
Favorable and unfavorable roles of microglia and macrophages in the pathologic central nervous system

Abstract
Resident microglia in the central nervous system (CNS) are activated rapidly in response to even minor pathologic changes in the CNS, releasing various cytokines, growth factors, reactive oxygen species and other bioactive substances, in addition to eliminating synapses and degenerating cells through phagocytosis. Monocytes in circulation invade the inflamed brain tissues and develop into macrophages that also produce several bioactive substances and engage in phagocytosis. This article introduces methods for distinguishing microglia and macrophages. The pathophysiological roles of resident microglia and macrophages are discussed in animal models with neuroinflammation in the brain either with or without disruption of the blood-brain barrier. Both cell types have ameliorating and aggravating effects on the pathologic CNS, and their different roles are addressed in this article. Furthermore, this article compares the effects of some pharmacological interventions to induce phenotypic cellular changes for improved outcomes of the pathologic CNS.
Keywords
Introduction
Microglia and blood-borne macrophages play major roles in the pathophysiological processes in various kinds of pathologies of the central nervous system (CNS) by releasing numerous bioactive substances, including cytokines, growth factors, and reactive oxygen/nitrogen species, and phagocytosing degenerating cells and materials[1-4]. In this article, we use “microglia” to denote resident microglia in the CNS, and “macrophages” to denote cells derived from circulating monocytes that have invaded (typically inflamed) CNS lesions with a disrupted blood-brain barrier (BBB). The two types of mesoderm-derived cells share many kinds of characteristics and surface antigens[5,6]. In particular, when microglia become activated in response to severe pathologic events, such as stroke or traumatic brain injury (TBI), which accompany BBB breakdown, both cell types resemble each other in terms of morphology, functions, and cell marker expression. The term “amoeboid microglia”[7] has long been used to denote extremely activated microglia displaying almost the same morphology as brain macrophages in severe lesions. Therefore, it is generally difficult to distinguish one from the other. This assumption originated from the historical observation by Rio-Hortega[8], who reported that ramified microglia in the normal mature brain can, in severely damaged brains, turn into phagocytes exhibiting spherical shapes of the same morphology as that of blood-borne macrophages[5]. Therefore, numerous studies have described CD11b-expresssing (in the case of the rat brain, a monoclonal antibody OX-42-immunoreactive) spherical cells in severely injured brains in the acute phase as activated microglia, although most of them should be recognized as invading neutrophils[9,10].
More recently, it is well-known that both cell types play major roles in pathophysiological processes[2,11,12]. The neuroinflammatory processes influence outcomes of CNS diseases or injuries in both favorable and unfavorable ways. In this review, “favorable” is used to describe microglia and macrophages that are neuroprotective cells bringing about the better outcome in the pathologic CNS than “unfavorable” ones that exert deleterious effects on the survival of neurons and other parenchymal cells. Therefore, various kinds of interventions, including pharmacological treatment and rehabilitation, have been studied in laboratory and clinical settings to determine if they enhance favorable responses while also suppressing the deleterious effects of microglia and macrophages[1,4,13,14]. This aim may be interpreted as attempting to induce M2-polarized or alternatively activated phenotypes of these cells[15-17]. However, phenotypes of activated microglia and macrophages cannot be classified clearly into M1-polarized or M2-polarized cells[18,19].
Diseases and injuries of the CNS can be categorized by the absence of BBB disruption[20,21]. In the absence of BBB disruption, the infiltration of circulating leukocytes is limited, and microglia play a central role as immune cells. When the BBB is disrupted, infiltrating leukocytes play much more significant roles[9,22]. In particular, invading monocytes increase hugely in number because of their strong proliferative nature and end up occupying almost the entire area of the lesion in either stroke or traumatic injury[11,12]. Marked accumulation of blood-borne macrophages is seen in malignant brain tumor masses, which are termed tumor-associated macrophages[23,24]. Tumor-associated macrophages contribute to tumor growth and angiogenesis by secreting many kinds of growth factors, including vascular endothelial growth factor. Tumor-associated macrophages are not discussed in this article.
Thus, microglia and macrophages are the critical cells in CNS diseases and injuries and have a profound impact on patient outcomes. Therefore, many studies have investigated interventions to both suppress the unfavorable effects of microglia and macrophages and induce their favorable functions. This article also deals with several interventions targeting microglia and macrophages. There are three types of resident macrophages that are distinct from microglia and the invaded blood-borne macrophages. Those are the meningeal, perivascular and choroid plexus macrophages. Although they play significant roles in health and pathology, they are not discussed in this review.
Distinguishing between microglia and macrophages
Discriminating microglia from macrophages has been considered to be very difficult. This is partly due to the old notion that microglia in the brain are derived from circulating monocytes[7,25]. Moreover, microglia have long been thought to be able to display almost completely spherical or amoeboid morphology when they become fully activated in the core of severe brain insults such as stroke or trauma[5]. Both cell types share numerous immune cell markers[26]. Therefore, distinguishing between them has been very problematic. However, many methods have been now established for this purpose.
Morphological characteristics
Despite the historical view, even highly activated microglia do not display spherical morphology[9,27]. In a rat stroke model prepared by transient occlusion of middle cerebral artery (tMCAO), activated microglia in the peri-ischemic regions exhibit enlarged somata and shortened processes that are distinct from spikes. The processes can be identified with immunoreactivity to microglia/macrophage markers such as Iba1 or CD11b. Conversely, blood-borne macrophages and neutrophils, the latter of which infiltrate in abundance, do not have processes even though they may have a polygonal shape and short spikes[27]. Thus, microglia can be distinguished from blood-borne cells via morphological observation[10]. This may be the simplest method for specific identification of microglia.
Specific markers to distinguish the two cell types in the pathologic brains
Iba1, CD11b, CD45, and CD68 have long been used to identify resident microglia; however, these markers are more strongly expressed by infiltrating macrophages than by activated microglia[9]. As CD11b is a marker for myeloid cells and CD45 is a marker for all of the leukocytes, they are not suitable for identifying microglia in the pathologic CNS. To examine specific roles of resident microglia in CNS pathology, they must be distinguished from blood-borne cells that are macrophages, lymphocytes, and neutrophils. Thus, considerable effort has been dedicated to finding microglia-specific markers that are not expressed by macrophages and other blood-borne cells[28]. Comprehensive gene analyses using RNAseq and/or microarray analyses have identified genes that are expressed predominantly by microglia rather than by macrophages such as Cx3cr1, Gpr34, P2ry12, P2ry13, Siglech, Tmem119, and Trem2[29-32]. Of the specific marker candidates, TMEM119 and Siglec-H may be the most promising for immunohistochemical discrimination of microglia from macrophages.
Transmembrane protein 119, commonly known as TMEM119, was identified by Bennet et al.[33] Specific expression of TMEM119 by microglia has been demonstrated by immunohistochemical staining, flow cytometry analyses, and in situ hybridization[33,34]. TMEM119 was identified originally as a type I transmembrane protein expressed by murine osteoblasts and is responsible for their differentiation[35]. It is not expressed by microglia in immature murine brains, but its expression increases along with their development or ramification[33]. In aged human brains either with or without Alzheimer’s disease (AD), microglia expressed TMEM119[34]. Activated microglia with enlarged somata in close proximity to amyloid plaques are less immunoreactive to TMEM119 antibody than are resting (or homeostatic) microglia. Moreover, TMEM119-expressing microglia in AD brains either do not or only weakly express the polarized markers CD80, CD163, or CD206. In a TBI model, ramified or homeostatic microglia express TMEM119 at higher levels than do activated microglia[10]. Therefore, TMEM119 is particularly suitable for identifying homeostatic microglia with a ramified shape in both the normal and the injured mature brain.
Sialic-acid-binding immunoglobulin-like lectin-H (Siglec-H) is another promising marker for immunohistochemical discrimination of microglia from macrophages[36]. Siglec-H is a single-pass transmembrane protein that was identified originally as a member of a CD33-related Siglec family. Siglec-H is barely expressed by circulating monocytes and their derived macrophages. In contrast to TMEM119, Siglec-H is expressed continually by activated microglia and by microglia in immature brains. Siglec-H may mediate signals necessary for phagocytosis by microglia[37].
