
An association between mitochondria and microglia effector function: what do we think we know?

Abstract
While resident innate immune cells of the central nervous system, the microglia, represent a cell population unique in origin, microenvironment, and longevity, they assume many properties displayed by peripheral macrophages. One prominent shared property is the ability to undergo a metabolic switch towards glycolysis and away from oxidative phosphorylation (OXPHOS) upon activation by the pro-inflammatory stimuli lipopolysaccharide. This shift serves to meet specific cellular demands and allows for cell survival, similar to the Warburg effect demonstrated in cancer cells. In contrast, normal surveillance phenotype or stimulation to a non-proinflammatory phenotype relies primarily on OXPHOS and fatty acid oxidation. Thus, mitochondria appear to function as a pivotal signaling platform linking energy metabolism and macrophage polarization upon activation. These unique shifts in cell bioenergetics in response to different stimuli are essential for proper effector responses at sites of infection, inflammation, or injury. Here, we present a summary of recent developments as to how these dynamics characterized in peripheral macrophages are displayed in microglia. The new insights provided by an increased understanding of metabolic reprogramming in macrophages may allow for translation to the central nervous system and a better understanding of microglia heterogeneity, regulation, and function.
Keywords
Introduction
The function of innate immune cells such as macrophages is to recognize and respond to a novel stimulus including microbial pathogens and sterile activators. These pro- and anti-inflammatory responses are major sources of soluble molecules, cytokines, hormones, and neuropeptides These factors provide tools to sense, process, and relay physiological signals beyond their canonical roles. Macrophages display a heterogeneous repertoire to fulfill a broad range of functions in host defense, including tissue homeostasis and repair, pathology, and development. To accomplish this, innate immune cells adopt various activation phenotypes. Precise regulation of such activation is essential for maintenance of tissue homeostasis with governance accomplished by a balance of stimulatory and inhibitory signals. Multiple lines of evidence suggest an interlinked relationship between innate immunity and the integrity and function of mitochondria serving to maintain this homeostatic balance. Metabolic pathways provide the necessary energy and serve to regulate phenotype and function. Pro-inflammatory macrophages {M[LPS(+IFNγ)]} display an enhanced glycolytic metabolism and impaired mitochondrial oxidative phosphorylation (OXPHOS). These energy shifts place mitochondria in a pivotal signaling role in macrophage response to stimuli and circumventing immune checkpoint signals[1-3]. The link between immunological and metabolic processes associated with mitochondria, immunometabolism, may influence activation states and polarization of myeloid cells to fine-tune their functions[4-7].
Of the specialized cells of the central nervous system (CNS), basic host defense mechanisms exist predominantly in microglia as resident macrophages. Microglia share many phenotypic characteristics with peripheral macrophages yet are unique in their origin and molecular or transcriptional profile[8-15]. The available literature on the immunometabolism of microglia, as compared to what is known of peripheral macrophages, is limited but growing to address questions of similarities and differences[6,16]. It may also allow for a framework to understand the various other tasks undertaken by microglia during development and chronic maintenance. Here, we present a summary of how these dynamics characterized in peripheral macrophages are displayed in microglia. While much of this work is still somewhat under a “work in progress” classification, even in the peripheral macrophage, new insights provided by an increased understanding of metabolic reprogramming foster a better understanding of macrophage and microglia regulation and function.
Microglia and immune cells in the CNS
The mechanical separation of the CNS from the circulation by the blood-brain barrier[17,18] influences immune responses[19,20] by excluding many peripherally derived innate and adaptive immune cells and inflammatory molecules[21]. However, infiltrating cells significantly contribute to any neuroinflammatory response following disruption of the blood-brain barrier, as can occur with physical injury or high levels of inflammation. In such cases, blood-borne monocytes are allowed to enter the brain parenchyma and, over time, can transition and assume a brain-specific phenotype[22-24]. Additionally, with a T cell-mediated neuroinflammation, peripheral innate immune cells enter the brain as a protective host defense against infection and facilitate repair following stroke or physical trauma[25,26]. In such a case, interactions between microglia and T cells can be signaled via interleukin (IL)-23 and IL-1β, leading to the production of granulocyte-macrophage colony-stimulating factor (GM-CSF) to facilitate microglia proliferation in a manner to promote an appropriate level of response to injury[27]. Recently, the identification of innate lymphoid cells in the brain suggests an additional innate immune cell population that may act to control neuroinflammation[28]. Thus, in such conditions, the macrophage population likely represents a combination of resident microglia and infiltrating monocytes. As a distinction between these two populations, it has been suggested that resident microglia focus on tasks related to maintaining tissue homeostasis while infiltrating cells are involved in severe inflammatory injuries[29,30].
