
Phf15 - a novel transcriptional repressor regulating inflammation in a mouse microglial cell line
 0
0Abstract
Aim: Excessive microglial inflammation has emerged as a key player in mediating the effects of aging and neurodegeneration on brain dysfunction. Thus, there is great interest in discovering transcriptional repressors that can control this process. We aimed to examine whether Phf15 - one of the top differentially expressed genes in microglia during aging in humans - could regulate transcription of proinflammatory mediators in microglia.
Methods: Real-time quantitative PCR was used to assess Phf15 mRNA expression in mouse brain during aging. Loss-of-function [short hairpin RNA (shRNA) -mediated knockdown (KD) and CRISPR/Cas9-mediated knockout (KO) of Phf15] and gain-of-function [retroviral overexpression (OE) of murine Phf15 cDNA] studies in a murine microglial cell line (SIM-A9) followed by immune activation with lipopolysaccharide were used to determine the effect of Phf15 on proinflammatory factor (Tnfα, IL-1β, and Nos2) mRNA expression. RNA sequencing was used to determine global transcriptional changes after Phf15 knockout under basal conditions and after lipopolysaccharide stimulation.
Results:Phf15 expression increases in mouse brain during aging, similar to humans. KD, KO, and OE studies determined that Phf15 represses mRNA expression levels of proinflammatory mediators such as Tnfα, IL-1β, and Nos2. Global transcriptional changes after Phf15 KO showed that Phf15 specifically represses genes related to the antiviral (type I interferon) response and cytokine production in microglia.
Conclusion: We provide the first evidence that Phf15 is an important transcriptional repressor of microglial inflammation, regulating the antiviral response and proinflammatory cytokine production. Importantly, Phf15 regulates both basal and signal-dependent activation and controls the magnitude and duration of the microglial inflammatory response.
Keywords
Introduction
Microglia are the resident myeloid-lineage cells of the brain. They actively provide homeostatic surveillance of the brain parenchyma, playing critical roles during development, maintenance, and repair throughout the life of an organism. As innate immune cells, however, microglia are also capable of mounting a full inflammatory response to environmental challenge in order to clear threats and restore homeostasis[1-6]. Microglia express pattern recognition receptors including Toll-like receptors to sense changes in their environment, such as infection by pathogens or endogenous danger signals. They can then respond by releasing proinflammatory mediators such as tumor necrosis factor alpha (TNFα), interleukin 1 beta (IL-1β), IL-6, reactive oxygen species, and reactive nitrogen species including nitric oxide (NO) to protect against threats[1,5,7].
Although beneficial when their production is tightly controlled, deregulated or sustained microglial production of inflammatory mediators can lead to collateral damage of surrounding neurons and other cells[5,7,8]. Thus, the transition to an activated state, as well as timely resolution of the inflammatory response, must be tightly regulated. Increasing evidence suggests that, during aging, microglia lose homeostatic function and acquire a proinflammatory phenotype that exacerbates aging-related brain dysfunction[9]. Indeed, aberrant microglia activation has been found in many types of age-related neurodegenerative conditions, for example Parkinson’s disease (PD) and Alzheimer’s disease (AD), which are marked by inflammatory processes involving glia, and microglia in particular[9-11].
Since excessive production of proinflammatory mediators is neurotoxic[8,12-14], various molecular mechanisms exist to regulate transcriptional repression of inflammatory gene expression. For example, basal state repression, that is, before the arrival of an activating signal, is generally carried out via recruitment of co-repressor complexes that prevent initiation of inflammatory gene transcription. After stimulation by an activating signal, additional mechanisms can maintain quiescence by restraining active transcription. Finally, numerous mechanisms mediate the timely resolution of the inflammatory response at the transcriptional level, including transrepression mechanisms that can remove transcription factors from inflammatory gene promoters[8,15-18].
Studies have also highlighted an important role for chromatin modifications in the transcriptional control of inflammatory gene expression[19,20]. A recent study by Soreq et al.[21], which compared transcriptional profiles of different brain cell types and regions throughout healthy human aging, found microglial gene expression profiles as being one of the most predictive markers of biological age in the brain. The same study identified a relatively unknown gene, PHD finger protein 15 (PHF15), among the top 25 differentially expressed genes in microglia during aging. Work in embryonic stem cells, as well as sequence and structural similarity to other members of the PHF family, indicate that PHF15 is a putative chromatin-mediated gene regulator[22].
Given that aging skews microglia towards a proinflammatory phenotype, and that PHF15 was found to be highly upregulated during non-pathological aging, we sought to determine whether Phf15 might regulate microglial inflammatory function. We found that Phf15 strongly represses proinflammatory gene expression, regulating both basal and signal-dependent activation and modulating the magnitude and duration of the mouse microglial inflammatory response. Importantly, Phf15 seems to regulate proinflammatory and interferon type I (IFN-I)-dependent gene expression. Increased IFN-I tone and proinflammatory cytokine expression are both hallmarks of the aging brain[23-26]. Our findings suggest that Phf15 is an important novel repressor of microglial inflammatory function that might work to counteract age-induced inflammation in the healthy, aging brain.
Methods
Animals
Adult male C57Bl6/J mice were purchased from The Jackson Laboratory and maintained on a 12-h/12-h light/dark cycle (lights on at 07:00) with ad libitum access to food and water and aged for ~2.5, ~14, or ~20 months. All animal care and procedures were approved by the University of California, Berkeley Animal Care and Use Committee.
shRNA-mediated knockdown of Phf15 in murine microglial cells
pGIPZ lentiviral mouse Phf15 shRNA constructs or a control scrambled shRNA were purchased from Dharmacon (Lafayette, CO). Lentivirus was packaged via co-transfection of each pGIPZ shRNA with pCMV-VSV-G (Addgene plasmid #8454)[27] and pCMV-dR8.2 (Addgene plasmid #8455)[27] into HEK 293T cells using Lipofectamine 3000 reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Viral supernatant was harvested after 48 h and incubated with SIM-A9 murine microglial cells in SIM-A9 complete medium [DMEM/F12 (Life Technologies, Carlsbad, CA), 10% fetal bovine serum (FBS; GE Healthcare Life Sciences, Chicago, IL), 5% horse serum (HS; GE Healthcare Life Sciences, Chicago, IL), and 1% Penicillin-Streptomycin (Life Technologies, Carlsbad, CA)]. After 48 h, GFP+ cells were sorted by Fluorescence-activated cell sorting (FACS) on an Aria Fusion (BD Biosciences, San Jose, CA; UC Berkeley Cancer Research Laboratory), expanded, and subcultured for immune stimulation experiments. Percent knockdown (KD) was determined via real-time quantitative PCR (RT-qPCR).