Bone marrow transplantation
Bone marrow transplantation (BMT) has long been a reliable method for identifying blood-borne cells in the CNS with BBB breakdown[28]. After ~10 Gy irradiation to cause near-total death of the host’s bone marrow, the bone marrow from transgenic animals that ubiquitously express fluorescent proteins such as enhanced green fluorescent protein is transplanted[12]. In the transplanted brain, blood-borne macrophages bear fluorescence but not resident microglia[27]. However, BMT usually leads to partial chimera, which can make it difficult to analyze the results. With reconstruction of the bone marrow, it is a long time before the anemia disappears. Radiation may cause degeneration of neural cells, such as NG2 glia and neurons, which may affect the results[38]. As radiation disrupts the BBB while increasing monocyte infiltration, BMT produces donor-derived microglia or microglia-like ramified cells in the brain parenchyma[28]. Thus, the BMT may provide firm evidence showing the presence of blood-borne cells in the CNS, but it should be noted that BMT itself will significantly change the brain functions due to the toxic effects on the neural cells.
Flow cytometry
Microglial cells belong to a group of myeloid leukocytes and express a macrophage colony-stimulating factor receptor known as CSF-1R. Therefore, microglia express a myeloid cell marker CD11b and a pan-leukocyte marker CD45. Based on this finding, microglia have been analyzed by flow cytometry using antibodies to CD11b and CD45[10,39]. Because of faint expression of CD45 by homeostatic microglia, immunohistochemical detection of this expression tends to be difficult. Conversely, macrophages and neutrophils express CD45 rather strongly[10]. However, weak CD45 expression can be detected easily by flow cytometry and is an advantage in flow cytometry analysis; microglia can be defined as CD11b+/CD45lo and macrophages as CD11b+/CD45hi. Even activated microglia express at lower levels of CD45 than do macrophages. Moreover, activated and homeostatic microglia can be distinguished by flow cytometry based on forward and side scatter value; activated microglia have larger somata (larger forward scatter values) and more intracellular organelles (larger side scatter values) than do homeostatic microglia[10]. Flow cytometry analyses can be used to isolate the cells by cell sorting[10,39]. If the cells are treated appropriately to prevent degradation of proteins, RNA, or DNA, the sorted cells can be used for either Western blotting or PCR.
Flow cytometry analyses of either cultured microglia or circulating monocytes are simple[40,41]. Nevertheless, analyses of microglia and macrophages in brain tissues have been difficult because of difficulties in dissociating the tissues into single cells. However, dissociation kits and apparatus are now available for preparing neural cell suspensions and are designed appropriately for dissociation of rodent and human brains[10,39]. The sorted cells can also be used for culturing and/or functional analyses.
Response of microglia in brain pathology in the absence of BBB breakdown
Microglia become activated in response to even minor pathological events that do not involve BBB disruption. This section discusses microglia in Parkinson’s disease (PD), peripheral nerve injury, and Carbon monoxide (CO) intoxication. Minute activation of homeostatic microglia accompanying circadian changes can be observed in the normal mature brain; microglia exhibit weakly activated phenotypes around the time of onset of sleep[39].
PD
PD is the second most frequent neurodegenerative disorder after AD. As BBB breakdown is not apparent in PD, infiltration of leukocytes, including monocytes, is not often seen. In PD pathophysiology, dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc) in the mesencephalon primarily undergo degeneration, leading to microglial activation in the vicinity of the degenerating neurons[42-44]. The activated microglia release potentially neurotoxic substances, such as either reactive oxygen/nitrogen species or glutamate[45,46]. The microglia-derived proinflammatory cytokines and chemokines may also contribute to aggravation. Injection of 6-hydorxydopamine (6-OHDA) into the striatum or medial forebrain bundle is used to prepare the PD rat model[47,48]. In the model, DA neurons in the SNc primarily undergo degeneration, and microglia then become activated in response to damage-associated molecular patterns (DAMPs), such as high mobility group box-1 protein (HMGB1) released from damaged neurons[49]. The activated microglia may exacerbate the degenerative processes of DA neurons. Another rat PD model is prepared via injection of lipopolysaccharide (LPS) either direct into the SNc or in its vicinity[48]. In this model, microglial activation is primarily induced followed by DA neuron degeneration, suggesting that activated microglia could be a key cause of neuronal degeneration.
The hematopoietic cytokines interleukin-3 (IL-3) and granulocyte/macrophage colony-stimulating factor (GM-CSF) have been shown to modulate the phenotype of microglia in the SNc while suppressing proinflammatory nature and increasing secretion of neurotrophic factors, insulin-like growth factor 1 (IGF-1), and hepatocyte growth factor (HGF)[44]. Following this, DA neurons increase the expression of anti-apoptotic factor Bcl-xL, and the symptoms of PD are ameliorated. This suggests that further investigation of microglial phenotypes would lead to a potential intervention for neurological disorders.
Microglia in the SNc have been the sole focus of studies investigating microglia in PD pathology. However, upon immunostaining sections of the mesencephalon of PD model rats with antibodies to microglia markers, activation of microglia in the substantia nigra pars reticulata (SNr) was more apparent than that of those in the SNc[47]. The activated microglia in the SNr bore large CD68+ phagosomes in their cytoplasm, in which synaptic proteins were included. In the PD pathology, glutamatergic neurons in the subthalamic nuclei (STN) become hyperactivated and release excess amounts of glutamate in the basal ganglia outputs that are the SNr and the internal segment of the globus pallidus (GPi). The change causes bradykinesia, rigidity, and other PD symptoms. Activated microglia with large phagosomes are present not only in the SNr but also in the GPi. They internalize the glutamatergic synapses from the hyperactive STN. As the neurological deficits do not manifest until most DA neurons are lost, it is likely that there are some significant compensatory mechanisms that prevent the symptoms from appearing[50]. Microglia should contribute to this compensation by eliminating hyperactive glutamatergic synapses. Administration of a single high dose of a synthetic glucocorticoid dexamethasone (Dex) to the PD model rats aggravated their motor deficits. Dex suppresses CD68 expression in the SNr and GPi, suggesting suppression of microglial phagocytosis.
Conversely, chronic administration of glucocorticoid[51,52] and other anti-inflammatory agents, such as bromovalerylurea (BU)[43], has been shown in laboratory settings to ameliorate the outcome of the motor deficits, likely because of suppression of proinflammatory activation of microglia in the SNc. These findings reveal that microglia play both ameliorative and detrimental roles. There appear to be two forms of microglia activation: one is characterized by the production of proinflammatory mediators found in the SNc[43,44], and the other is characterized by enhanced phagocytic ability in the SNr and GPi[47]. The dual role of microglia in the PD pathophysiology is summarized in a schematic diagram [Figure 1].
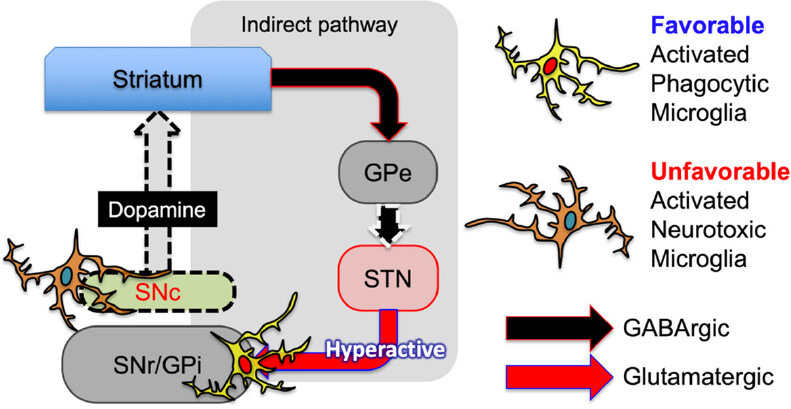
Figure 1. Both favorable and unfavorable activated microglia participate in the pathophysiology of PD. Unfavorable microglia present in the SNc release neurotoxic proinflammatory mediators while accelerating the degeneration of DA neurons. Favorable microglia present in the SNr and GPi engage in eliminating hyperactive glutamatergic synapses from the STN in the indirect pathway of the basal ganglia. This figure is based primarily on the study by Aono et al.[47]. PD: Parkinson’s disease; SNc: substantia nigra pars compacta; DA: dopaminergic; SNr: substantia nigra pars reticulata; GPi: globus pallidus; STN: subthalamic nuclei; GPe: globus pallidus pars externa
Despite significant evidence showing the involvement of microglia in the DA neuron loss in animal PD models, it is not clear whether microglia actually affect the pathology of human PD cases. A few clinical trials have shown the positive effects of antiinflammatory drugs[53], and activated microglia are found in PD patients’ brains by in vivo imaging with positron emission tomography[54]. However, there is still no firm evidence demonstrating that microglia actually induce DA neuron death in human PD cases. Most anti-inflammatory interventions are not ameliorative[55,56]. This contrasts with animal model cases, in which anti-inflammatory interventions markedly suppress DA neuron death. The discrepancy between animal PD models and human cases may be partially attributable to the speed of the pathological processes. The animal model is acutely prepared, whereas human PD is a chronic disease. Furthermore, when PD is diagnosed based on motor symptoms, more than 60 % of DA neurons in the SNc are degenerated. In animal models, anti-inflammatory drugs are often given simultaneously with or even before administration of neurotoxic substances causing DA neuron loss. Even if anti-inflammatory treatments could delay or prevent progressive neuronal loss in PD pathology, it is a prerequisite for the treatments that diagnosis is made at much earlier time points when most DA neurons in the SNc are still viable.