Sensing and responding to the environment
While microglia appear to be tightly adapted to the specific requirements within brain regions, they all function in a surveillance mode with mobile processes extending into the surrounding microenvironment to detect tissue changes[31,32]. Upon sensing such changes, microglia respond to their environment via several “sensome” genes, allowing them to sense and interact with their local environment[8,14,33]. These sensome genes include those for putative purinergic receptors, P2ry12 and P2ry13, transmembrane protein 119 (Tmem119), G-protein coupled receptor 34 (Gpr34), the C-type lectin receptor, the fractalkine receptor, Cx3cr1, sialic acid-binding immunoglobulin-type lectin H (Siglec-h), and triggering receptor expressed on myeloid cells 2 (Trem2). Siglec proteins contribute to immune regulation by binding sialic acid residues on neurons[34] and TREM2 contributes via recruitment of the immunoreceptor tyrosine-based activation motif-containing adapter protein, DAP-12[35]. The final response of the cell is dictated by the overall pattern of sensome gene activation.
The microglia host-response begins with the recognition of pathogen-associated molecular patterns (PAMPs) such as bacterial, viral, and protozoal products (protein lipid, nucleic acid, and carbohydrate). This occurs via pattern recognition receptors on the plasma membrane or in the endosomal compartments[36], or by binding phagocytic scavenger receptors[37] and macrophage antigen complex I (MAC1, CD11b/CD18), which is a pattern recognition receptor linked to the superoxide-generating enzyme NADPH oxidase[38]. In the absence of microorganisms, a similar but sterile inflammatory response occurs often as a result of trauma, ischemia-reperfusion injury, or chemical exposure[39-41]. Activation in the absence of microbial compounds occurs by endogenous molecules called danger-associated molecular patterns (DAMPs)[42]. Molecules that function as DAMPs include nucleic acids, lipids, and proteins that normally are not present to immune cells until released or unmasked during cell death due to tissue injury. In the CNS, microglia responding in various neurodegenerative diseases in the absence of pathogen have been termed disease-associated microglia (DAM). Intracellular DAMPs include high mobility group box 1 (HMGB1) and peroxiredoxin family proteins. These damage signals can activate immune cells through three major families of intracellular recognition receptors: toll-like receptors, nucleotide-binding domain leucine-rich repeat containing proteins (also known as NOD-like receptors), and Rig1-like receptors. Receptor activation induces specific pathways and the release of cytokines that contribute to injury mitigation[43]. Microglia are influenced by a plethora of factors including receptor agonists[44-48] and transcription factor inducers[49]. It is thought that microglial receptors[50-52] can act as molecular switches to control microglial responses and that many of these actions function through alterations in calcium signals[53]. In all cases, the immediate response upon sensing DAMPs, PAMPs, or other damaging events requires a robust increase in metabolic demand to support actions that initially are beneficial to the homeostatic balance of the nervous system.
Injury-induced inflammatory processes are dynamic and demonstrate spatial and temporal heterogeneity[54-56]. In general, characterization of the macrophage response is based on the nature of the activating stimulus and the resulting production of factors[57]. A conceptual framework has been proposed that suggests the nature of the activating stimulus can drive a range of activation phenotypes[58-62], and it has been used as a basis for characterizing cellular responses[63-70]. While phenotypic activation-state distinctions are currently under scrutiny[67,71], it has been shown that classically activated microglia associated with inflammation can be produced upon stimulation with agonists for toll-like receptors (e.g., lipopolysaccharide, LPS) or IFNγ receptors. In contrast, different aspects of the immune response that do not involve the classical response can be observed upon stimulation by IL-4 or IL-13 with the expression of anti-inflammatory cytokines (IL-4, IL-10, IL-13, and TGF-β), arginase-1 (Arg1), CD206, and Chitinase-3-like-3 (Ym1 in rodents)[72-76]. It is considered that the different phenotypes may be related, yet have different roles in host defense, wound healing, and resolution of inflammation[57,60]. Differences in metabolic processes have been identified across these different activation inducers, suggesting a role for mitochondria in phenotypic outcome[77].
Stimulus-driven metabolic re-programming of microglia
In a normal “resting” cell, energy demands are addressed with the conversion of glucose to pyruvate with entry into glycolysis. Pyruvate in the cytosol can be taken up by mitochondria and enters the tricarboxylic acid (TCA) cycle where it is oxidized to generate ATP. This provides a total energy gain of approximately 36 ATP per one molecule of glucose. In contrast, with hypoxia or anoxia, the cell has the ability to divert pyruvate away from mitochondria OXPHOS, allowing for ATP generation during low oxygen conditions. In this case, one glucose molecule will generate two pyruvate molecules that will be converted to lactate by lactate dehydrogenase in the cytosol[78,79]. While this reaction generates significantly fewer molecules of ATP, glycolysis proceeds due to the production of NAD+. While less efficient, a beneficial effect of a shift to glycolysis is that it can be very quickly induced to meet cellular demands in cells with high glucose capacity[80]. The importance of this shift was initially demonstrated in cancer cells in what is known as the Warburg effect[81,82]. In cancer, malignant cells shift their demand for biosynthetic precursors and energy change and change their metabolic profile from a relatively low rate of glycolysis and the oxidation of pyruvate by the TCA cycle. The shift in metabolic profile is characterized by a lower rate of OXPHOS, high rate of glycolysis, and elevated lactic acid production. The high glycolytic rate induced during the Warburg effect is driven by the need to meet the increased demand for production of nucleotides and amino acids. While this effect was initially identified and characterized in cancer cells, a similar ability to utilize such a metabolic switch has been demonstrated in immune cells to meet increased energy demands when responding to infection or injury[1].