Overexpression of Phf15 in murine microglial cells
A Phf15 overexpression (OE) vector was constructed by cloning the full length Phf15 cDNA (Mus musculus PHD finger protein 15, mRNA cDNA clone MGC:143877 IMAGE:40094330) obtained from Dharmacon (Lafayette, CO) into a pMYs-IRES-GFP retroviral vector (Cell Biolabs Inc., San Diego, CA). Viruses expressing the full length Phf15 cDNA or empty vector control were co-transfected with pCL-10 A1 (Addgene plasmid #15805)[28] in HEK 293T cells using Lipofectamine 3000 (Life Technologies, Carlsbad, CA) reagent according to the manufacturer’s instructions. SIM-A9 cells were incubated with virus for 24 h and then sorted via FACS on an Aria Fusion, expanded, and subcultured for immune stimulation experiments. Fold OE was verified via RT-qPCR.
Generation of Phf15 knockout microglia
Phf15 knockout (KO) SIM-A9 cells were generated using the Alt-R CRISPR-Cas9-mediated gene editing system (guide RNA sequence ACTACATCCTGGCGGACCCGTGG) from IDT (Coralville, IA) using CRISPRMAX Lipofectamine reagent (IDT) as per the manufacturer’s instructions. ATTO 550+ cells were single-cell sorted on an Aria Fusion. Clones were screened for Phf15 deletion using PCR (primers Forward: agcacacttgtaaccctcct and Reverse: gaccaatgtctgttgttgttcg) followed by restriction digest with BtgI (New England Biolabs, Ipswich, MA). Percent decrease in Phf15 mRNA transcript expression was determined via RT-qPCR. Primer sequences are listed in Supplementary Table 1.
Immune stimulation
For all immune stimulation time course experiments, cells (KD, KO, and OE, and respective controls) were subcultured in 24-well plates at a density of 0.05 × 106 cells/well (in triplicate) and stimulated with lipopolysaccharide (LPS; final concentration of 100 ng/mL; Sigma Aldrich, St. Louis, MO), CpG oligodeoxynucleotide (CpG ODN; final concentration of 2.5 μmol/L; InvivoGen, San Diego, CA), or polyinosinic:polycytidylic acid [Poly(I:C); final concentration of 25 μmol/L; Sigma Aldrich, St. Louis, MO] for 1, 6, 12, or 24 h. No stimulation controls received an equivalent volume of sterile 1× PBS (Invitrogen, Carlsbad, CA).
RNA extraction
Mice were sacrificed according to the approved protocol. Brains were quickly isolated and frontal cortical areas were dissected, flash frozen, and stored at -80 °C. RNA was extracted using a bead homogenizer (30 s, setting “5”; Bead Mill, VWR) in Trizol reagent (ThermoFisher, Waltham, MA). Total RNA was extracted using the Direct-zol RNA miniprep kit (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. For cell lines, after immune stimulation, media was aspirated and wells were washed 2 × with ice-cold 1 × PBS (Invitrogen, Carlsbad, CA). RNA was extracted using the Direct-zol RNA miniprep kit (Zymo Research, Irvine, CA).
RT-qPCR
cDNA was reversed transcribed from total RNA using the SuperScriptTM III First-Strand Synthesis System kit (ThermoFisher, Waltham, MA) following the manufacturer’s instructions. RT-qPCR was run using SYBR green (Roche, Pleasanton, CA) on a QuantStudio 6 (ThermoFisher, Waltham, MA) real-time PCR machine. All RT-qPCR primers were specific to the desired template, spanned exon-exon junctions, and captured all transcript variants for the specific gene under study. Ct values were normalized to the housekeeping gene hypoxanthine phosphoribosyltransferase (Hprt). Primer sequences used in this study are listed in Supplementary Table 1.
RNA-seq library preparation and analysis
RNA was extracted from a total of n = 3 replicates per condition (Phf15 KO or control) and was used to prepare libraries for RNA sequencing using the mRNA HyperPrep Kit according to the manufacturer’s instructions (KAPA Biosystems, Wilmington, MA). Libraries were quality control checked via Qubit (ThermoFisher, Waltham, MA) and via RT-qPCR with a next generation sequencing library quantification kit (Zymo Research, Irvine, CA). RNA sequencing (one lane) was performed on a HiSeq4000 sequencing system (Illumina Inc., San Diego, CA; UC Berkeley Genomics Sequencing Laboratory). Sequencing reads were aligned to the Mus musculus reference genome assembly GRCm38 (mm10) using Spliced Transcripts Alignment to a Reference (STAR) aligner[29]. Count data were analyzed with Hypergeometric Optimization of Motif EnRichment (HOMER) software for next-generation sequencing analysis (http://homer.ucsd.edu/homer/ngs/index.html), which uses the R/Bioconductor package DESeq2[30] to perform differential gene expression analysis. To adjust for multiple comparisons, DESeq2 uses the Benjamini-Hochberg procedure to control the false discovery rate and returned false discovery rate adjusted P values and log2-fold expression changes between Phf15 KO and control conditions for each gene. Genes were filtered by adjusted P value (adjusted P < 0.01 for upregulated genes or 0.05 for downregulated genes) and log2-fold change in expression (greater than 1.5 log2-fold change for upregulated genes and less than -1.5 for downregulated genes). Too few downregulated genes (< 200) passed the more stringent adjusted P < 0.01 cutoff for robust downstream biological function analysis, thus the adjusted P value threshold was lowered to P adj < 0.05. Results were visualized using the R package EnhancedVolcano[31]. Lists of upregulated and downregulated genes were input into Metascape[32], a gene annotation and analysis tool, to determine enriched biological themes within the gene lists.
Motif enrichment
Transcription factor binding site (“motif”) enrichment was analyzed using HOMER (http://homer.ucsd.edu/homer/ngs/index.html).
Statistical analysis
Relative mRNA expression of Phf15 in mouse frontal cortical areas was analyzed using ordinary one-way ANOVA with post hoc Tukey’s multiple comparisons to compare expression levels across age. Percent KD and time course experiments measuring expression levels of inflammatory markers [Tnfα, nitric oxide synthase, inducible (Nos2), and IL-1β] between control and Phf15 shRNAs shPhf15-1 and shPhf15-2 after immune stimulation [with LPS, CpG-ODN, or Poly(I:C)] were analyzed via ordinary one-way ANOVA with Dunnett’s multiple comparisons between each shRNA versus control shRNA within timepoint. Fold OE or percent reduction for Phf15 OE and KO cell lines, respectively, were analyzed using unpaired t-tests (OE or KO vs. respective control). Time course experiments for Phf15 OE and KO cell lines were analyzed using unpaired t-tests with Holm-Sidak correction for multiple comparisons. RNA-seq data were analyzed as described above. P < 0.05 was considered significant in all experiments.
Results
Aging increases Phf15 expression in mouse brain
To investigate whether Phf15 increases in mouse brains similar to humans[21], we measured Phf15 mRNA expression in mouse frontal cortical brain areas across age. We were interested in frontal cortical regions because of their involvement in mediating various aspects of cognitive function and because they are selectively affected in several aging-related neurodegenerative conditions, e.g., PD, AD, and frontotemporal dementia[33,34].