Peripheral nerve injury
Peripheral nerve injuries induce activation of microglia in close proximity to damaged neurons. In particular, facial nerve transection has been used frequently to observe the response of microglia[57]. Microglia become activated and proliferate while attaching intimately to the axotomized neurons. Direct contact with neurons may be one of the direct causes of the microglial activation[58]. The activated microglia detach afferent axonal endings through “synaptic stripping”[57,59]. In the axotomy model, the activated microglia may be neuroprotective through releasing a plethora of neuroprotective factors[60]. Axotomy and spinal cord ischemia both caused similar changes of microglia around the alpha motoneurons in the anterior horn[61]. Synapses surrounding the neurons disappeared when activated microglia attached intimately to damaged neurons.
Constriction injury of the sciatic nerve is another model that is used often to study the responses of microglia in the spinal cord. This model is well known to cause chronic neuropathic pain[62]. Microglia exhibit activated morphology in the posterior horn, and they phagocytose myelin elements[63]. Constriction injury-induced hyperalgesia and the activation of microglia in the posterior horn are sustained chronically. Microglia in the anterior horn also become activated while surrounding damaged motoneurons and remove afferent synapses, as do microglia do in the facial nerve axotomy model. Different from the chronic sensory impairment, Constriction injury-induced motor deficits become ameliorated quite rapidly, indicating that the activated microglia in the anterior horn may be neuroprotective. Thus, microglia could become either protective/favorable or destructive/unfavorable cells.
CO intoxication
CO intoxication causes serious adverse effects in brain functions that are known as delayed encephalopathy. Compared with hypoxia-induced disorder, CO intoxication causes more severe memory impairment and more aggravated degeneration of neuronal cells in the hippocampus[64]. Moreover, CO intoxication causes damage of oligodendrocytes, myelin and NG2 glia. Notably, CO intoxication induces profound loss of microglia. Expression of neurotrophic factors, such as IGF-1, HGF, platelet-derived growth factor, and basic fibroblast growth factor (bFGF) is reduced considerably, whereas expression of pro-apoptotic factors, such as Bid, Bad, and Bax, is increased, leading to marked loss of the hippocampal neurons. These findings indicate that microglia contribute significantly to supporting neuronal survival.
Response of microglia and macrophages in the brain with ischemic and traumatic injuries that cause BBB breakdown
Severe damage to brains and spinal cords disrupts the BBB, resulting in massive infiltration of blood-borne cells, such as monocytes and neutrophils[9,22]. Discrimination of the roles of these immune cell types in pathophysiological processes is becoming possible, although further study is necessary[10]. Different responses of microglia and macrophages are seen in the ischemic lesion core and the ischemic penumbra or peri-ischemic region[11,27]. It should be noted that microglia are very vulnerable to various kinds of severe brain insults, including CO intoxication, as mentioned above[64]. Six hours after reperfusion in tMCAO (90 min-occlusion) in a rat model of severe stroke, microglia undergo apoptotic degeneration in the lesion core, whereas neurons appear unchanged[9,11]. Therefore, blood-borne macrophages and neutrophils are the main immune cells in the lesion cores and during the acute phase. However, microglia are both viable and become activated in the peri-region neighboring the core of the lesion and modulate pathological processes.
Roles of microglia
The activated microglia in the peri-ischemic regions bear large phagosomes that can be recognized by immunohistochemical staining of CD68[27]. Moreover, they frequently express NG2 chondroitin sulfate proteoglycan (NG2), which may be another marker for phagocytosing microglia and macrophages[27]. CD68+-phagocytosing microglia in the SNr and GPi in the rat PD model brains also express NG2[47]. Such NG2+/CD68+ microglia engage in phagocytosis of degenerating neurons in the very limited region located along the border zone delineating the ischemic core and the penumbra[27]. The region is termed the demarcation zone[65] and is characterized by high expression of NG2. Neurodegeneration, known as delayed neuronal death, is still progressing in the zone in the subacute phase, which has long been a therapeutic target in stroke research to ameliorate the outcome of stroke.
Microglia are assumed to eliminate still viable neurons via phagocytosis in the ischemic penumbra[66,67]. Because of decreased blood flow, neurons reduce their ATP synthesis and, thus, frequently externalize phosphatidylserine (PS) on their surface. PS is a typical eat-me-signal molecule that is recognized by molecules, such as either Milk fat globule EGF-like factor 8 (MFG-E8) or protein S, which are, in turn, recognized by either vitronectin receptor or Mer receptor tyrosine kinase (MerTK) expressed by microglia. Expression of MFG-E8 and MerTK is enhanced either in activated microglia or in macrophages in the ischemic lesions. Knocking out expression of MerTK or MFG-E8 by microglia prevented the delayed neuronal loss considerably. These findings may indicate that CD68+/NG2+-phagocytosing microglia are aggravating cells in stroke pathology.
Administration of the CSF1R antagonist PLX3397 to mice depletes microglia. This pharmacological intervention was used to study the overall effects of microglia on the outcome of ischemic brain insults. Eliminating microglia increases infarct volume, indicating that the overall effects of microglia on the ischemic brain are ameliorative. Microglia in the non-ischemic regions may maintain the neuronal circuitry, suppress proinflammatory activation of astrocytes, and prevent infiltration by various leukocytes, such as T cells, monocytes, and granulocytes[68,69].
Activated microglia in the ischemic brain release a considerable quantity of transforming growth factor β1 (TGFβ1)[27], which is a strong immunosuppressive cytokine. Ischemic brain lesions should contain abundant DAMPs, such as HMGB1[70] and peroxiredoxin[71], which are potential ligands for toll-like receptors (TLRs). DAMPs cause proinflammatory activation of microglia as lipopolysaccharide (LPS). However, expression of proinflammatory cytokine by both microglia and macrophages in the ischemic brain is not very remarkable[72]. TLR ligands strongly induce expression of inducible nitric oxide synthase (iNOS) by microglia in culture. However, microglia in the ischemic brain either do not express iNOS protein or do so very faintly. TGFβ1 expression increases gradually until 7 days after the ischemic insults. Either LPS or DAMPs induce phosphorylation of IκB kinase (IKK), causing degradation of IκB, enabling the major proinflammatory transcription factor NFκB to translocate into nuclei and resulting in increased transcription of mRNA for proinflammatory mediators[73]. However, after being incubated with TGFβ1 for ~24 h, primary cultured microglia do not respond to LPS treatments, even when the TGFβ1 is removed from the culture media. The TGFβ1-treated microglia cannot be classified into M2 polarized cells as they do not express M2 markers. TGFβ1 also inhibits phosphorylation of signal transducers and activators of transcription 1 (STAT1) and expression of interferon regulatory factor 1 (IRF1), both of which augment proinflammatory reactions of microglia in a sustained manner. When injected into the parenchyma of the ischemic brain, both microglia and macrophages lose their immunoreactivity to phosphorylated IKK (pIKK). Expression of other anti-inflammatory cytokines, such as IL-4, IL-10, or IL-13, is much weaker than is that of TGFβ1. These findings indicate that TGFβ1 is a key ameliorating factor released by both microglia and macrophages.