There is now evidence suggesting a role for metabolic reprogramming by mitochondria in the maintenance and establishment of innate and adaptive immune responses[75,83-96]. Given that immune cell populations depend on unique effector functions in response to distinct stimuli that often require production and secretion of high amounts of signaling factors and antimicrobial agents, it follows that changes in mitochondria function to meet these demands are crucial for efficient response to distinct contexts[97-101]. It was initially observed that, upon activation, macrophages increase glycolysis and decrease oxygen consumption[102,103]. It was further demonstrated that macrophage phenotype can be shifted by reprogramming glucose metabolism[104,105], which helps meet energy demands required for shifting cell function and survival[106].
Under normal conditions, microglia exist in a surveillance phenotype for constant monitoring of the parenchyma[107,108] and preferentially rely on oxidative metabolism[90,109,110]. Upon activation by LPS[94], amyloid-b[111], and iron loading[112,113], microglia switch their reliance on OXPHOS metabolism[69,110,114,115] towards glycolytic metabolism to maintain mitochondrial function and ensure cell survival[91,94,95,116]. Voloboueva et al.[94] showed that, upon stimulation by LPS, BV-2 microglia increased lactate production and decreased mitochondria oxygen consumption and ATP production. This shift was reported to be modulated by mitochondrial glucose-regulated protein 75/mortalin[94]. Exposure to a combination of LPS and IFN-γ increases nitric oxide formation, glucose consumption, hexokinase activity, glucose-6-phosphate dehydrogenase activity, phosphofructokinase-1 activity, lactate dehydrogenase activity, and lactate release, suggesting potentiated glycolysis[94]. Similar findings were reported by Orihuela et al.[69]: following LPS, BV2 microglia and primary murine microglia shifted from a primary oxidative metabolic towards glycolytic metabolism with no evidence of cell death. An increase in microglial mitochondria has been observed with activation[117,118], implicating an association with mitochondria biogenesis. Recent studies have suggested that a shift in glycolysis in microglia is accompanied by an increase in the enzyme PFKFB3, which is responsible for activation of phosphofructokinase[119]. Additionally, this metabolic shift has been found to be regulated by the anti-inflammatory cytokine IL-10 for aerobic glycolysis inhibition and OXPHOS[120].
In a non-classical activation state, macrophages use oxidative metabolism for functions involved in normal maintenance functions, tissue repair, and wound healing[73,121,122]. In IL-4 stimulated macrophages [M(IL-4)], the Krebs cycle and OXPHOS remain intact. In addition, the cells are able to utilize fatty acid oxidation and oxidative respiration for energy production[98,123-125] and arginine metabolism is shifted to ornithine and polyamines[126]. Work by Ferger et al.[118] suggested that the stability of the electron transport chain in mitochondria plays a more substantial and critical role for the microglia response to IL-4 as compared to the response to LPS. In microglia, exposure to LPS induced a rapid and transitory decrease in the mitochondrial uncoupling protein-2 (UCP-2) levels accompanied by increased mitochondrial reactive oxygen species (mtROS) production. In UCP-2-silenced microglia, the response to LPS was exacerbated and a response to IL-4 was eliminated[127]. An earlier study examining the translation of responses in macrophages to microglia reported reduced glucose consumption and lactate production in BV-2 cells exposed to IL-4[109]. It was suggested that this phenotype was associated with phagocytosis of debris and the reduced need for anabolic reactions. Similar findings were reported by Orihuela et al.[69] with exposure of BV-2 cells or primary murine microglia to IL-4/IL-13 in that the cells remained within an oxidative metabolic state with OCR and ECAR levels similar to non-stimulated cells. There were also elevations in mRNA levels for Ym1, Il4, Cd163, and Arg1, but no induction of Tnfa or Il1[69]. The lack of a demonstrated metabolic shift with IL-4/IL-13 stimulation is in contrast to observations in peripheral macrophages of stimulated glucose uptake in addition to fatty acid metabolism and shift in mitochondrial biogenesis[125].