We found that compared to young (~2.5-month-old) mice, old (~20-month-old) mice had significantly elevated Phf15 mRNA levels in frontal cortical areas [Figure 1]. Middle-aged (~14-month-old) mice showed a trend towards increased Phf15 mRNA expression that did not reach statistical significance. Our data suggest that Phf15 expression increases in mouse frontal cortical regions upon normal aging, similar to what was previously reported in humans[21].
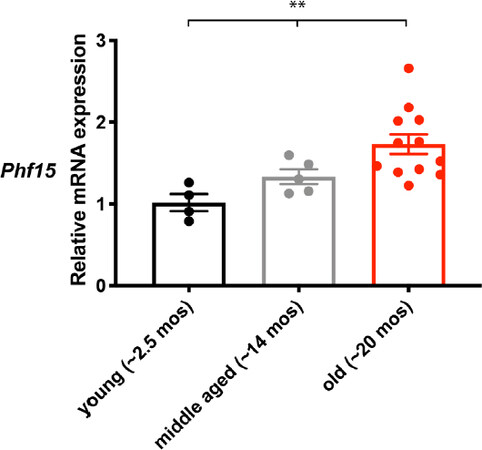
Figure 1. Phf15 expression increases in aged mouse frontal cortical areas. Phf15 mRNA expression was significantly elevated in frontal cortical areas of old (~20-month-old; red bar) mice compared to young (~2.5-month-old; black bar) mice. Data are mean ± SEM (n = 4 young, n = 5 middle aged, n = 12 old). One-way ANOVA with Tukey’s post hoc comparisons between age groups: **P < 0.01
Knockdown of Phf15 increases the magnitude of the microglial inflammatory response
To determine whether Phf15 regulates microglial inflammatory function, we performed loss-of-function studies via shRNA-mediated KD in a murine microglial cell line, SIM-A9, followed by immune activation with LPS, a component of Gram-negative bacterial cell walls and TLR4 agonist. SIM-A9 cells are an established murine microglial cell line and their original characterization has been previously published[35]. In short, SIM-A9 cells are a spontaneously transformed microglial cell line isolated from cultured primary glia from postnatal murine cerebral cortices. They display key characteristics of cultured primary microglia; for example, they express the microglia/macrophage-specific proteins cluster of differentiation 68 (CD68) and ionized calcium-binding adapter molecule 1 (IBA1) and they are responsive to immune stimulation with LPS, triggering nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) inflammatory signaling cascades, increase of protein levels of the proinflammatory factors NOS2 (the enzyme that catalyzes the production of NO) and cyclooxygenase 2b (COX2), and secretion of TNFα. Importantly, they are also responsive to the anti-inflammatory effects of interleukin 4 and increasing levels of Arginase 1, demonstrating that SIM-A9 cells can switch between pro- and anti-inflammatory states. Finally, they also exhibit phagocytic uptake of bacterial particles and fluorescently-labeled amyloid-β (Aβ).
Additionally, we chose LPS as the immune stimulant because: (1) intraperitoneal and/or intracranial administration of LPS in mice led to increased microglial activation, neuroinflammation, neuronal loss including loss of dopaminergic neurons in the substantia nigra in a mouse model of PD[8], and cognitive and neurological deficits[36]; (2) aged individuals show increased systemic levels of LPS in the bloodstream[37], which are associated with increased inflammation and microglial activation[38]; and (3) in humans, TLR4 activation is linked to age-related pathologies such as PD and AD[39-41]; thus, LPS serves as a relevant aging-related physiological immune stimulant.
KD of Phf15 resulted in a significant reduction in Phf15 mRNA transcript levels of 52% and 60% for cell lines shPhf15-1 and shPhf15-2, respectively [Figure 2A], as well as significantly increased mRNA expression of Tnfα, a proinflammatory cytokine, after KD with shPhf15-2 at 0, 1, 6, and 12 h after LPS stimulation [Figure 2B].
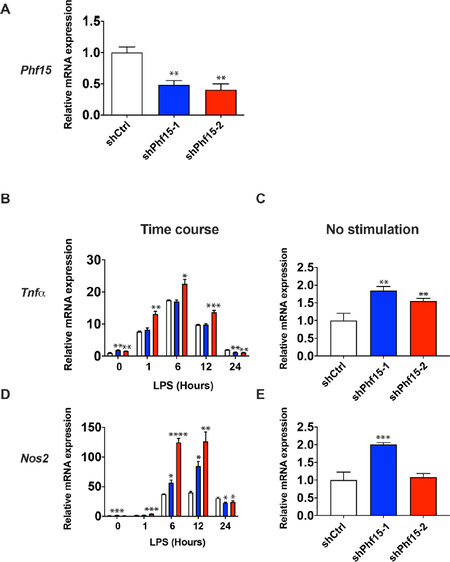
Figure 2. Knockdown of Phf15 increases the magnitude of the microglial inflammatory response. (A) Knockdown efficiency for anti-Phf15 shRNAs shPhf15-1 (blue bar, 52% knockdown) and shPhf15-2 (red bar, 60% knockdown). Data are mean ± SEM (n = 3 per condition). One-way ANOVA with Dunnett’s multiple comparisons between shPhf15-1 or shPhf15-2 and shCtrl cells: **P < 0.01. Twenty-four-hour time course experiments showing relative mRNA expression levels of Tnfα (B) and Nos2 (D) after LPS stimulation of shRNAs shPhf15-1 and shPhf15-2 compared to shCtrl (control scrambled shRNA). No stimulation (0-h time point) is shown for Tnfα (C) and Nos2 (E). Data are mean ± SEM (n = 3 per condition). One-way ANOVA with Dunnett’s multiple comparisons for shPhf15-1 or shPhf15-2 relative to shCtrl cells for individual timepoints: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. LPS: lipopolysaccharide; Tnfα: tumor necrosis factor alpha; Nos2: nitric oxide synthase, inducible
Similarly, mRNA levels of Nos2 were significantly elevated at 1, 6, and 12 h post stimulation for shPhf15-2 and 0, 6, and 12 h for shPhf15-1 [Figure 2D]. Overall, our experiments show that ~50%-60% KD, the equivalent of a “heterozygous” condition, results in increased expression of proinflammatory mediators over a 12-h time course that resolves and falls below control levels by 24 h after immune stimulation. Importantly, microglial inflammatory function was elevated in the absence of immune stimulation (0 h time point, Figure 2B and D, and no stimulation condition, Figure 2C and E), suggesting a loss of repressive mechanisms that inhibit basal state inflammatory gene transcription.