Roles of macrophages
Macrophages accumulate densely in the core of the ischemic and traumatic lesions where almost all microglia disappear[11,12,19,74]. Therefore, macrophages play central roles in the immunological modulation of pathophysiological processes in these lesions. As most of the accumulated macrophages express NG2[74], they have been called brain Iba1+/NG2+ cells (BINCs)[11]. At least some are involved in removing degenerated materials in the core. The macrophage-precursor monocytes invade inflamed brain tissues in ischemic and traumatic injuries through recognizing chemokines that are typically either CCL2 or monocyte chemoattractant protein 1 and either CX3CL1 or fractalkine[22]. Kinetic study in the rat tMCAO models showed that expression of CCL2 and CX3CL1 disappeared rapidly after tMCAO, within 2 days. When macrophage proliferation was inhibited by a single administration of the anti-cancer drug 5-fluorouracil 48 h after tMCAO, accumulation of macrophages at 7 dpr did not occur[12]. Therefore, proliferation of monocytes that infiltrate within 2 days of the onset of the events causes the massive accumulation of macrophages in the core of the lesion[11]. The reduction in number of the accumulated macrophages often causes death of the model rats. When BINCs were isolated from ischemic rat brains and transplanted into the core lesions of other stroke model rats, the BINCs proliferate hugely and the outcomes were ameliorated greatly. BINCs express various neuroprotective factors, such as IGF-1 and HGF, that may contribute to improved outcomes. Although BINCs were also found in lesions in aged human brains of stroke cases, however, the density was considerably less than that in the young rat model lesions[12]. This may be one reason why human stroke cases become more severe than do the young rat models. Overall, the collective effects of macrophages on ischemic brains appear to be ameliorative.
However, the effects of monocyte-derived macrophages may not be favorable during the acute phase. Microglia and macrophages were isolated individually from TBI model rats at 1.5 days after injury using a fluorescence-activated cell sorter based on the different levels of CD45 expression[10]. In this TBI model, oxidative injury may play a significant detrimental role in inducing neuronal degeneration, as an oxidative product 8-hydroxydeoxyguaine (8-OHdG) was found in neuronal nuclei. Reactive oxygen species (ROS) is the cause for this product, and most of the ROS may be derived from the mitochondria of macrophages[75]. Infiltrating macrophages produce much higher levels of mitochondrial ROS than do microglia. ROS is also produced by NADPH oxidase activity, and macrophages express much higher levels of NADPH oxidase than do microglia. Moreover, expression of IL-1β and iNOS is more significant in macrophages than in microglia. Although NO is occasionally described as a neuroprotective factor[76], it can bind to superoxide O2- and form the highly toxic molecule peroxynitrite (ONOO-)[77]. Conversely, microglia can save neurons from degeneration induced by oxidative stress[78,79]. Collectively, the roles of blood-borne macrophages are more detrimental than are those of microglia in the earliest phase of severe brain injuries.
Some different phenotypes of macrophages accumulate in the traumatic and ischemic lesion cores[10,19]. During the acute phase, 2 days after the onset of stroke, most macrophages exhibit a rather neurotoxic phenotype characterized by expression of IL-1β and iNOS and release of ROS[10]. However, most macrophages exhibit neuroprotective phenotypes characterized by expression of IGF-1 and HGF. This does not imply that a single population of macrophages turns into the two different (neuroprotective and neuro-destructive) populations depending on the pathological processes. As the neuroprotective macrophages characteristically express NG2, they are termed BINCs. However, BINCs cannot be classified as M2-polarized macrophages; they express a typical M1 marker CD86 and not an M2 marker CD163, whereas they do not express IL-1β and iNOS in the ischemic lesion core. There is a minor subpopulation of Iba1+/CD68+ macrophages; though they do not express either NG2 or CD86, they express IL-1β, CCL2, and iNOS, in addition to TLR4, which may be the source of the proinflammatory factors. Characteristically, they express CD200[19,80], which mediates immunosuppressive signals primarily to myeloid cells expressing its receptor CD200R. Though the CD200+/NG2- macrophages are likely the predominant population during the acute phase of severe brain injury, they undergo gradual apoptotic degeneration due to the oxidative injury, as evidenced by the accumulation of 8OHdG+ materials in their nuclei[10]. Importantly, as they are not proliferative, they decline in number along with the pathological processes[19]. Alternatively, BINCs may be a small population during the acute phase, but they are highly proliferative and become the main macrophage population in the lesion core. Figure 2 summarizes the two types of macrophages.
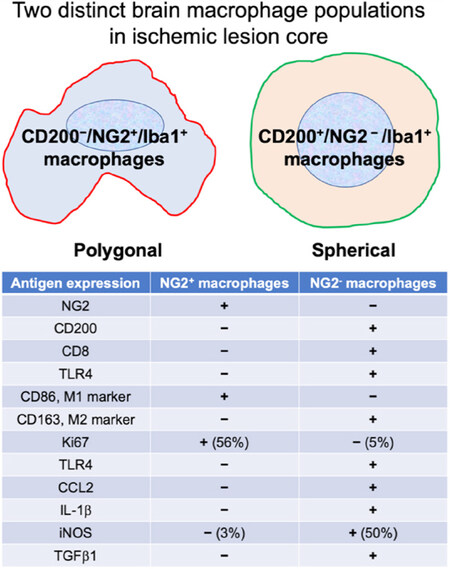
Figure 2. Distinctions between two populations of blood-borne macrophages accumulated in ischemic core lesions of a rat stroke model prepared by tMCAO. CD200-/NG2+/Iba1+ macrophages (BINCs) are the dominant populations occupying the most ischemic core regions. The two populations cannot be classified into M1 and M2 categories. The data come mainly from the data Matsumoto et al.[19] (2015)
In conclusion, macrophages also play both favorable and unfavorable roles in brain pathology. However, unlike the case with microglia, there are different macrophage populations that exert favorable and unfavorable effects independently.
Pharmacological interventions
The term “double-edged sword” has been used frequently to describe the favorable and unfavorable effects of microglia and macrophages[81-83]. Pharmacological interventions have long been sought that would either induce or strengthen the favorable effects of these cells[15,84]. However, in the case of macrophages, it may be more likely that there are different subpopulations with different effects on neurons rather than there being opposing effects of a single population of macrophages. Nevertheless, numerous pharmacological interventions have been shown to modulate the natures of both microglia and macrophages. Some of these are discussed below. Here it is better to once again confirm the notion that the favorable and unfavorable phenotypes for neuronal survival are incompatible with the M1 and M2 classifications[18,19,72].
Glucocorticoids
Glucocorticoids have strong immunosuppressive effects on many kinds of cells and may be the most often-used anti-inflammatory agent in laboratory and clinical settings. Glucocorticoids may have stronger anti-inflammatory effects on microglia than do adrenergic agonists, minocycline, troglitazone, and antidepressants [Figure 3]. They cause microglial cells to shrink, reduce lysosomal enzyme activities, and suppress proliferation of primary cultured rat microglia[85]. They suppress expression of the proinflammatory mediators by LPS-treated cultured microglia primarily at the transcription level[43]. Glucocorticoids can bind not only to the glucocorticoid receptor but also to the mineralocorticoid receptor[85]. Mineralocorticoid receptor may mediate activating effects on microglia rather than inhibitory effects. As the major rodent glucocorticoid corticosterone has an affinity for both mineralocorticoid receptor and glucocorticoid receptor, it exerts biphasic effects on the microglial proinflammatory actions - activation at low concentrations and inhibition at high concentrations.