Mitochondria and free radical production
A key feature of classically activated macrophages is their ability to produce reactive oxygen species (ROS) to facilitate killing of phagocytized bacteria[128]. Stimulation of macrophages with LPS and IFN-γ increases inducible nitric oxide synthase (iNOS), generating nitric oxide (NO), a reactive nitrogen species that can inhibit mitochondrial respiration by nitrosylating iron-sulfur proteins in electron transport chain complexes and cytochrome c oxidase[129,130]. It is considered that iNOS and NO-mediated inhibition of mitochondrial metabolism in macrophages is essential for the metabolic switch activated by LPS. This is not as well established in microglia, especially given that, while nitric oxide production is often linked with pro-inflammatory cytokines, such cytokines can be stimulated by sterile activators in the absence of NO. In microglia, it has been proposed that activation of the rapamycin (mTOR) pathway may actively contribute to this process[131] as well as pro-inflammatory cytokine production and phagocytic activity[132-134]. The resulting elevated thiamin pyrophosphate activity increased production of purines and pyrimidines, which yield nicotinamide adenine dinucleotide phosphate (NADPH) for the NADPH oxidase enzyme and ROS production[135] implicated in the transition of microglia to a pro-inflammatory phenotype[136-139]. It has been proposed that glycolytic ATP production may utilize the electron transport chain to compensate for this shift towards ROS production[128]. It is known that superoxide produced by NADPH oxidase is predominantly extracellular. In vivo, extracellular superoxide dismutase 3 (SOD3) forms membrane permeable H2O2. Studies have suggested that H2O2, rather than SOD, serves as the primary ROS involved in mediating microglial activation and proliferation in response to pro-inflammatory stimuli[136,140,141]. H2O2 has also been implicated in the increase in CD11b expression both in vitro and in vivo[142], as well as in persistent neuroinflammation related to impaired NF-kB p50 function[143]. Superoxide anion is the primary ROS produced by mitochondria and mitochondria-derived H2O2 and, in addition to NADPH oxidase, may contribute to a pro-inflammatory phenotype of microglia such as that observed with the mitochondrial toxin, rotenone[144]. With a response sufficient to result in ROS production, the associated intracellular damage is limited by increased generation of NADPH required for maintenance of reduced glutathione and nitric oxide production[145,146].
Glucose
In addition to the critical role that glucose plays in energy metabolism, it serves as an exclusive substrate for the hexose monophosphate shunt, which produces NADPH that is required by glutathione reductase to convert oxidized glutathione (GSSG) back to reduced glutathione (GSH). It also serves to quench ROS and repair oxidative damage through glutathione- and thioredoxin-coupled pathways[147]. Glucose metabolism influences microglial activation through an NADH-sensitive co-repressor termed C-terminal binding protein (CtBP). Slowed glucose flux through glycolysis reduces NADH levels and reduce NADH:NAD+ ratio[148]. In both microglia and macrophage RAW264.7 cells, glucose flux regulates iNOS expression and other pro-inflammatory genes through effects on cytosolic NADH:NAD+ ratio and CtBP[149].
Several glucose transporters such as GLUT1[150], GLUT3[151], and GLUT5[152] are expressed in microglia. Acute fluctuation of available glucose impacts microglia activity with an elevated response to LPS upon shifting from a normal to high glucose level. Shifting from a high to normal glucose level can also induce metabolic stress[153]. Glucose levels can influence pro-inflammatory gene transcription by several mechanisms. One such mechanism relies on the formation of advanced glycation end-products (AGE). These products consist of modified proteins and lipids as a result of non-enzymatic reactions with sugars. It is known that microglia express receptors of AGE and, upon activation, pro-inflammatory signaling pathways are stimulated[127,154]. In peripheral macrophages, it has been reported that a shift in the cell’s energy source induced by glucose deprivation results in an altered response to a pro-inflammatory stimulus[155-157]. Multiple studies have reported an inability of microglia to respond appropriately to LPS under oxygen and glucose deprivation or with 2-DG inhibition of glucose metabolism[109]. However, there is evidence that microglia are capable of functioning with alternative energy sources to adequately respond to an inflammatory challenge. Choi et al.[158] reported an increase in mRNA and protein levels for IL-6 in microglia after 7 h of glucose and serum free medium. Upon stimulation with LPS, glucose-deprived microglia retained their normal ability to respond with elevations in nitrite, IL-1β, and TNFα[159]. Primary rat microglia shifted to glucose-free medium for 1 h to LPS showed an exacerbated release of NO within 24 h and similar elevations in TNFα and IL-1β as compared to non-glucose-deprived cells. Glucose deprivation for 24 h prior to LPS exposure increased release of IL-1β with no deficits in NO or TNFα. The authors suggested that microglia were able to mobilize fatty acids from intracellular lipid droplets as an energy source. The majority of studies examining the effects of glucose deprivation have focused on relatively short-term exposures, within 1-24 h. While these studies demonstrated that both peripheral macrophages and microglia can shift their response to a pro-inflammatory stimulus in a selective manner, the question remains as to whether such a response would be altered when the cells were forced to a more prolonged shift in energy metabolism. When RAW 264.7 [Figure 1] or BV-2 [Figure 2] cells were maintained for three days under culture conditions to force cells to rely on galactose as an alternative energy source, the cells were able to normally respond to LPS stimulation. However, the diminished pro-inflammatory cytokine response observed when 2-DG was used in previous studies to inhibit glycolysis may have been related to the lower basal OXPHOS induced[160]. The differences across these studies likely lie with the method of depleting glucose: removing glucose from the medium; the addition of 2-DG, which in and of itself can lower basal induction of OXPHOS[160]; or the combination of glucose deprivation with hypoxia. In RAW cells, the morphological changes observed with LPS activation have been demonstrated to be diminished under galactose, suggesting a requirement of glucose to facilitate cell spreading[161]. This was not clearly observed in the current study where similar LPS-induced morphological patterns were observed in the absence of pyruvate [Figure 1]. In BV-2 cells, a slight morphological shift was observed with the low level of LPS stimulation with minimal induction of nitrate and an elevation in TNFα and IL-1 protein.