We repeated the immune activation time course experiments in Phf15 KD cells using two separate immune stimulants specific to two distinct Toll-like receptors to test the pathway specificity of the inflammatory response: CpG ODN, a synthetic bacterial and viral DNA mimic that targets TLR9, and Poly(I:C), a synthetic viral dsRNA mimic that targets TLR3. While TLR4 uses both the myeloid differentiation primary response 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF) downstream adapters to transduce its inflammatory cascade, TLR9 and TLR3 utilize MyD88 and TRIF, respectively [Supplementary Figure 1][42,43].
Immune stimulation with CpG ODN and Poly(I:C) both yielded similar results to those obtained with LPS stimulation [Supplementary Figures 2 and 3, respectively], denoting no adapter selectivity and confirming that Phf15 antagonizes inflammatory gene expression downstream of both the MyD88 and TRIF signaling pathways.
Genetic deletion of Phf15 increases the magnitude and prolongs the duration of the microglial inflammatory response
Since our KD strategy resulted in ~50% reduction in Phf15 mRNA expression, we next performed CRISPR/Cas9-mediated genetic deletion of Phf15 in SIM-A9 microglial cells followed by immune activation with LPS. KO of Phf15[Figure 3A] resulted in significantly increased LPS-induced expression of Tnfα[Figure 3B], IL-1β[Figure 3D], and Nos2, albeit to a lesser extent [Figure 3F], over a 24-h time course. Importantly, mRNA levels of both Tnfα and IL-1β remained elevated at 24 h compared to control cells, denoting a prolonged inflammatory response and failure to return to steady-state. mRNA expression of Nos2 showed a significant upregulation over 12 h (0-, 1-, and 12-h timepoints) but had returned to control levels by 24 h [Figure 3F]. Notably, basal expression of all three genes was significantly elevated, with a 4-fold increase in Tnfα, 14-fold increase in IL-1β, and 32-fold increase in Nos2 when comparing KO to control cells [Figure 3C, E and G].
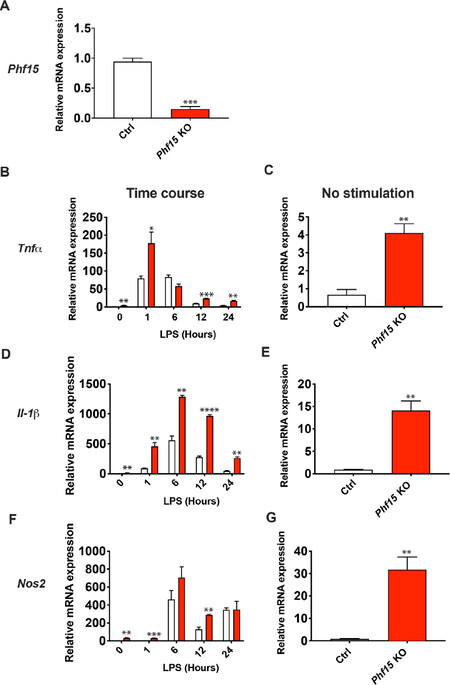
Figure 3. Knockout of Phf15 increases the magnitude and duration of inflammatory gene expression. (A) Percent reduction in Phf15 transcript expression in Phf15 knockout SIM-A9 microglia (Phf15 KO, red bar) compared to control (Ctrl, open bar). Data are mean ± SEM (n = 3 per condition). Unpaired t-test between Phf15 KO and control cells. Twenty-four-hour time course experiments showing relative mRNA expressions levels of Tnfα (B), IL-1β (D), and Nos2 (F) after LPS stimulation. No stimulation (0-h time point or baseline) expressions of Tnfα (C), IL-1β (E), and Nos2 (G) are also shown. Data are mean ± SEM (n = 3 per condition). Unpaired t-tests with Holm-Sidak correction for multiple comparisons between Phf15 KO and control cells within timepoint: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. LPS: lipopolysaccharide; KO: knockout; Tnfα: tumor necrosis factor alpha; inducible; IL-1β: interleukin 1 beta; Nos2: nitric oxide synthase, inducible
Time course experiments after stimulation of TLR9 with CpG-ODN [Supplementary Figures 4] and TLR3 with Poly(I:C) [Supplementary Figures 5] in Phf15 KO cells again yielded similar results to LPS stimulation in Phf15 KO microglial cells, denoting no difference in downstream adapter selectivity and confirming our prior KD results.
Overall, KO of Phf15 resulted in a more severe phenotype compared to our KD results, increasing the magnitude and prolonging the duration of the microglial inflammatory response. Taken together, our KD and KO results indicate that Phf15 functions to restrict microglial inflammatory output, regulating the magnitude and duration, as well as basal inhibition of the inflammatory response.
Overexpression of Phf15 in microglia results in a dampened inflammatory response
To further test the role of Phf15 as a repressor of proinflammatory genes, we carried out gain-of-function studies of Phf15 in SIM-A9 cells. OE via retroviral delivery of the full-length murine Phf15 cDNA [Figure 4A] resulted in significantly decreased expression of Nos2 at 0 and 24 h [Figure 4F]. mRNA expression levels of Tnfα[Figure 4B], IL-1β[Figure 4D], and Nos2[Figure 4F] were decreased at 6 h post LPS stimulation at an adjusted P = 0.0501, which did not reach statistical significance. Notably, basal levels (no stimulation) of Nos2 were also significantly decreased [Figure 4E, G].
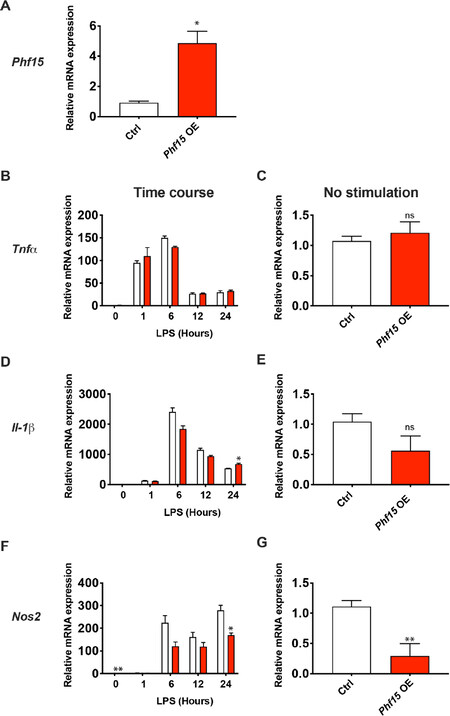
Figure 4. Phf15 overexpression decreases the microglial inflammatory response. (A) Fold OE of Phf15 in SIM-A9 microglia (red bar) versus control cells (Ctrl, open bar). Data are mean ± SEM (n = 3 per condition). Unpaired t-test between Phf15 OE and control cells. Twenty-four-hour time course experiments showing relative mRNA expression levels of Tnfα (B), IL-1β (D), and Nos2 (F) after LPS stimulation. Baseline (0-h time point, no stimulation) expressions of Tnfα (C), IL-1β (E), and Nos2 (G) are displayed separately from time course experiments. Data are mean ± SEM (n = 3 per condition). Unpaired t-tests with Holm-Sidak correction for multiple comparisons between Phf15 OE and control cells within timepoint: *P < 0.05, **P < 0.01. OE: overexpression; LPS: lipopolysaccharide; Tnfα: tumor necrosis factor alpha; inducible; IL-1β: interleukin 1 beta; Nos2: nitric oxide synthase, inducible
Time course experiments following stimulation with CpG-ODN [Supplementary Figures 6] and Poly(I:C) [Supplementary Figures 7] showed a similar but stronger repressive phenotype compared to LPS stimulation, with decreased expression levels of Tnfα[Supplementary Figures 6A, B and Figures 7A, B], IL-1β[Supplementary Figures 6C, D and Figures 7C, D], and Nos2[Supplementary Figures 6E, F and Figures 7E, F], over the time course, as well as under basal (no stimulation) conditions, confirming our previous results. Taken together, our OE results show a dampened microglial inflammatory response, revealing a reciprocal response phenotype compared to our KD and KO experiments. Collectively, these results confirm that Phf15 functions to repress both basal and stimulus-dependent inflammatory gene expressions in microglia.