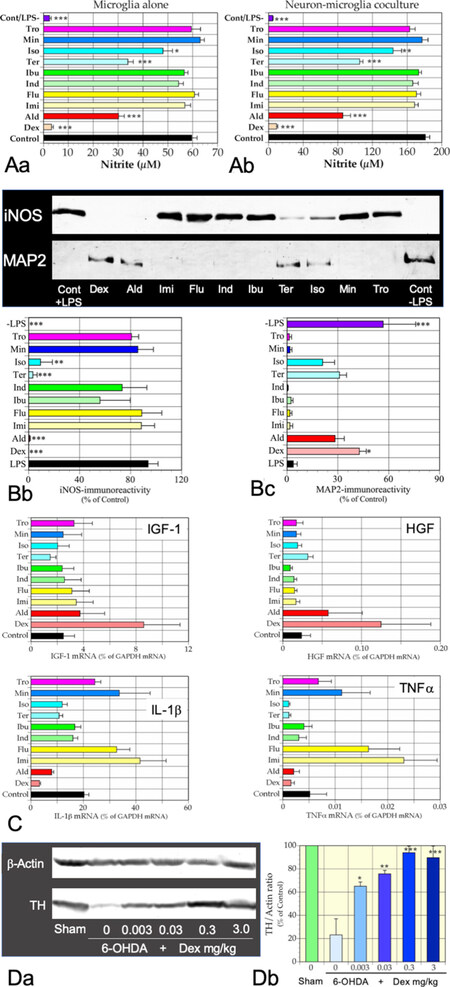
Figure 3. Immunomodulatory effects of a synthetic glucocorticoid Dex on microglia. A: effects of 10 agents on NO release by rat primary microglia (A-a) and rat primary cortical neuron-microglia coculture (A-b) incubated for 48 h with LPS. Nitrite levels in conditioned media were determined. Dex suppressed NO release most effectively. *P < 0.05, **P < 0.01, ***P < 0.001, respectively vs. LPS. B: effects of LPS and the agents on LPS-treated neuron-microglia cocultures. B-a: representative immunoblots showing the contents of MAP2 and iNOS proteins in the cocultures. (-LPS) denotes an absence of LPS; B-b: statistical comparison of suppressive effects on LPS-induced iNOS expression; B-c: neuroprotective effects of the 10 agents on the LPS-treated coculture. Strong iNOS expression was correlated with loss of MAP2-immunoreactivity. Only Dex protected neurons significantly from microglial neurotoxicity. Data from four independent cultures are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, respectively, vs. LPS. C: microglia were incubated with an indicated agent for 16 h in the absence of LPS. Only Dex increased expression of mRNAs encoding IGF-1 and HGF significantly while suppressing expression of IL-1β and TNFα mRNAs. n = 4, *P < 0.05, **P < 0.01, ***P < 0.001, respectively, vs. control. D: dose-dependent ameliorative effects of Dex on a rat 6-OHDA-induced PD model. D-a: representative immunoblots showing dose-dependent effects of Dex on the content of TH, a DA neuron marker; D-b: statistical analyses of the dose-dependent effects of Dex. A dose 0.003 µg/kg weight of Dex significantly prevented the reduction in TH immunoreactivity. Data from four rats for each dose were expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, respectively vs. 6-OHDA/Dex 0. The data are from unpublished ones by Wada et al. (unpublished data). Dex: dexamethasone; Ald: aldosterone; Imi: imipramine; Flu: fluvoxamine; Ind: indomethacin; Ibu: ibuprofen; Ter: terbutaline; Iso: isoproterenol; Min: minocycline; Tro: troglitazone; LPS: lipopolysaccharide; TH: tyrosine hydroxylase; DA: dopaminergic; PD: Parkinson’s disease; HGF: hepatocyte growth factor; IGF-1: insulin-like growth factor 1; 6-OHDA: 6-hydorxydopamine; iNOS: inducible nitric oxide synthase
A synthetic glucocorticoid dexamethasone (Dex), a specific ligand for glucocorticoid receptor, strongly inhibits LPS-induced NO release and expression of mRNA for IL-1β and TNFα[43]. When primary cortical neurons were cocultured with microglia in the presence of LPS, neurons were degenerated. As this LPS-induced neurodegeneration was prevented by an NOS inhibitor, L-NMMA, the neurodegeneration is caused by NO derived from activated microglia. However, Dex, rather than the NOS inhibitor, suppresses the release of NO more strongly and prevents the degeneration almost completely [Figure 3]. Moreover, Dex increases the expression of mRNA for the neuroprotective factors HGF and IGF-1. The findings indicate that glucocorticoids can induce a neuroprotective microglia phenotype even in the presence of LPS in culture or of DAMPs in the damaged brain parenchyma.
Because of such favorable effects, glucocorticoids have been studied in various animal brain disease models with neuroinflammation. As mentioned, in the rat 6-OHDA-induced PD model, microglia in the SNc are activated in response to degeneration of DA neurons. Chronic administration of glucocorticoids appears to inhibit microglia activation and enhance both the viability of DA neurons and the amount of tyrosine hydroxylase, a rate-limiting enzyme for DA synthesis [Figure 3]. However, glucocorticoids have several adverse effects, including induction of osteoporosis, impaired glucose tolerance, and increased susceptibility to infection. Moreover, glucocorticoids potentially impair cognitive functions by damaging the circuitry of the hippocampus, in which glucocorticoid receptor is expressed abundantly[86,87]. Nevertheless, as shown in Figure 3, chronic oral administration of glucocorticoid at a rather low dose of 3 µg/kg body weight for 6-OHDA-induced PD model rats prevented the reduction in tyrosine hydroxylase immunoreactivity in the SNc to a significant extent. It may be worth administering this dose in a clinical setting.
As mentioned above, microglia play a favorable role by eliminating hyperactive glutamatergic synapses from STN in the PD pathophysiology[47]. When 6-OHDA-induced PD model rats are administered a high dose of glucocorticoid, the motor deficits are aggravated. This may be one example demonstrating that strong immunosuppressive interventions for activated microglia and macrophages have an aggravating effect, as they have both favorable and unfavorable roles.
Noradrenaline and other cAMP-elevating agents
As shown in Figure 3, noradrenaline (NA) and related agents have significant inhibitory effects on activated microglia. These effects have been attributed mainly to the adrenergic β2 receptor that increases levels of intracellular cAMP[73,88]. The β2 agonist terbutaline strongly suppressed LPS-induced proinflammatory activation of microglia. cAMP-elevating agents, such as phosphodiesterase (PDE) inhibitors also exhibited strong inhibitory effects on LPS-treated microglia[89]. Rolipram, an inhibitor of the cAMP-selective hydrolase PDE4, may be the PDE inhibitor with the strongest inhibitory effects on rat primary microglia. A cAMP-analogue, 8-bromo cAMP, and forskolin, an activator for adenylate cyclase, also have strong inhibitory effects on microglia in vitro. Furthermore, the cAMP-elevating agents inhibit proliferation of microglia in vitro[90]. However, it should be noted that adrenergic α1 receptor agonists such as phenylephrine can inhibit microglial activation to an extent similar to that of terbutaline[73,88]. The inhibitory effects of NA cannot be eliminated by an inhibitor for cAMP-dependent protein kinase[73]. NA, α1, and β2 agonists strongly prevent LPS-induced translocation into the nuclei of NFκB[73]. This effect is mediated mainly by suppression of IκB degradation. NA and the agonists suppressed LPS-induced phosphorylation of STAT1 and expression of IRF1. IRF1 may contribute significantly to microglia activation[43]. However, the precise molecular mechanisms underlying the inhibitory effects of NA, in addition to the agonists, remain to be identified.
These studies may indicate that BBB-permeable adrenergic agonists are promising agents for suppressing neuroinflammatory processes in pathologic brains. The curative effects of BBB-permeable β2 agonists, while preventing the proinflammatory nature of microglia in the SNc, have been shown in murine PD models[91]. In AD pathology, NA neurons in the locus ceruleus, which is the most important NA source in the brain, undergo degeneration leading to microglia activation. This subsequently results in further neuronal degeneration[92]. However, there are many conflicting studies demonstrating the stimulatory actions of NA and the adrenergic agonists on microglia in the brain[93].
TGFβ1
TGFβ1 may be the most abundantly released cytokine from microglia and macrophages in severely damaged brains[27,72]. TGFβ1 suppresses iNOS expression by LPS-treated primary rat microglia almost completely in culture at both mRNA and protein levels. The inhibitory effect of TGFβ1 is as strong as is 100 nM of Dex in culture experiments[72]. As mentioned above, once incubated with TGFβ1, LPS cannot induce NFκB translocation into nuclei in microglia. TGFβ1 also inhibits LPS-induced phosphorylation of STAT1 and expression of IRF1. Although TGFβ1 does not increase expression of the M2 markers CD206 and Ym1, it increases expression of the neuroprotective factors HGF and bFGF. When TGFβ1 was injected into ischemic tissues 48 h after MCAO, the immunoreactivity of pIKK in the tissue surrounding the injection site was suppressed markedly in a dose-dependent manner.
Severely damaged brain tissues contain DAMPs that should induce proinflammatory activation of microglia and macrophages. DAMPs bind to TLRs on both microglia and macrophages, resulting in translocation of NFκB into nuclei. Microglia and macrophages should then display proinflammatory phenotypes expressing proinflammatory mediators. However, there were very few pIKK-bearing or iNOS-expressing microglia/macrophages in the lesion, especially during the subacute phase when TGFβ1 expression is high. Interventions to increase the actions of TGFβ1 for stroke models have been shown to ameliorate the outcomes for severely damaged brains[94]. Thus, TGFβ1 may ameliorate severe CNS damage through inducing anti-inflammatory phenotypes of microglia and macrophages.