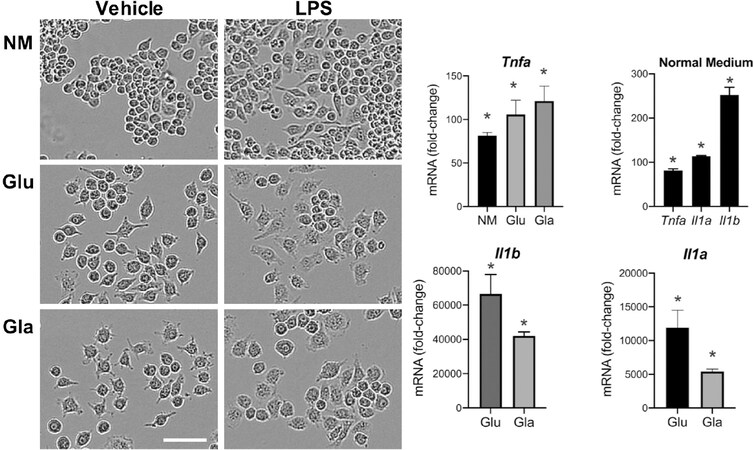
Figure 1. Response of RAW 264.7 cells to LPS under glucose or galactose medium conditions. RAW 264.7 cells were plated in 6-well tissue culture plates (Corning, Corning, NY) and maintained in normal growth medium (NM) [DMEM (Gibco, ThermoFisher, Waltham, MA) containing 4.5 g/L glucose, 2 mM L-glutamine, sodium pyruvate, supplemented with 100 U/mL penicllin/ streptomycin (Sigma-Aldrich, Burlington, MA) and 10% fetal bovine serum (FBS #1 00-106, 0.25 endotoxin units/mL; Gemini Bio-Products, Sacramento, CA) and allowed to reach 85% confluence over 3-5 days. Cells were maintained at 37 °C, 5% CO2/5% O2, 90% humidity (Nu-5831 tri-gas incubator, Nuaire, Plymouth, MN). NM was changed to phenol-free NM medium or phenol-free DMEM medium containing 2 mM L-glutamine and 100 U/mL penicillin/streptomycin supplemented with (2) high glucose (25 mM; Glu), or 3] galactose (10 mM; Gla). Cells were maintained in the experimental medium for 3 days following which, under the same media conditions, cells were exposed to lipopolysaccharide (LPS; 1 μg/mL; Sigma) for up to 18 h and monitored using a live cell imaging system (IncuCyte, Sartorius) under normal incubator conditions. (A) Representative images (20x) of cell morphology under normal medium (NM), high glucose (Glu), or galactose (Gla). Scale bar = 25 microns. (B) Samples were collected at 3 h post-LPS and mRNA isolated using TRIzol® Reagent (Invitrogen, Carlsbad, CA), and 2.5 μL cDNA was used for qRT-PCR for Tnfa, II 1a, II 1b, and cyclophilin A using TaqManTM. Individual gene expression levels were normalized to cyclophilin A and presented as fold-change from vehicle controls in each medium condition. Data were analyzed by 2-way ANOVA followed by Dunnett’s test for independent group mean comparisons. Data represent mean +/- SEM (n = 3-4). *Significance level as compared to vehicle control set at P < 0.05
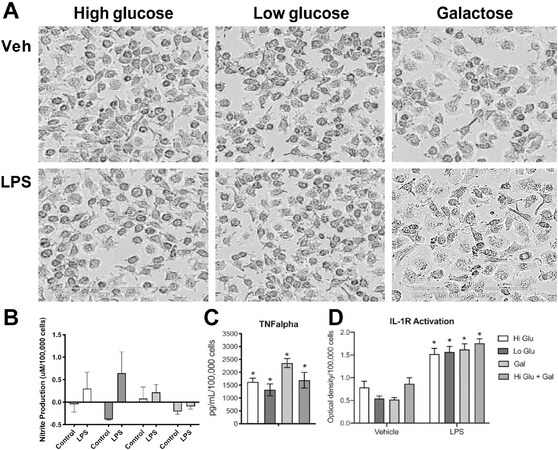
Figure 2. Response of BV-2 cells to LPS under glucose or galactose medium conditions. BV-2 murine microglia cells were plated in 6-well tissue culture plates (Corning, Corning, NY) and maintained in normal growth medium (DMEM (Gibco, ThermoFisher, Waltham, MA) containing 4.5 g/L glucose, 2mM L-glutamine, supplemented with 100 U/mL penicillin/streptomycin (Sigma- Aldrich, Burlington, MA) and 10% fetal bovine serum (FBS #100-106, 0.25 endotoxin units/mL; Gemini Bio-Products, Sacramento, CA) and allowed to reach 85% confluence over 3-5 days. Cells were maintained at 37 °C, 5% CO2/5% O2, 90% humidity (Nu-5831 tri gas incubator, Nuaire, Plymouth, MN). The normal growth medium was changed to phenol-free complemented DMEM medium containing 2mM L-glutamine, supplemented with 100 U/mL penicllin/ strepto supplemented with 1) high glucose (25 mM), 2) low glucose (10 mM), 3) galactose (10mM), or 4) high glucose + galactose). Cells were maintained in the experimental medium for 3 days following which, under the same media conditions, cells were exposed to lipopolysaccharide (LPS; 100 ng/mL; Sigma) monitorec using a live cell imaging system (IncuCyte) under normal incubator conditions for up to 18 h. A: Representative images (20x) of cell morphology under high glucose (Hi Glu), low glucose (Lo Glu), or galactose (Gal). Scale bar = 200 microns; B: Nitrite accumulation in culture medium was measured as an indirect indicator of nitric oxide synthesis using a Greiss Reagent kit (Promega, Madison, WI) following manufacturer s instructions. Estimates were