Loss of Phf15 affects global expression of genes involved in antiviral responses and regulation of inflammatory processes
To examine global transcriptional changes as a result of Phf15 deletion in microglia, we carried out RNA-sequencing (RNA-seq) on Phf15 KO SIM-A9 cells under no stimulation conditions and 6 h post LPS stimulation. We chose to examine the no stimulation condition (0-h time point) based on our KD and KO time course results showing that baseline is one of the most consistently and strongly deregulated time points. Importantly, elevated or “leaky” proinflammatory mediator expression at baseline might result in chronic inflammation leading to neurodegeneration. Similarly, 6 h after LPS stimulation corresponded to the peak of the transcriptional inflammatory response, with large increases in magnitude for both IL-1β and Nos2.
Differential gene expression analysis revealed that 466 genes with log2-fold change > 1.5 and P adj < 0.01 were upregulated and 309 genes with log2-fold change < -1.5 and P adj < 0.05 were downregulated [Figure 5A]. Biological theme enrichment analysis using Metascape[32] on the upregulated genes revealed that the most enriched biological process categories under basal conditions were “response to virus” and “cytokine production” [Figure 5B and C]. Under the “response to virus” category, there was significant upregulation of various interferon-stimulated genes (ISGs), for example Isg15, interferon induced protein with tetratricopeptide repeats 1 (Ifit1), Ifit3, interferon regulatory factor 7 (Irf7), 2’-5’-oligoadenylate synthetase 2 (Oas2), and Oas-like 2 (Oasl2) [Figure 5C]. The downregulated genes showed more variability in the types of pathways affected, largely involving growth, differentiation, and glial cell migration processes [Figure 5A and Supplementary Figures 8A].
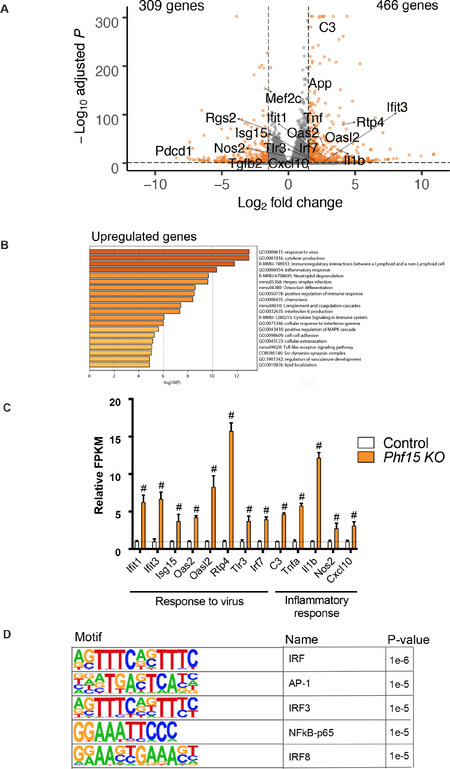
Figure 5. Loss of Phf15 affects the expression of genes involved in viral response and regulation of inflammatory processes in the absence of immune stimulation. A: Volcano plot representing the RNA-seq results. Orange dots represent differentially expressed genes in Phf15 knockout microglia compared to control (upregulated genes at a cutoff of log2-fold change > 1.5 and P adj < 0.01; downregulated genes at a cutoff of log2-fold change < -1.5 and P adj < 0.05); B: GO analysis for significantly upregulated genes showing biological process categories related to “response to virus” and “inflammatory response”; C: Upregulated genes associated with “response to virus” and “inflammatory response” in the no stimulation (baseline) condition. Relative FPKM values were obtained by normalizing FPKM values of Phf15 knockout SIM-A9 microglia to control FPKM values for each gene (n = 3 per condition). Statistics are by DESeq2: #P < 0.0001; D: Top 5 enriched transcription factor binding motifs for the set of upregulated genes in the no stimulation (baseline) condition. KO: knockout; FPKM: Fragments per kilobase million; GO: Gene ontology
Motif analysis for transcription factor binding sites enriched in the promoters of the upregulated genes at baseline revealed consensus motifs for interferon regulatory factors (IRFs), i.e., the interferon (IFN) stimulated response element (ISRE) and motifs for IRF3 and IRF8 specifically in the top 5 best matches. Activator protein 1 (AP-1) and NF-κB p65 subunit (NF-κB-p65) motifs were also enriched. Both can regulate expression of canonical proinflammatory cytokines such as TNFα and IL-1β[44,45][Figure 5D]. Motif enrichment for the set of downregulated genes revealed motifs for twist-related protein 2 (Twist2) and Class A basic helix-loop-helix protein 15 (BHLHA15). Twist2 has been shown to mediate cytokine downregulation after chronic nucleotide-binding oligomerization domain-containing protein 2 (NOD2, a bacterial peptidoglycan sensor) stimulation[46]. BHLHA15 has been shown to induce and maintain secretory architecture in cells specialized for secretion[47][Supplementary Figures 8B].
Differential gene expression analysis after 6 h of LPS stimulation in KO versus control cells revealed 576 upregulated genes (log2-fold change > 1.5 and P adj < 0.01) and 322 downregulated genes (log2-fold change < -1.5 and P adj < 0.05) [Figure 6A]. Interestingly, by 6 h after LPS administration, some of the most enriched biological process categories in KO cells were related to “cytokine secretion” and “immunoregulatory interaction” [Figure 6B, C], denoting a strong increase in magnitude of expression of genes involved in regulating the secretion of proinflammatory mediators. The downregulated genes at 6 h after LPS stimulation in KO cells relative to control again displayed more variability, but did show decreases in biological process categories related to “regulation of defense response” and “cytokine production”, indicating negative regulation of these processes in Phf15 KO cells compared to control [Supplementary Figures 9A].