Bromovalerylurea
Bromovalerylurea (BU; C6H11BrN2O2, CAS: 496-67-3) is a hypnotic/sedative that was developed more than a century ago[95]. It is not prescribed currently because of its weak actions as a hypnotic/sedative compared with newer agents such as benzodiazepines and also due to dependency. Recently, it has been found to have strong anti-inflammatory effects[10,43,96,97]. The effects of BU on LPS-treated primary rat microglia are as strong as those of Dex[43]. Like Dex, BU increases the expression of neuroprotective factors such as HGF or IGF-1 by LPS-treated primary microglia while inhibiting the expression both of proinflammatory factors and of the proinflammatory transcription factors IRF1, IRF7, and IRF8[43,98]. Although BU does not suppress LPS-induced NFκB translocation into nuclei, its inhibitory effects are exerted at the transcriptional level. BU inhibited ATP synthesis in mitochondria; this may be related to its anti-inflammatory effects[10,96]. Moreover, BU inhibits Janus kinase 1 activity, thus suppressing the phosphorylation of STAT1 and the subsequent expression of IRF1[43].
BU prevented the death of rats with cecum ligation and puncture-induced sepsis by suppressing the proinflammatory activation of peritoneal macrophages[97]. Oral administration of BU ameliorated PD in rat models while inhibiting the expression of proinflammatory mediators in the ventral midbrain[43]. When BU is added to the LPS-treated microglia/neuron coculture, it can almost completely inhibit neuronal death by almost completely suppressing the release of NO. In rat TBI models, BU ameliorated neurological deficits considerably while inhibiting expression of chemokine CCL2 and suppressed monocyte infiltration of the lesion[10]. Furthermore, BU inhibited mitochondrial ROS release by macrophages strongly. On the other hand, BU did not affect the accumulatio of favorable macrophages at later time points.
As BU is a hypnotic/sedative, it crosses the BBB easily. It may hold promise as an agent for ameliorating a range of brain diseases and injuries. However, BU is associated with marked dependency, and it has been used for suicide. Nevertheless, recent research has shown that the sleep-inducing effects of BU necessitate a higher dose than that required to produce the anti-inflammatory effects. As a hypnotic, BU increases the total sleeping period while reducing rapid eye movement sleep at a dose of 250 mg/kg for rats (Takeda et al., unpublished observation). Conversely, BU has been administered to sepsis, TBI, and PD model rats at a dose of 50 mg/kg. The findings suggest that BU could be used as a specific anti-inflammatory drug without significant dependency.
Agents that elevate expression of antiapoptotic factors in neurons
Microglia and macrophages can be activated by the presence of damaged neurons because of released DAMPs. Moreover, damaged neurons will reduce the activity of flippase, which is required for asymmetric distribution of phospholipids. Flippase reverses the translocation of PS to the exoplasmic face of the plasma membrane in an ATP-dependent manner. Therefore, even weak neuron damage that reduces intracellular ATP results in translocation of PS to the exoplasmic surface of the plasma membrane. The PS will be recognized by proteins such as MFG-E8 and also by complement C1[99]. These changes stimulate phagocytic elimination of neurons by microglia and macrophages in a process called phagoptosis[66]. Therefore, either suppression of the apoptotic changes or mitochondrial damage will suppress the unfavorable activation of microglia and macrophages[100,101].
A hematopoietic cytokine IL-3 has been shown to increase Bcl-xL expression in neurons in ischemic hippocampi[102] and in the SNc of PD model rats[44]. Moreover, GM-CSF also increases Bcl-xL expression[103]. Simultaneous administration of IL-3 and GM-CSF shows more marked ameliorative effects in TBI and PD model rats. The mixture of the cytokines also inhibited neuronal loss in a rat stroke model prepared by MCAO. Subcutaneous administration of the cytokine mixture to PD model rats inhibited IL-1β and TNFα expression in the ventral midbrain, whereas the expression of IGF-1 and HGF was increased. The favorable changes in microglia may be mediated by the ameliorated survival of neurons. The cytokines also have specific direct effects on microglia and macrophages[13,104]. Addition of cytokines to the culture of isolated macrophages (BINCs) from TBI lesions increased HGF and IGF-1 expression, whereas it did not affect IL-1β expression[13]. This indicates that inhibition of the proinflammatory nature of the cytokines may be attributed to ameliorated neuronal survival rather than the direct effects on macrophages and microglia. Thus, an agent that can increase expression of anti-apoptotic factors in neurons would increase the number of favorable microglia and macrophages.
Ginsenosides are natural products isolated from the plant ginseng. Among the ginsenosides, ginsenoside Rb1 and its derivatives strongly ameliorate the outcome of stroke[105] and TBI[106] models, while increasing Bcl-xl expression. Along with their direct effects on neurons, ginsenosides have been shown to inhibit the proinflammatory activation of microglia. It remains to be determined whether they inhibit LPS-induced proinflammatory activation of microglia and macrophages[107,108].
Conclusion
Microglia and macrophages have profound effects on the pathophysiological processes of several brain pathologies. They become activated in response to pathological changes of neurons that produce DAMPs and express PS on their surface. Microglia and macrophages can have ameliorative and/or deleterious effects on the CNS depending on the severity of the disease or injuries, time course, and BBB disruption. Even within the same pathology, the cells exert both different effects in completely different ways, as described in the cases of PD and the spinal cord of the peripheral nerve injury model. The development of pharmacological interventions to regulate the response of microglia and macrophages has long been anticipated. However much more research into their responses is required before that goal can be attained.
Declarations
AcknowledgmentsThe author would like to thank Enago (www.enago.jp) for the English language review.
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Jian Z, Liu R, Zhu X, Smerin D, Zhong Y, et al. The involvement and therapy target of immune cells after ischemic stroke. Front Immunol 2019;10:2167.
2. Sevenich L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front Immunol 2018;9:697.
3. Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, et al. Microglial and macrophage polarization - new prospects for brain repair. Nat Rev Neurol 2015;11:56-64.
4. Flores JJ, Klebe D, Tang J, Zhang JH. A comprehensive review of therapeutic targets that induce microglia/macrophage-mediated hematoma resolution after germinal matrix hemorrhage. J Neurosci Res 2020;98:121-8.
5. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci 1996;19:312-8.
6. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011;91:461-553.
7. Ling EA, Wong WC. The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia 1993;7:9-18.
9. Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Shudou M, et al. Antibodies to CD11b, CD68, and lectin label neutrophils rather than microglia in traumatic and ischemic brain lesions. J Neurosci Res 2007;85:994-1009.
10. Abe N, Choudhury ME, Watanabe M, Kawasaki S, Nishihara T, et al. Comparison of the detrimental features of microglia and infiltrated macrophages in traumatic brain injury: a study using a hypnotic bromovalerylurea. Glia 2018;66:2158-73.
11. Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Shudou M, et al. Accumulation of macrophage-like cells expressing NG2 proteoglycan and Iba1 in ischemic core of rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab 2008;28:149-63.
12. Smirkin A, Matsumoto H, Takahashi H, Inoue A, Tagawa M, et al. Iba1(+)/NG2(+) macrophage-like cells expressing a variety of neuroprotective factors ameliorate ischemic damage of the brain. J Cereb Blood Flow Metab 2010;30:603-15.
13. Nishihara T, Ochi M, Sugimoto K, Takahashi H, Yano H, et al. Subcutaneous injection containing IL-3 and GM-CSF ameliorates stab wound-induced brain injury in rats. Exp Neurol 2011;229:507-16.
14. Taguchi S, Choudhury ME, Miyanishi K, Nakanishi Y, Kameda K, et al. Aggravating effects of treadmill exercises during the early-onset period in a rat traumatic brain injury model: when should rehabilitation exercises be initiated? IBRO Rep 2019;7:82-9.
15. Hu X, Li P, Guo Y, Wang H, Leak RK, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012;43:3063-70.
16. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 2009;29:13435-44.
17. Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J Neuroinflammation 2014;11:98.