determined relative to standard curve; C: Estimates of the release of IL-1 into the medium were obtained using HEX BlueTM IL-1R cells (InvivoGen, San Diego, CA) following manufacturer’s instructions. From 20 μL aliquot of the medium, after a 1.5 h incubation at 37 °C, absorbance at 620 nm was measured using a BioTeck Synergy 4 plate reader. Data were calculated relative to background control; D: TNFα protein levels were determined by Mouse TNFα ELISA MAX kit (BioL egend, San Diego, CA) with BD OptEIA Reagent Set B according to manufacturer’ instructions. Absorbance was measured at 450nm with a 570nm background subtraction. Protein levels were determined based on standard curve and calculated relative to total cell number as determined using IncuCyte software. Data represent mean +/- SEM (n = 5-6). Data were analyzed by 2-way ANOVA followed by Bonferroni post-hoc comparisons. *Significance level as compared to vehicle control set at P < 0.05
Glutamine
Macrophages utilize glutamine at high rates to synthesize amino acids, nucleotides, NADPH, and energy production and are dependent upon extracellular sources of the amino acid[103]. Channeling of glutamine into the Krebs cycle is a primary route to promote succinate synthesis in macrophages. This occurs with glutamine being used for synthesis of glutamate, GABA, and succinate, bypassing the TCA cycle[155]. This stabilizes hypoxia-inducible factor 1-alpha (HIF-1α), an oxygen-sensitive transcription factor that allows the cell to adapt to a hypoxic environment[162]. It has been suggested that HIFs function to facilitate cross-talk between inflammation and metabolism[163]. HIF-1a can induce the expression of pro-inflammatory cytokines and has been proposed to serve a role in shifting glycolytic pathways to favor anaerobic metabolism[114]. With classic activation, succinate regulates HIF-1a to drive a sustained production of IL-1β[100] and the subsequent cell actions serve to maintain the macrophage survival. For the non-classical phenotype, glutamine metabolism acts at multiple levels including the generation of a-ketoglutarate and serves as a substrate for UDP-GlcNAc synthesis[164].
NLRP3 inflammasome
One biological response to an inflammatory event that is critically dependent upon metabolic regulation is inflammasome activation. This is especially relevant for inflammasomes that require prior cell priming for full activation upon a secondary stimulus. For example, glycolytic rate can influence formation of NLRP1 and NLRP3 inflammasomes in macrophages[165,166]. Inflammasomes are multiprotein complexes formed in the cytosol of immune and neural cells in response to pathogenic and danger signals. They consist of a cytosolic sensor belonging to the AIM2 (absent in melanoma 2), or NLR, an adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD), and an effector caspase, primarily caspase-1. There are a variety of inflammasomes, most of which fall into the NLR domain[167]. NLRP1 and AIM2 inflammasomes have been characterized in neurons[168-170] and the NLRC4, NLRP2, and NLRP3 inflammasomes in astrocytes[171,172]. Components for multiple inflammasomes are expressed in microglia[173,174]. The NLRP3 inflammasome responds to a number of activators, including sterile activators, such as asbestos, silica crystals, aluminum salts, and polystyrene nanoparticles[175-178], and aberrant proteins, such as extracellular Aβ[179], thereby contributing to a broad range of common inflammatory pathologies and chronic inflammation.