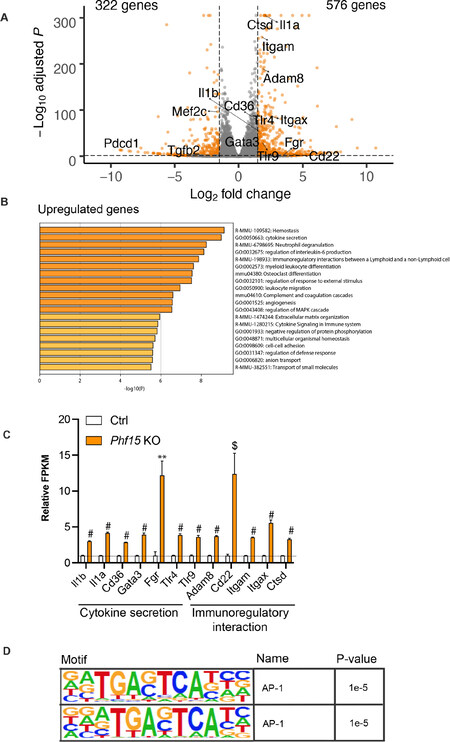
Figure 6. Knockout of Phf15 affects the expression of genes involved in inflammatory factor secretion and immunoregulatory processes after LPS stimulation. A: volcano plot representing the RNA-seq results. Orange dots represent differentially expressed genes in Phf15 knockout microglia 6 h after LPS administration compared to control (upregulated genes at a cutoff of log2-fold change > 1.5 and Padj < 0.01; downregulated genes at a cutoff of log2-fold change < -1.5 and Padj < 0.05); B: GO analysis for upregulated genes shows biological process categories associated with “cytokine secretion” and “immunoregulatory interaction”; C: Upregulated genes associated with “cytokine secretion” and “immunoregulatory interaction” biological process categories 6 h post LPS stimulation. Relative FPKM values were obtained by normalizing FPKM values of Phf15 knockout SIM-A9 microglia to control FPKM values for each gene (n = 3 wells per condition). Statistics are by DESeq2: **P < 0.01, $P < 0.001, #P < 0.0001; D: Transcription factor binding motifs for the set of upregulated genes 6 h after LPS stimulation are enriched for activator protein 1 (AP-1). LPS: lipopolysaccharide; KO: knockout; FPKM: Fragments per kilobase million; GO: Gene ontology
Motif enrichment analysis for transcription factor binding sites enriched in the promoters of upregulated genes at the 6-h time point revealed consensus sequences for AP-1, a key regulator of microglia reactivity in inflammation[48][Figure 6D]. Motif enrichment for the set of downregulated genes revealed motifs for IRFs (the ISRE) and motifs for IRF1 and IRF3 specifically [Supplementary Figures 9B], supporting the observation that there is a negative “regulation of defense response” by 6 h post stimulation. It is interesting to note that a functional transition from cytokine production to cytokine secretion seems to occur in the 6-h period after LPS activation.
Taken together, our RNA-seq results confirm that Phf15 is a repressor of microglial inflammatory gene expression, regulating the antiviral responses - specifically, IFN-I-dependent responses - as well as processes related to proinflammatory cytokine production and release.
Discussion
Our results show that Phf15 inhibits microglial expression of proinflammatory mediators under basal and signal-dependent activation, regulating both the magnitude and duration of the inflammatory response. Genetic deletion of PhfF15 in a microglial cell line followed by stimulation with LPS led to an exaggerated proinflammatory response with increased production of Tnfα, IL-1β, and Nos2 over a time course of 24 h. Importantly, levels of proinflammatory factors remained elevated at 24 h, demonstrating a sustained and prolonged response. Consistent with our LPS stimulation of TLR4 results, similar results were obtained after TLR9 and TLR3 activation, confirming that Phf15 is a general negative regulator and controls both the MyD88 and TRIF downstream signal transduction pathways [Supplementary Figures 1]. Overexpression of Phf15 showed a dampened microglial inflammatory response, highlighting a reciprocal response phenotype that further supports our loss-of-function results.
Prolonged inflammation can damage surrounding healthy tissue, eventually resulting in neuronal degeneration and loss, and negatively affecting brain function. For example, levels of TNFα are seen to rapidly rise in experimental models of PD and are highly toxic to dopaminergic neurons[13,14,49]. Similarly, high levels of TNFα are a hallmark of PD in humans[50-52]. Additionally, both TNFα and IL-1β are involved in maintaining proper synaptic plasticity at physiological levels[53,54] and overproduction of these cytokines can result in neuronal death via excitotoxicity and cognitive dysfunction[55,56].
Our studies further demonstrate that Phf15 can regulate both basal and signal-dependent microglial inflammatory gene expression. KD and KO of Phf15 in microglial cell lines resulted in significantly increased levels of proinflammatory cytokine gene expression - without stimulation and after immune activation - while OE had the reverse effect. The inflammatory response is a tightly controlled process in immune cells in order to protect against unintended damage to healthy tissue. Even in aged microglia, where production and secretion of proinflammatory mediators is generally increased, this process is dependent upon treatment with immune stimulants[9,57,58]. Increased proinflammatory cytokine gene expression without stimulation denotes constitutive or “leaky” expression of inflammatory mediators, simulating a state of low-grade but constant activation. Similarly, hyperresponsiveness to immune stimuli combined with a lack of resolution of the inflammatory response can lead to a state of chronic inflammation. All three can trigger pathological chronic inflammation in the brain, which is detrimental to brain function.
Importantly, distinct molecular mechanisms regulate transcriptional control of different phases (“modules”) of the inflammatory response and it is noteworthy that Phf15 might be involved in regulating several of these. Basal inflammatory function, for example, is generally regulated by co-repressors such as nuclear receptor co-repressor (NCOR), silencing mediator of retinoid and thyroid receptors (SMRT), and RE1 silencing transcription factor (REST) co-repressor 1 (RCOR1 or CoREST) that block poised promoters from active transcription, preventing “leaky” expression of primary response genes (e.g., TNFα, Type I IFNs, IL-1β, etc.) (for review, see[17]). Significantly increased inflammatory gene transcription under baseline conditions, as observed in our Phf15 KD and KO experiments, suggests a loss of this repressive mechanism.
