strategy in both clinical trials is similar to reMATCH, dose-intensive temozolomide (temodarTM, Merck, Kenilworth, NJ) (TMZ) is used in these two studies as both adjuvant chemotherapy post standard chemo-radiotherapy (concurrent TMZ) and as a lymphodepletive agent prior to HSC and ttRNA DC + -x-ALT and during maintenance monthly ttRNA DCs. We have introduced a few changes in these two trials including (1) obtain autologous lymphocytes after 3 bi-weekly ttRNA DCs following chemo-radiotherapy; (2) ex-vivo expansion using REP; and (3) administer up to a total of 10 ttRNA DCs.
Conclusion
The field of adoptive T-cell therapy using x-ALT + tt-RNA DCs in children with brain tumors is evolving and appears, in our preliminary experience, to have provided sustained benefit in a handful of patients with recurrent medulloblastoma without undue toxicity. It is obvious that success in a larger proportion of treated children is unlikely to equal what has been observed in adults with metastatic melanomas. This might be related to the differences in the degree of immunogenicity and mutational load seen in tumors like melanomas that is hard to match in pediatric brain tumors that appear to have an immunosuppressive microenvironment. It is intriguing to speculate whether the rare population of children with germline p53 mutations with medulloblastoma[105] or MMR deficiency malignant gliomas[30] might be a more suitable population to evaluate this therapeutic approach given the exceptionally high mutational load in these tumors compared to wild type counterparts. For most pediatric CNS tumors with an immunosuppressed landscape as previously discussed due to decreased MHC class I expression, decreased or absent TILs, high programmed cell death-1 (PD-1) or programmed cell death ligand-1 (PD-L1) expression, or increased MDSC infiltration, significant refinements need to be made to improve immune responses and outcome. It is also entirely possible that utilizing this strategy in the upfront setting in treatment-naïve patients might provide better outcomes due to lack of prior treatment related immune suppression and minimal tumor burden. Radiotherapy, typically given at diagnosis, will further reduce tumor bulk and augment immune responses through multiple mechanisms[106]. The role of the microbiome in affecting outcome following immune checkpoint inhibitor therapy has been confirmed recently in pre-clinical studies of mice bearing melanoma and non-small lung cancer tumors[107,108]. Whether such optimization of fecal microbiome in patients receiving ALT will prove beneficial remains be evaluated. Using NGS methodology and HLA-typing to improve prediction of MHC-I class and MHC-II class binding epitopes to create a robust neoantigen predominant transcriptome for electroporation into DCs[47], nanoparticle vaccines[109], and/or the use of immune checkpoint inhibitors[30,110] are other potential strategies to enhance efficacy in an adjuvant setting or relapse following x-ALT + ttRNA DCs. With these refinements and more this form of ACT promises to be an important therapeutic approach in the management of pediatric brain tumors.
Declarations
Authors’ contributionsConceived and wrote the manuscript: Gururangan S
Helped with critical review and revisions: Sayour E, Mitchell DA
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Centers for Disease Control and Prevention. Available from:
2. Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol 2017;19:v1-88.
3. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 2008;8:299-308.
5. Jackson CM, Lim M, Drake CG. Immunotherapy for brain cancer: recent progress and future promise. Clin Cancer Res 2014;20:3651-9.
7. Vivier E, Malissen B. Innate and adaptive immunity: specificities and signaling hierarchies revisited. Nat Immunol 2005;6:17-21.
9. Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol 2004;5:971-4.
10. Palucka K, Banchereau J. Human dendritic cell subsets in vaccination. Curr Opin Immunol 2013;25:396-402.
11. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012;12:265-77.
13. Steinman RM, Pack M, Inaba K. Dendritic cell development and maturation. Adv Exp Med Biol 1997;417:1-6.
14. Maraskovsky E, Daro E, Roux E, Teepe M, Maliszewski CR, et al. In vivo generation of human dendritic cell subsets by Flt3 ligand. Blood 2000;96:878-84.
15. Shastri N, Cardinaud S, Schwab SR, Serwold T, Kunisawa J. All the peptides that fit: the beginning, the middle, and the end of the MHC class I antigen-processing pathway. Immunol Rev 2005;207:31-41.
16. Steinman RM, Dhodapkar M. Active immunization against cancer with dendritic cells: the near future. Int J Cancer 2001;94:459-73.
17. Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol 2001;19:47-64.
18. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996;14:233-58.
19. Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol 2003;3:939-51.
20. Chen L, Ashe S, Brady WA, Hellström I, Hellström KE, et al. Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell 1992;71:1093-102.
21. Shahinian A, Pfeffer K, Lee KP, Kündig TM, Kishihara K, et al. Differential T cell costimulatory requirements in CD28-deficient mice. Science 1993;261:609-12.
22. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol 2011;11:852-63.
23. Apetoh L, Smyth MJ, Drake CG, Abastado JP, Apte RN, et al. Consensus nomenclature for CD8+ T cell phenotypes in cancer. Oncoimmunology 2015;4:e998538.
24. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015;28:690-714.
25. Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, et al. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res 2011;71:4809-20.
26. Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol 2002;2:251-62.
27. Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev 2006;211:214-24.
28. Rosati E, Dowds CM, Liaskou E, Henriksen EKK, Karlsen TH, et al. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol 2017;17:61.
29. Mack SC, Northcott PA. Genomic analysis of childhood brain tumors: methods for genome-wide discovery and precision medicine become mainstream. J Clin Oncol 2017;35:2346-54.
30. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 2016;34:2206-11.
31. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321-7.
32. Mackay A, Burford A, Molinari V, Jones DTW, Izquierdo E, et al. Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade glioma from the HERBY phase II randomized trial. Cancer Cell 2018;33:829-42.
33. Yang I, Han SJ, Sughrue ME, Tihan T, Parsa AT. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: evidence of distinct immunological microenvironments that reflect tumor biology. J Neurosurg 2011;115:505-11.
34. Mehling M, Simon P, Mittelbronn M, Meyermann R, Ferrone S, et al. WHO grade associated downregulation of MHC class I antigen-processing machinery components in human astrocytomas: does it reflect a potential immune escape mechanism? Acta Neuropathol 2007;114:111-9.
35. Vermeulen JF, Van Hecke W, Adriaansen EJM, Jansen MK, Bouma RG, et al. Prognostic relevance of tumor-infiltrating lymphocytes and immune checkpoints in pediatric medulloblastoma. Oncoimmunology 2017;7:e1398877.
36. Smith C, Santi M, Rushing EJ, Cornelison R, MacDonald TJ, et al. Characterization of signaling function and expression of HLA class I molecules in medulloblastoma. J Neurooncol 2011;103:197-206.
37. Raffaghello L, Nozza P, Morandi F, Camoriano M, Wang X, et al. Expression and functional analysis of human leukocyte antigen class I antigen-processing machinery in medulloblastoma. Cancer Res 2007;67:5471-8.
38. Thompson YY, Ramaswamy V, Diamandis P, Daniels C, Taylor MD. Posterior fossa ependymoma: current insights. Childs Nerv Syst 2015;31:1699-706.
39. Mack SC, Pajtler KW, Chavez L, Okonechnikov K, Bertrand KC, et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2018;553:101-5.
40. Griesinger AM, Birks DK, Donson AM, Amani V, Hoffman LM, et al. Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol 2013;191:4880-8.
41. Lin GL, Nagaraja S, Filbin MG, Suvà ML, Vogel H, et al. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol Commun 2018;6:51.
42. McNamara MA, Nair SK, Holl EK. RNA-based vaccines in cancer immunotherapy. J Immunol Res 2015;2015:794528.
43. Holl EK, Brown MC, Boczkowski D, McNamara MA, George DJ, et al. Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models. Oncotarget 2016;7:79828-41.
44. Benitez-Ribas D, Cabezón R, Flórez-Grau G, Molero MC, Puerta P, et al. Immune response generated with the administration of autologous dendritic cells pulsed with an allogenic tumoral cell-lines lysate in patients with newly diagnosed diffuse intrinsic pontine glioma. Front Oncol 2018;8:127.
45. Mitchell D, Archer G, Bigner D, Friedman A, Friedman H, et al. RNA-loaded dendritic cells targeting cytomegalovirus in patients with malignant glioma. Neuro Oncol 2007;9:509.
46. Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015;519:366-9.
47. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol 2018;18:168-82.
48. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol 2011;13:324-33.
50. Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E. Induction of tumor immunity and cytotoxic T lymphocyte responses using dendritic cells transfected with messenger RNA amplified from tumor cells. Cancer Research 2000;60:1028-34.
51. Heiser A, Maurice MA, Yancey DR, Wu NZ, Dahm P, et al. Induction of polyclonal prostate cancer-specific CTL using dendritic cells transfected with amplified tumor RNA. J Immunol 2001;166:2953-60.
52. Nair SK, Morse M, Boczkowski D, Cumming RI, Vasovic L, et al. Induction of tumor-specific cytotoxic T lymphocytes in cancer patients by autologous tumor RNA-transfected dendritic cells. Ann Surg 2002;235:540-9.
53. Learn CA, Grossi PM, Schmittling RJ, Xie W, Mitchell DA, et al. Genetic analysis of intracranial tumors in a murine model of glioma demonstrate a shift in gene expression in response to host immunity. J Neuroimmunol 2007;182:63-72.
54. Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest 2005;115:3623-33.
55. Ashley DM, Faiola B, Nair S, Hale LP, Bigner DD, et al. Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J Exp Med 1997;186:1177-82.
56. Bryant CE, Sutherland S, Kong B, Papadimitrious MS, Fromm PD, et al. Dendritic cells as cancer therapeutics. Semin Cell Dev Biol 2018; doi: 10.1016/j.semcdb.2018.02.015.
57. Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res 2009;15:2531-40.
58. Shi Y, Liu CH, Roberts AI, Das J, Xu G, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res 2006;16:126-33.
59. Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, et al. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol 2007;18:226-32.
60. Gupta R, Emens LA. GM-CSF-secreting vaccines for solid tumors: moving forward. Discov Med 2010;10:52-60.
61. Mitchell DA, Sayour EJ, Reap E, Schmittling R, DeLeon G, et al. Severe adverse immunologic reaction in a patient with glioblastoma receiving autologous dendritic cell vaccines combined with GM-CSF and dose-intensified temozolomide. Cancer Immunol Res 2015;3:320-5.
62. Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 1976;193:1007-8.
63. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 2012;12:269-81.
64. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002;298:850-4.
65. Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin Cancer Res 2006;12:4294-305.
66. Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res 2007;13:2158-67.
67. Mitchell DA, Fecci PE, Sampson JH. Immunotherapy of malignant brain tumors. Immunol Rev 2008;222:70-100.
68. Hess PR, Boczkowski D, Nair SK, Snyder D, Gilboa E. Vaccination with mRNAs encoding tumor-associated antigens and granulocyte-macrophage colony-stimulating factor efficiently primes CTL responses, but is insufficient to overcome tolerance to a model tumor/self antigen. Cancer Immunol Immunother 2006;55:672-83.
69. Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med 1996;184:465-72.
70. Nair SK, Boczkowski D, Morse M, Cumming RI, Lyerly HK, et al. Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfected with RNA. NatBiotechnol 1998;16:364-9.
71. Nair SK, Heiser A, Boczkowski D, Majumdar A, Naoe M, et al. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat Med 2000;6:1011-7.
72. Nair SK, Hull S, Coleman D, Gilboa E, Lyerly HK, et al. Induction of carcinoembryonic antigen (CEA)-specific cytotoxic T-lymphocyte responses in vitro using autologous dendritic cells loaded with CEA peptide or CEA RNA in patients with metastatic malignancies expressing CEA. Int J Cancer 1999;82:121-4.
73. Thornburg C, Boczkowski D, Gilboa E, Nair SK. Induction of cytotoxic T lymphocytes with dendritic cells transfected with human papillomavirus E6 and E7 RNA: implications for cervical cancer immunotherapy. J Immunother 2000;23:412-8.
74. Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 2003;26:332-42.
75. Tanchot C, Rosado MM, Agenes F, Freitas AA, Rocha B. Lymphocyte homeostasis. Semin Immunol 1997;9:331-7.
76. Grossman Z, Paul WE. Self-tolerance: context dependent tuning of T cell antigen recognition. Semin Immunol 2000;12:197-203.
77. Cho BK, Rao VP, Ge Q, Eisen HN, Chen J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med 2000;192:549-56.
78. Dummer W, Niethammer AG, Baccala R, Lawson BR, Wagner N, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest 2002;110:185-92.
79. Asavaroengchai W, Kotera Y, Mulé JJ. Tumor lysate-pulsed dendritic cells can elicit an effective antitumor immune response during early lymphoid recovery. Proc Natl Acad Sci U S A 2002;99:931-6.
80. Rapoport AP, Stadtmauer EA, Aqui N, Badros A, Cotte J, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med 2005;11:1230-7.
81. Hu HM, Poehlein CH, Urba WJ, Fox BA. Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res 2002;62:3914-9.
82. Khoruts A, Fraser JM. A causal link between lymphopenia and autoimmunity. Immunol Lett 2005;98:23-31.
83. Krupica T Jr, Fry TJ, Mackall CL. Autoimmunity during lymphopenia: a two-hit model. Clin Immunol 2006;120:121-8.
84. Rosenberg SA. Development of effective immunotherapy for the treatment of patients with cancer. J Am Coll Surg 2004;198:685-96.
85. Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol 2004;173:7125-30.
86. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 2005;23:2346-57.
87. Zhou J, Dudley ME, Rosenberg SA, Robbins PF. Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J Immunother 2005;28:53-62.
88. Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, et al. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol 2005;175:7046-52.
89. Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer 2003;3:666-75.
90. Restifo NP, Rosenberg SA. Use of standard criteria for assessment of cancer vaccines. Lancet Oncol 2005;6:3-4.
91. Wrzesinski C, Paulos CM, Gattinoni L, Palmer DC, Kaiser A, et al. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J Clin Invest 2007;117:492-501.
92. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol 2008;26:5233-9.
93. Wang LX, Li R, Yang G, Lim M, O’Hara A, et al. Interleukin-7-dependent expansion and persistence of melanoma-specific T cells in lymphodepleted mice lead to tumor regression and editing. Cancer Res 2005;65:10569-77.
94. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 2005;202:907-12.
95. Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol 2005;26:111-7.
96. Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest 2007;117:2197-204.
97. Wrzesinski C, Restifo NP. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumor immunotherapy. Curr Opin Immunol 2005;17:195-201.
98. Finlay JL. The role of high-dose chemotherapy and stem cell rescue in the treatment of malignant brain tumors: a reappraisal. Pediatr Transplant 1999;3:87-95.
99. Flores C, Pham C, Snyder D, Yang S, Sanchez-Perez L, et al. Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology 2015;4:e994374.
100. Wildes TJ, Grippin A, Dyson KA, Wummer BM, Damiani DJ, et al. Cross-talk between T cells and hematopoietic stem cells during adoptive cellular therapy for malignant glioma. Clin Cancer Res 2018;24:3955-66.
101. Heimberger AB, Crotty LE, Archer GE, McLendon RE, Friedman A, et al. Bone marrow-derived dendritic cells pulsed with tumor homogenate induce immunity against syngeneic intracerebral glioma. J Neuroimmunol 2000;103:16-25.
102. Liau LM, Black KL, Prins RM, Sykes SN, DiPatre PL, et al. Treatment of intracranial gliomas with bone marrow-derived dendritic cells pulsed with tumor antigens. J Neurosurg 1999;90:1115-24.
103. Heimberger AB, Archer GE, Crotty LE, McLendon RE, Friedman AH, et al. Dendritic cells pulsed with a tumor-specific peptide induce long-lasting immunity and are effective against murine intracerebral melanoma. Neurosurgery 2002;50:158-64.
104. Gururangan S, Grant G, Driscoll T, Archer G, Herndon J, et al. IMMU-27. Re-MATCH protocol: phase I study of autologous tumor specific lymphocyte transfer (ALT) + DC vaccine (DCV) during recovery from myeloablative chemotherapy (MAC) and autologous stem cell rescue (HDC + ASCR) or non-myeloablative chemotherapy (NMAC) in patients with recurrent central PNETs (r-PNETs). Neuro Oncol 2018;20:i104.
105. Rausch T, Jones DT, Zapatka M, Stütz AM, Zichner T, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012;148:59-71.
106. Ishihara D, Pop L, Takeshima T, Iyengar P, Hannan R. Rationale and evidence to combine radiation therapy and immunotherapy for cancer treatment. Cancer Immunol Immunother 2017;66:281-98.
107. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015;350:1084-9.
108. Pitt JM, Vétizou M, Gomperts Boneca I, Lepage P, Chamaillard M, et al. Enhancing the clinical coverage and anticancer efficacy of immune checkpoint blockade through manipulation of the gut microbiota. Oncoimmunology 2016;6:e1132137.
109. Sayour EJ, De Leon G, Pham C, Grippin A, Kemeny H, et al. Systemic activation of antigen-presenting cells via RNA-loaded nanoparticles. Oncoimmunology 2016;6:e1256527.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Gururangan S, Sayour E, Mitchell DA. Total tumor RNA pulsed dendritic cells plus adoptive transfer of ex-vivo enriched autologous T-lymphocytes in the treatment of children with primary brain tumors. Neurosciences 2018;5:45. http://dx.doi.org/10.20517/2347-8659.2018.44
AMA Style
Gururangan S, Sayour E, Mitchell DA. Total tumor RNA pulsed dendritic cells plus adoptive transfer of ex-vivo enriched autologous T-lymphocytes in the treatment of children with primary brain tumors. Neuroimmunology and Neuroinflammation. 2018; 5: 45. http://dx.doi.org/10.20517/2347-8659.2018.44
Chicago/Turabian Style
Gururangan, Sridharan, Elias Sayour, Duane A. Mitchell. 2018. "Total tumor RNA pulsed dendritic cells plus adoptive transfer of ex-vivo enriched autologous T-lymphocytes in the treatment of children with primary brain tumors" Neuroimmunology and Neuroinflammation. 5: 45. http://dx.doi.org/10.20517/2347-8659.2018.44
ACS Style
Gururangan, S.; Sayour E.; Mitchell DA. Total tumor RNA pulsed dendritic cells plus adoptive transfer of ex-vivo enriched autologous T-lymphocytes in the treatment of children with primary brain tumors. Neurosciences. 2018, 5, 45. http://dx.doi.org/10.20517/2347-8659.2018.44
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.