The NLRP3 inflammasome responds to metabolic regulation[180] and has been increasingly recognized as a bridge between mitochondrial damage sensing and pro-inflammatory signaling within monocytes, including microglia[181,182]. Unlike most inflammasomes, NLRP3 typically requires a two-step activation and it is this process for which there is mounting evidence that mitochondrial damage plays a contributing role[101]. Activation of TLR, tumor necrosis factor receptor, or interleukin-1 receptor (IL-1R) initiates an intracellular cascade of effects, including activation of NF-kB. This upregulates NLRP3 and pro-IL-1β within the cell and facilitates post-transcriptional changes to NLRP3 to free ubiquitinated binding sites by BRCC3 (BRCA1/2-containing complex subunit 3)[176,183,184]. Delivery of a secondary “trigger” such as PAMPs, DAMPs, or intact pathogens to the “primed” cell causes the release of the repressed state of NLRP3. Upon release, NLRP3 activates the inflammasome forming a multiprotein complex comprised of the cytosolic sensor NLRP3, ASC, and caspase 1[185]. Caspase 1 facilitates the cleavage of the pro-forms of IL-1β and IL-18[186], resulting in the release of mature protein[187]. The release of active IL-1 family cytokines is normally related to pyroptotic cell death; however, in the absence of cell death, hyperactivity of cells and the recruitment of a process dependent on plasma membrane-localized pores can result in similar protein release[188,189]. While inflammasome activation is an efficient producer of mature IL-1β, inflammasome independent mechanisms exist, including cathepsin B or caspase 11 dependent pathways[190,191], bacterial pore-forming toxins, and extracellular ATP[177]. Thus, an upregulation of mature IL-1β does not automatically indicate an inflammasome mechanism. In addition to the release of inflammatory factors, the physical release of ASC specks into the extracellular environment represents a stimulus for activating phagocytic cells in the immediate environment, thus contributing to a prolonged propagation of inflammation[192] or other biological responses[193].
Induction of mitophagy, the process by which cells clear damaged mitochondria, has been implicated in inhibition of NLRP3 signaling[194]. Release of oxidized mitochondrial DNA (mtDNA) produced during the priming stage[195,196] can interact with the NLRP3 receptor and induce inflammasome activation. Nakahira et al.[196] reported that inhibition of mitophagy in macrophages heightened the NLRP3 inflammasome activation in parallel with uncleared mitochondrial DNA released into the cytosol. The mitochondrial cytidine/uridine monophosphate kinase-2 (CMPK2) is a nucleotide kinase required for mtDNA synthesis and production of oxidized mtDNA fragments. These fragments can act as activating ligands for the NLRP3 inflammasome complex[197]. In addition, the release of mtROS triggered by small molecule inhibition of complex I and III has been associated with NLRP3 inflammasome activation[198]. The association between mtROS as a trigger for NLRP3 inflammasome activation remains controversial given potential off-target effects of mtROS inhibitors. While studies have reported a role for mtROS in NLRP3 inflammasome activation, other conflicting studies have been reported. At least one study reported that mtROS inhibitors do not block the secondary activation step, but rather the initial priming step[199]. Apart from acting as an activator of NLRP3, mitochondria can act as a docking system for inflammasome assembly. This interaction is driven by the externalization of mitochondrial lipid cardiolipin from the inner membrane to the outer membrane, which then independently interacts with caspase-1 and NLRP3[200].
NLRP3 inflammasome activation in microglia has gained attention as a contributing mechanism in several neuroinflammatory disease pathologies including Alzheimer’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and Parkinson’s disease[201-205]. While much is similar between the biochemistries of microglia and macrophages, differences in inflammasome activation have been reported. For example, exposure of microglia cells to the antioxidant NAC did not affect LPS priming yet inhibited Aβ 1-42 peptide stimulation of caspase-1 dependent IL-1β secretion[202]. While microglia show similar expression of inflammasome components and response to stimulus, the dependency on caspase-1 for IL-1β secretion is only partial and a higher level of mature IL-1β secretion is observed with longer periods of priming than in hematopoietic macrophages. Prolonged IL-1β secretion from microglia likely occurs as a result of a deficit in negative regulation mechanisms as compared to macrophages. As an additional consideration, activation of the inflammasome in peripheral macrophages serves in a regulatory capacity in the induction of pyroptotic cell death to remove the damaging immune cell. How this translates to microglia remains in question given the long-lived nature of these cells.
Conclusion
The interest in metabolic functions of microglia has evolved from our knowledge of mitochondrial demands and responses of peripheral macrophages in their various effector functions. Recent findings have set the framework for an association between the metabolic status of immune cells with the characteristics of an immune response against pathogens. The majority of studies have relied on different pro-inflammatory stimuli, such as virus, GM-CSF, LPS, LPS + INFγ, or IL-4, to induce cells to examine macrophage metabolism in vitro. However, the resulting phenotype and metabolic profiles can differ with not all stimuli leading towards glycolysis. Conversely, the diverse non-inflammatory stimuli normally examined, i.e., IL-10, glucocorticoids, IL-13, M-CSF, and IL-4, are grouped together[74]; however, similar to the pro-inflammatory stimuli, the phenotypic change may differ. This is not unexpected given that macrophage activation states display multiple profiles depending on the initiating stimuli[206]. However, examination of metabolic adaptations of macrophages has demonstrated that such adaptations are critical factors regulating a variety of immune cell responses. The need to rapidly modulate cellular responses to pathogen or inflammatory signals demands a remodeling of the metabolic pathways to execute such actions. While many of the basic responses translate across peripheral macrophages and microglia, the uniqueness of microglia suggests that this may not be a complete translation across cells. Additionally, the limited range of inducing stimuli examined in microglia cells raises the question of how the cells will compare given a broader range of stimuli. Further exploration of similarities and uniqueness will contribute to our understanding of the interplay between metabolism and immune cell responses as they apply to the nervous system. It may also offer a framework from which to address issues of translation between experimental animal data to human disease conditions that involve the innate immune system[207-209]. Understanding the mitochondrial-related characteristics of microglia will likely be critical in identifying successful therapeutic approaches to the detrimental effects of neuroinflammation or in facilitating repair.