19. Matsumoto S, Tanaka J, Yano H, Takahashi H, Sugimoto K, et al. CD200+ and CD200- macrophages accumulated in ischemic lesions of rat brain: the two populations cannot be classified as either M1 or M2 macrophages. J Neuroimmunol 2015;282:7-20.
20. da Fonseca ACC, Matias D, Garcia C, Amaral R, Geraldo LH, et al. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci 2014;8:362.
21. Pun PBL, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res 2009;43:348-64.
22. Tei N, Tanaka J, Sugimoto K, Nishihara T, Nishioka R, et al. Expression of MCP-1 and fractalkine on endothelial cells and astrocytes may contribute to the invasion and migration of brain macrophages in ischemic rat brain lesions. J Neurosci Res 2013;91:681-93.
23. Inoue A, Tanaka J, Takahashi H, Kohno S, Ohue S, et al. Blood vessels expressing CD90 in human and rat brain tumors. Neuropathology 2016;36:168-80.
24. Kobayashi K, Yano H, Umakoshi A, Matsumoto S, Mise A, et al. A truncated form of CD200 (CD200S) expressed on glioma cells prolonged survival in a rat glioma model by induction of a dendritic cell-like phenotype in tumor-associated macrophages. Neoplasia 2016;18:229-41.
25. Perry VH, Hume DA, Gordon S. Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience 1985;15:313-26.
26. Perry VH, Gordon S. Macrophages and microglia in the nervous system. Trends Neurosci 1988;11:273-7.
27. Sugimoto K, Nishioka R, Ikeda A, Mise A, Takahashi H, et al. Activated microglia in a rat stroke model express NG2 proteoglycan in peri-infarct tissue through the involvement of TGF-beta1. Glia 2014;62:185-98.
28. Cronk JC, Filiano AJ, Louveau A, Marin I, Marsh R, et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med 2018;215:1627-47.
29. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 2014;17:131-43.
30. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, et al; Immunological Genome Consortium. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 2012;13:1118-28.
31. DePaula-Silva AB, Gorbea C, Doty DJ, Libbey JE, Sanchez JMS, et al. Differential transcriptional profiles identify microglial- and macrophage-specific gene markers expressed during virus-induced neuroinflammation. J Neuroinflammation 2019;16:152.
32. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014;159:1327-40.
33. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 2016;113:E1738-46.
34. Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016;36:39-49.
35. Kanamoto T, Mizuhashi K, Terada K, Minami T, Yoshikawa H, et al. Isolation and characterization of a novel plasma membrane protein, osteoblast induction factor (obif), associated with osteoblast differentiation. BMC Dev Biol 2009;9:70.
36. Konishi H, Kobayashi M, Kunisawa T, Imai K, Sayo A, et al. Siglec-H is a microglia-specific marker that discriminates microglia from CNS-associated macrophages and CNS-infiltrating monocytes. Glia 2017;65:1927-43.
37. Siew JJ, Chern Y. Microglial lectins in health and neurological diseases. Front Mol Neurosci 2018;11:158.
38. Kumar MAS, Peluso M, Chaudhary P, Dhawan J, Beheshti A, et al. Fractionated radiation exposure of rat spinal cords leads to latent neuro-inflammation in brain, cognitive deficits, and alterations in apurinic endonuclease. PLoS One 2015;10:e0133016.
39. Choudhury ME, Miyanishi K, Takeda H, Islam A, Matsuoka N, et al. Phagocytic elimination of synapses by microglia during sleep. Glia 2020;68:44-59.
40. Yokoyama A, Yang L, Itoh S, Mori K, Tanaka J. Microglia, a potential source of neurons, astrocytes, and oligodendrocytes. Glia 2004;45:96-104.
41. Umakoshi K, Choudhury ME, Nishioka R, Matsumoto H, Abe N, et al. B lymphocytopenia and Bregs in a not-to-die murine sepsis model. Biochem Biophys Res Commun 2020;523:202-7.
43. Higaki H, Choudhury ME, Kawamoto C, Miyamoto K, Islam A, et al. The hypnotic bromovalerylurea ameliorates 6-hydroxydopamine-induced dopaminergic neuron loss while suppressing expression of interferon regulatory factors by microglia. Neurochem Int 2016;99:158-68.
44. Choudhury ME, Sugimoto K, Kubo M, Nagai M, Nomoto M, et al. A cytokine mixture of GM-CSF and IL-3 that induces a neuroprotective phenotype of microglia leading to amelioration of (6-OHDA)-induced Parkinsonism of rats. Brain Behav 2011;1:26-43.
46. Takeuchi H, Jin S, Suzuki H, Doi Y, Liang J, et al. Blockade of microglial glutamate release protects against ischemic brain injury. Exp Neurol 2008;214:144-6.
47. Aono H, Choudhury ME, Higaki H, Miyanishi K, Kigami Y, et al. Microglia may compensate for dopaminergic neuron loss in experimental Parkinsonism through selective elimination of glutamatergic synapses from the subthalamic nucleus. Glia 2017;65:1833-47.
48. Miyanishi K, Choudhury ME, Watanabe M, Kubo M, Nomoto M, et al. Behavioral tests predicting striatal dopamine level in a rat hemi-Parkinson’s disease model. Neurochem Int 2019;122:38-46.
49. Kumar V. Toll-like receptors in the pathogenesis of neuroinflammation. J Neuroimmunol 2019;332:16-30.
50. Bezard E, Gross CE, Brotchie JM. Presymptomatic compensation in Parkinson’s disease is not dopamine-mediated. Trends Neurosci 2003;26:215-21.
51. Kurkowska-Jastrzebska I, Litwin T, Joniec I, Ciesielska A, Przybylkowski A, et al. Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson’s disease. Int Immunopharmacol 2004;4:1307-18.
52. Castano A, Herrera AJ, Cano J, Machado A. The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma. J Neurochem 2002;81:150-7.
53. Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol 2005;58:963-7.
54. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 2006;21:404-12.
55. Gagne JJ, Power MC. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology 2010;74:995-1002.
56. Leal MC, Casabona JC, Puntel M, Pitossi FJ. Interleukin-1beta and tumor necrosis factor-alpha: reliable targets for protective therapies in Parkinson’s Disease? Front Cell Neurosci 2013;7:53.
58. Sudo S, Tanaka J, Toku K, Desaki J, Matsuda S, et al. Neurons induce the activation of microglial cells in vitro. Exp Neurol 1998;154:499-510.
59. Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron 2013;77:10-8.
61. Nakata T, Kawachi K, Nagashima M, Yasugi T, Izutani H, et al. Transient ischemia-induced paresis and complete paraplegia displayed distinct reactions of microglia and macrophages. Brain Res 2011;1420:114-24.
62. Tsuda M, Beggs S, Salter MW, Inoue K. Microglia and intractable chronic pain. Glia 2013;61:55-61.
63. Nishihara T, Tanaka J, Sekiya K, Nishikawa Y, Abe N, et al. Chronic constriction injury of the sciatic nerve in rats causes different activation modes of microglia between the anterior and posterior horns of the spinal cord. Neurochem Int 2020;134:104672.
64. Sekiya K, Nishihara T, Abe N, Konishi A, Nandate H, et al. Carbon monoxide poisoning-induced delayed encephalopathy accompanies decreased microglial cell numbers: Distinctive pathophysiological features from hypoxemia-induced brain damage. Brain Res 2019;1710:22-32.
65. Claus HL, Walberer M, Simard ML, Emig B, Muesken SM, et al. NG2 and NG2-positive cells delineate focal cerebral infarct demarcation in rats. Neuropathology 2013;33:30-8.
67. Neher JJ, Emmrich JV, Fricker M, Mander PK, Thery C, et al. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A 2013;110:E4098-107.
68. Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 2016;7:11499.
69. Jin WN, Shi SX, Li Z, Li M, Wood K, et al. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab 2017;37:2224-36.
70. Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, et al. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 2006;26:6413-21.
71. Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med 2012;18:911-7.
72. Islam A, Choudhury ME, Kigami Y, Utsunomiya R, Matsumoto S, et al. Sustained anti-inflammatory effects of TGF-beta1 on microglia/macrophages. Biochim Biophys Acta 2018;1864:721-34.
73. Ishii Y, Yamaizumi A, Kawakami A, Islam A, Choudhury ME, et al. Anti-inflammatory effects of noradrenaline on LPS-treated microglial cells: suppression of NFkappaB nuclear translocation and subsequent STAT1 phosphorylation. Neurochem Int 2015;90:56-66.