After stimulation by an activating signal, additional mechanisms can maintain quiescence by restraining active transcription. For example, nuclear receptors such as peroxisome proliferator-activated receptor-γ, glucocorticoid receptor, and liver X receptors can inhibit the signal-activated exchange of co-repressors for co-activators at poised promoters, inhibiting the initiation of transcription[15,17]. Lastly, several mechanisms regulate resolution of inflammation at the transcriptional level, including transrepression mechanisms that can remove transcription factors such as NF-κB from inflammatory gene promoters, effectively blocking expression of secondary response genes, that is, genes which require chromatin-modification as well as protein synthesis for their induction (e.g., Nos2 and ISGs)[8,16,17]. Timely resolution of an inflammatory response is crucial in order to limit cellular and tissue damage caused by prolonged or chronic inflammation. Our results suggest that Phf15 may be involved in regulating all three of the abovementioned mechanisms.
However, how might Phf15 be involved in regulating transcriptional repression of the inflammatory response in microglia? PHF15 was first described in embryonic stem cells as an E3 ligase that directly targets Lysine-specific demethylase 1 (LSD1, Kdm1a) - a key demethylase of histone 3 lysine 4 - for degradation[22]. LSD1 has been identified as a member of the CoREST co-repressor complex[59,60], which is required for transcriptional repression of inflammation in microglia[8]. We therefore initially hypothesized that increased levels of Phf15 upon aging might lead to decreased levels of LSD1 and increased microglial inflammatory output. Our results, however, demonstrate that Phf15 itself inhibits microglial inflammatory function; thus, its purported mechanism for inhibition is likely not via degradation of LSD1.
Interestingly, the global transcriptional changes caused by Phf15 deletion are highly similar to previously reported age-associated transcriptional changes in microglia[9,61,62]. In particular, a study by Deczkowska et al.[63], found “immune system process” and specifically “response to virus” among the most highly upregulated biological categories for differentially expressed genes in microglia of young (2-month old) versus aged (22-month old) mice, consistent with our results in Phf15 KO microglia. Notably, a study by Hammond et al.[62], which used single-cell RNAseq to look at microglia profiles throughout the mouse lifespan, found subpopulations in aged (P540) mouse brains which were largely: (1) inflammatory, that is, they upregulated IL-1β, Tnfα, and other cytokines; or (2) IFN-I-responsive, upregulating Irf7 and ISGs, particularly Ifit3, Isg15, Oasl2, interferon induced transmembrane protein 3 (Ifitm3), and receptor transporter protein 4 (Rtp4), compared to younger adult (P100) brains. Similarly, a recent study from the Tabula Muris Consortium[64], which produced a single-cell transcriptomic atlas of 23 tissues and organs across the Mus musculus life span, confirmed that microglia in the aged (P540 and P720) brain are enriched for IFN-I-responsive genes and upregulate a similar set of genes including Ifit3, Irf7, Isg15, Oasl2, Ifitm3, and Rtp4. The genes upregulated by the interferon-responsive microglia clusters in both studies are highly similar to those upregulated in our Phf15 KO cells under basal conditions [see Figure 5A and C]. Because ISGs can modulate inflammation[23], it is possible that interferon-responsive microglia could play a role in contributing to the inflammatory signature found in the aged brain. Interestingly, among the set of downregulated genes in Phf15 KO cells at baseline and 6 h after LPS stimulation is myocyte enhancer factor 2C (Mef2C). MEF2C is an important checkpoint inhibitor that restrains microglial activation in response to proinflammatory insults and is lost in brain aging via IFN-I mediated downregulation[63,65]. Thus, an increase in Phf15 expression in microglia during healthy aging could putatively work to counteract not only microglial activation but increased IFN-I in the aged brain as well.
Notably, a recent study by Readhead et al.[66] found that several virus species are commonly present in the aged human brain. Among them, human herpesvirus 6A and 7 (HHV-6A and HHV-7) were highly upregulated in the brain of AD patients and were found to modulate host genes associated with AD risk, for example amyloid precursor protein (APP) processing. APP is the precursor molecule whose proteolysis forms Aβ and formation of Aβ plaques has long been thought of as the driving force behind Alzheimer’s disease[67]. Aβ has more recently been found to have antimicrobial properties[68], conferring increased resistance against infection from both bacteria and viruses[69]. App is among the significantly upregulated genes under basal conditions in our Phf15 KO cells (log2-fold change = 1.492 and P adj < 0.0001; see Figure 5A). Upregulation of App due to loss of Phf15 in mouse microglia is thus consistent with our data showing Phf15 regulation of the antiviral microglial response.
Altogether, our results show that Phf15 is a novel repressor of microglial inflammatory gene expression, regulating both the magnitude and time-to-resolution of the inflammatory response. Importantly, Phf15 also serves to repress baseline inflammatory output in the absence of immune activation. Putatively, increases in Phf15 during healthy aging could help counteract brain inflammation and protect brain health.
Future studies will determine the mechanism of action of Phf15. For example, the identity of its binding partner proteins and its genome-wide binding sites and associated histone marks will be elucidated to determine the specific gene regulatory regions it interacts with (e.g., active enhancers or promoters). Additionally, studies in Phf15 KO mice will determine whether loss of Phf15-mediated repression of proinflammatory factors is sufficient to induce cognitive decline or exacerbate LPS-induced neurotoxicity of dopaminergic neurons in the substantia nigra.
Declarations
AcknowledgmentsWe thank Prof. Ellen Robey for helpful comments on the manuscript and Wendy Yan for technical assistance.
Authors’ contributionsDesigned and performed experiments, analyzed data, and wrote the manuscript: Muroy SE
Performed experiments and analyzed data: Timblin GA, Preininger MK
Performed experiments: Cedillo P
Designed experiments and wrote the manuscript: Saijo K
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Berkeley Fellowship to S.E.M., ADA Postdoctoral fellowship to G.A.T., NSF GRFP to M.K.P., and R01HD092093 and Pew Scholarship to K.S.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateAll procedures were approved by the Animal Care and Use Committee of the University of California, Berkeley (Animal Use Protocol AUP-2017-02-9539).
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011;91:461-553.
2. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005;308:1314-8.
3. Parkhurst CN, Gan WB. Microglia dynamics and function in the CNS. Curr Opin Neurobiol 2010;20:595-600.
4. Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Ann Rev Immunol 2009;27:119-45.
5. Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 2011;11:775-87.
6. Wake H, Moorhouse AJ, Miyamoto A, Nabekura J. Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci 2013;36:209-17.
7. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 2007;10:1387-94.
8. Saijo K, Winner B, Carson CT, Collier JG, Boyer L, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009;137:47-59.
9. Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol 2014;88:594-604.
10. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci 2018;21:1359-69.
11. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med 2017;23:1018-27.
12. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell 2010;140:918-34.
13. McCoy MK. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J Neurosci 2006;26:9365-75.
14. Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, et al. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson’s disease. FASEB J 2002;16:1474-6.
15. Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol 2005;6:44-55.
16. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol 2010;10:365-76.
17. Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol 2009;9:692-703.
18. Smale ST, Natoli G. Transcriptional control of inflammatory responses. Cold Spring Harb Perspect Biol 2014;6:a016261.