74. Yokoyama A, Sakamoto A, Kameda K, Imai Y, Tanaka J. NG2 proteoglycan-expressing microglia as multipotent neural progenitors in normal and pathologic brains. Glia 2006;53:754-68.
75. Gao L, Laude K, Cai H. Mitochondrial pathophysiology, reactive oxygen species, and cardiovascular diseases. Vet Clin North Am Small Anim Pract 2008;38:137-55.
76. Sinz EH, Kochanek PM, Dixon CE, Clark RS, Carcillo JA, et al. Inducible nitric oxide synthase is an endogenous neuroprotectant after traumatic brain injury in rats and mice. J Clin Invest 1999;104:647-56.
77. Hall ED, Wang JA, Miller DM. Relationship of nitric oxide synthase induction to peroxynitrite-mediated oxidative damage during the first week after experimental traumatic brain injury. Exp Neurol 2012;238:176-82.
78. Toku K, Tanaka J, Yano H, Desaki J, Zhang B, et al. Microglial cells prevent nitric oxide-induced neuronal apoptosis in vitro. J Neurosci Res 1998;53:415-25.
79. Tanaka J, Toku K, Zhang B, Ishihara K, Sakanaka M, et al. Astrocytes prevent neuronal death induced by reactive oxygen and nitrogen species. Glia 1999;28:85-96.
80. Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Takahashi H, et al. Expression of CD200 by macrophage-like cells in ischemic core of rat brain after transient middle cerebral artery occlusion. Neurosci Lett 2007;418:44-8.
81. Dudvarski Stankovic N, Teodorczyk M, Ploen R, Zipp F, Schmidt MHH. Microglia-blood vessel interactions: a double-edged sword in brain pathologies. Acta Neuropathol 2016;131:347-63.
82. Jiang Z, Jiang JX, Zhang GX. Macrophages: a double-edged sword in experimental autoimmune encephalomyelitis. Immunol Lett 2014;160:17-22.
83. Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol 2013;5:73-90.
84. Xu H, Wang Z, Li J, Wu H, Peng Y, et al. The polarization states of microglia in TBI: a new paradigm for pharmacological intervention. Neural Plast 2017;2017:5405104.
85. Tanaka J, Fujita H, Matsuda S, Toku K, Sakanaka M, et al. Glucocorticoid- and mineralocorticoid receptors in microglial cells: the two receptors mediate differential effects of corticosteroids. Glia 1997;20:23-37.
86. Kvarta MD, Bradbrook KE, Dantrassy HM, Bailey AM, Thompson SM. Corticosterone mediates the synaptic and behavioral effects of chronic stress at rat hippocampal temporoammonic synapses. J Neurophysiol 2015;114:1713-24.
87. Smith MA. Hippocampal vulnerability to stress and aging: possible role of neurotrophic factors. Behav Brain Res 1996;78:25-36.
88. Mori K, Ozaki E, Zhang B, Yang L, Yokoyama A, et al. Effects of norepinephrine on rat cultured microglial cells that express alpha1, alpha2, beta1 and beta2 adrenergic receptors. Neuropharmacology 2002;43:1026-34.
89. Zhang B, Yang L, Konishi Y, Maeda N, Sakanaka M, et al. Suppressive effects of phosphodiesterase type IV inhibitors on rat cultured microglial cells: comparison with other types of cAMP-elevating agents. Neuropharmacology 2002;42:262-9.
90. Fujita H, Tanaka J, Maeda N, Sakanaka M. Adrenergic agonists suppress the proliferation of microglia through beta 2-adrenergic receptor. Neurosci Lett 1998;242:37-40.
91. Qian L, Wu HM, Chen SH, Zhang D, Ali SF, et al. beta2-adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. J Immunol 2011;186:4443-54.
92. Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, et al. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci U S A 2010;107:6058-63.
93. Sugama S, Takenouchi T, Hashimoto M, Ohata H, Takenaka Y, et al. Stress-induced microglial activation occurs through beta-adrenergic receptor: noradrenaline as a key neurotransmitter in microglial activation. J Neuroinflammation 2019;16:266.
94. Dhandapani KM, Brann DW. Transforming growth factor-beta: a neuroprotective factor in cerebral ischemia. Cell Biochem Biophys 2003;39:13-22.
95. Eeckhout Avd. Studien über die hypnotische Wirkung in der Vaïerian-süuregruppe. Arc Exp Path Pharmak 1907;57:338-57.
96. Kawasaki S, Abe N, Ohtake F, Islam A, Choudhury ME, et al. Effects of hypnotic bromovalerylurea on microglial BV2 cells. J Pharmacol Sci 2017;134:116-23.
97. Kikuchi S, Nishihara T, Kawasaki S, Abe N, Kuwabara J, et al. The ameliorative effects of a hypnotic bromvalerylurea in sepsis. Biochem Biophys Res Commun 2015;459:319-26.
98. Tanaka T, Murakami K, Bando Y, Yoshida S. Interferon regulatory factor 7 participates in the M1-like microglial polarization switch. Glia 2015;63:595-610.
99. Gyorffy BA, Kun J, Torok G, Bulyaki E, Borhegyi Z, et al. Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc Natl Acad Sci U S A 2018;115:6303-8.
100. Nakatsuka H, Ohta S, Tanaka J, Toku K, Kumon Y, et al. Cytochrome c release from mitochondria to the cytosol was suppressed in the ischemia-tolerance-induced hippocampal CA1 region after 5-min forebrain ischemia in gerbils. Neurosci Lett 2000;278:53-6.
101. Nakatsuka H, Ohta S, Tanaka J, Toku K, Kumon Y, et al. Histochemical cytochrome c oxidase activity and caspase-3 in gerbil hippocampal CA1 neurons after transient forebrain ischemia. Neurosci Lett 2000;285:127-30.
102. Wen TC, Tanaka J, Peng H, Desaki J, Matsuda S, et al. Interleukin 3 prevents delayed neuronal death in the hippocampal CA1 field. J Exp Med 1998;188:635-49.
103. Schabitz WR, Kruger C, Pitzer C, Weber D, Laage R, et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF). J Cereb Blood Flow Metab 2008;28:29-43.
104. Fujita H, Tanaka J, Toku K, Tateishi N, Suzuki Y, et al. Effects of GM-CSF and ordinary supplements on the ramification of microglia in culture: a morphometrical study. Glia 1996;18:269-81.
105. Zhang B, Hata R, Zhu P, Sato K, Wen TC, et al. Prevention of ischemic neuronal death by intravenous infusion of a ginseng saponin, ginsenoside Rb(1), that upregulates Bcl-x(L) expression. J Cereb Blood Flow Metab 2006;26:708-21.
106. Sakanaka M, Zhu P, Zhang B, Wen TC, Cao F, et al. Intravenous infusion of dihydroginsenoside Rb1 prevents compressive spinal cord injury and ischemic brain damage through upregulation of VEGF and Bcl-XL. J Neurotrauma 2007;24:1037-54.
107. Li DW, Zhou FZ, Sun XC, Li SC, Yang JB, et al. Ginsenoside Rb1 protects dopaminergic neurons from inflammatory injury induced by intranigral lipopolysaccharide injection. Neural Regen Res 2019;14:1814-22.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Tanaka J. Favorable and unfavorable roles of microglia and macrophages in the pathologic central nervous system. Neurosciences 2020;7:73-91. http://dx.doi.org/10.20517/2347-8659.2020.04
AMA Style
Tanaka J. Favorable and unfavorable roles of microglia and macrophages in the pathologic central nervous system. Neuroimmunology and Neuroinflammation. 2020; 7(2): 73-91. http://dx.doi.org/10.20517/2347-8659.2020.04
Chicago/Turabian Style
Tanaka, Junya. 2020. "Favorable and unfavorable roles of microglia and macrophages in the pathologic central nervous system" Neuroimmunology and Neuroinflammation. 7, no.2: 73-91. http://dx.doi.org/10.20517/2347-8659.2020.04
ACS Style
Tanaka, J. Favorable and unfavorable roles of microglia and macrophages in the pathologic central nervous system. Neurosciences. 2020, 7, 73-91. http://dx.doi.org/10.20517/2347-8659.2020.04
About This Article
Copyright

Data & Comments
Data


 Cite This Article 15 clicks
Cite This Article 15 clicks

 Like This Article 34
likes
Like This Article 34
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.