20. Smale ST, Tarakhovsky A, Natoli G. Chromatin contributions to the regulation of innate immunity. Ann Rev Immunol 2014;32:489-511.
21. Soreq L, Rose J, Soreq E, Hardy J, Trabzuni D, et al. Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep 2017;18:557-70.
22. Han X, Gui B, Xiong C, Zhao L, Liang J, et al. Destabilizing LSD1 by Jade-2 promotes neurogenesis: an antibraking system in neural development. Mol Cell 2014;55:482-94.
23. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014;346:89-93.
24. Gabuzda D, Yankner BA. Physiology: inflammation links ageing to the brain. Nature 2013;497:197-8.
25. Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, et al. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J 2005;19:1329-31.
26. Sparkman NL, Johnson RW. Neuroinflammation associated with aging sensitizes the brain to the effects of infection or stress. Neuroimmunomodulation 2008;15:323-30.
27. Stewart SA. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003;9:493-501.
28. Sena-Esteves M, Tebbets JC, Steffens S, Crombleholme T, Flake AW. Optimized large-scale production of high titer lentivirus vector pseudotypes. J Virol Methods 2004;122:131-9.
29. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2012;29:15-21.
30. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:31-21.
31. Blighe K, Rana S, Lewis M. emphEnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling n.d. Available from: https://doi.org/10.18129/B9.bioc.EnhancedVolcano [Last accessed on 6 Mar 2020].
32. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 2019;10:1523.
33. Craik FIM, Salthouse TA. The handbook of aging and cognition. 1st ed. Psychology Press; 2011.
34. Simen AA, Bordner KA, Martin MP, Moy LA, Barry LC. Cognitive dysfunction with aging and the role of inflammation. Ther Adv Chronic Dis 2011;2:175-95.
35. Nagamoto-Combs K, Kulas J, Combs CK. A novel cell line from spontaneously immortalized murine microglia. J Neurosci Methods 2014;233:187-98.
36. Zhao J, Bi W, Xiao S, Lan X, Cheng X, et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci Rep 2019;9:5790.
37. Glaros TG, Chang S, Gilliam EA, Maitra U, Deng H, et al. Causes and consequences of low grade endotoxemia and inflammatory diseases. Front Biosci (Schol Ed) 2013;5:754-65.
38. Sandiego CM, Gallezot JD, Pittman B, Nabulsi N, Lim K, et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci U S A 2015;112:12468-73.
40. Chen YC, Yip PK, Huang YL, Sun Y, Wen LL, et al. Sequence variants of toll like receptor 4 and late-onset Alzheimer’s disease. PLoS One 2012;7:e50771.
41. Perez-Pardo P, Dodiya HB, Engen PA, Forsyth CB, Huschens AM, et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut 2019;68:829-43.
45. McDonald PP. Transcriptional regulation in neutrophils: teaching old cells new tricks. Adv Immunol 2004;82:1-48.
46. Zheng S, Hedl M, Abraham C. Twist1 and Twist2 contribute to cytokine downregulation following chronic NOD2 stimulation of human macrophages through the coordinated regulation of transcriptional repressors and activators. J Immunol 2015;195:217-26.
47. Lo HYG, Jin RU, Sibbel G, Liu D, Karki A, et al. A single transcription factor is sufficient to induce and maintain secretory cell architecture. Genes Dev 2017;31:154-71.
48. Kaminska B, Mota M, Pizzi M. Signal transduction and epigenetic mechanisms in the control of microglia activation during neuroinflammation. Biochim Biophys Acta 2016;1862:339-51.
49. Nagatsu T, Sawada M. Inflammatory process in Parkinson’s disease: role for cytokines. Curr Pharm Des 2005;11:999-1016.
50. Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, et al. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci Lett 1994;172:151-4.
51. Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, et al. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett 1994;165:208-10.
52. Qin XY, Zhang SP, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease. JAMA Neurol 2016;73:1316-9.
53. Rizzo FR, Musella A, De Vito F, Fresegna D, Bullitta S, et al. Tumor necrosis factor and interleukin-1β modulate synaptic plasticity during neuroinflammation. Neural Plast 2018;2018:1-12.
55. Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 2006;281:21362-8.
56. Ye L, Huang Y, Zhao L, Li Y, Sun L, et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem 2013;125:897-908.
57. Denver P, McClean P. Distinguishing normal brain aging from the development of Alzheimer’s disease: inflammation, insulin signaling and cognition. Neural Regen Res 2018;13:1719-12.
58. Njie eMalick G, Boelen E, Stassen FR, Steinbusch HWM, Borchelt DR, et al. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging 2012;33:195.e1-12.
59. Foster CT, Dovey OM, Lezina L, Luo JL, Gant TW, et al. Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol Cell Biol 2010;30:4851-63.
60. Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005;437:432-5.
61. Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 2016;19:504-16.
62. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 2019;50:253-71.e6.
63. Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, et al. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun 2017;8:717.
64. The Tabula Muris Consortium, Pisco AO, McGeever A, Schaum N, Karkanias J, Neff NF, et al. A single cell transcriptomic atlas characterizes aging tissues in the mouse. bioRxiv; 2019. Available from: https://doi.org/10.1101/661728 [Last accessed on 6 Mar 2020].
65. Deczkowska A, Amit I, Schwartz M. Microglial immune checkpoint mechanisms. Nat Neurosci 2018;21:779-86.
66. Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 2018;99:64-82.e7.
68. Vijaya Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 2016;8:340ra72.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Muroy SE, Timblin GA, Preininger MK, Cedillo P, Saijo K. Phf15 - a novel transcriptional repressor regulating inflammation in a mouse microglial cell line. Neurosciences 2020;7:166-82. http://dx.doi.org/10.20517/2347-8659.2020.16
AMA Style
Muroy SE, Timblin GA, Preininger MK, Cedillo P, Saijo K. Phf15 - a novel transcriptional repressor regulating inflammation in a mouse microglial cell line. Neuroimmunology and Neuroinflammation. 2020; 7(2): 166-82. http://dx.doi.org/10.20517/2347-8659.2020.16
Chicago/Turabian Style
Muroy, Sandra E., Greg A. Timblin, Marcela K. Preininger, Paulina Cedillo, Kaoru Saijo. 2020. "Phf15 - a novel transcriptional repressor regulating inflammation in a mouse microglial cell line" Neuroimmunology and Neuroinflammation. 7, no.2: 166-82. http://dx.doi.org/10.20517/2347-8659.2020.16
ACS Style
Muroy, SE.; Timblin GA.; Preininger MK.; Cedillo P.; Saijo K. Phf15 - a novel transcriptional repressor regulating inflammation in a mouse microglial cell line. Neurosciences. 2020, 7, 166-82. http://dx.doi.org/10.20517/2347-8659.2020.16
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 11 clicks
Cite This Article 11 clicks

 Like This Article 11
likes
Like This Article 11
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.