Declarations
AcknowledgmentsThe authors acknowledge Drs. Christopher McPherson, Christian Lefebvre d’Hellencourt, and Negin Martin for providing reviewer comments on the manuscript.
Authors’ contributionsContributed to the conceptualization, design and interpretation of the experiments and in manuscript preparation: Childers G
Generated the original data: Lopez Hernandes I, Giridharan S
Contributed to the conceptualization of the manuscript, data interpretation, and writing of the final manuscript: Harry GJ
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe research was supported by NIH intramural research funding ES021164.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 2011;32:157-64.
2. Boyapati RK, Tamborska A, Dorward DA, Ho GT. Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. F1000Res 2017;6:169.
3. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, et al. Oxidized mitochondrial DNA activated the NRLP3 inflammasome during apoptosis. Immunity 2012;36:401-14.
4. O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol 2016;16:553-65.
5. Russell DG, Huang L, VanderVen BC. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol 2019;19:291-304.
6. Lynch MA. Can the emerging field of immunometabolism provide insights into neuroinflammation? Prog Neurobiol 2020;184:101719.
7. Ghosh M, Xu Y, Pearse DD. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J Neuroinflammation 2016;13:9.
8. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 2014;17:131-43.
9. Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 2012;60:717-27.
10. Finsen B, Owens T. Innate immune responses in central nervous system inflammation. FEBS Lett 2011;585:3806-12.
11. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010;330:841-5.
12. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 2013;16:1896-905.
13. Holtman IR, Skola D, Glass CK. Transcriptional control of microglia phenotypes in health and disease. J Clin Invest 2017;127:3220-9.
14. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest 2012;122:1164-71.
15. Crotti A, Ransohoff RM. Microglial physiology and pathophysiology: insights from genome-wide transcriptional profiling. Immunity 2016;44:505-15.
16. Borst K, Schwabenland M, Prinz M. Microglia metabolism in health and disease. Neurochem Int 2019;130:104331.
17. Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 2005;57:173-85.
18. Hermann DM, El-Ali A. The abluminal endothelial membrane in neurovascular remodeling in health and disease. Sci Signal 2012;5:re4.
19. Ifergan I, Kébir H, Bernard M, Wosik K, Dodelet-Devillers A, et al. The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain 2008;131:785-99.
20. Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, Del Zoppo GJ, et al. Immunologic privilege in the central nervous system and the blood-brain barrier. Cereb Blood Flow Metab 2013;33:13-21.
21. Banerjee S, Bhat MA. Neuron-glial interactions in blood-brain barrier formation. Annu Rev Neurosci 2007;30:235-58.
22. King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 2009;113:3190-7.
23. Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci 2007;12:1544-53.
24. Vallieres L, Sawchenko PE. Bone marrow-derived cells that populate the adult mouse brain preserve their hematopoietic identity. J Neurosci 2003;23:5197-207.
25. Trifilo MJ, Montalto-Morrison C, Stiles LN, Hurst KR, Hardison JL, et al. CXC chemokine ligand 10 controls viral infection in the central nervous system: evidence for a role in innate immune response through recruitment and activation of natural killer cells. J Virol 2003;78:585-94.
26. González H, Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflam 2014;11:201.
27. Li J1, Gran B, Zhang GX, Ventura ES, Siglienti I, et al. Differential expression and regulation of IL-23 and IL-12 subunits and receptors in adult mouse microglia. J Neurol Sci 2003;215:95-103.
28. Romero-Suárez S, Del Rio Serrato A, Bueno RJ, Brunotte-Strecker D, Stehle C, et al. The central nervous system contains ILC1s that differ from NK cells in the response to inflammation. Front Immunol 2019;10:2337.
29. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 2014;14:392-404.
30. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 2014;211:1533-49.
31. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005;8:752-8.
32. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005;308:1314-8.
33. Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 2013;4:385-401.
34. Angata T, Kerr SC, Greaves DR, Varki NM, Crocker PR, et al. Cloning and characterization of human Siglec-11. A recently evolved signaling molecule that can interact with SHP-1 and SHP-2 and is expressed by tissue macrophages, including brain microglia. J Biol Chem 2002;277:24466-74.
35. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 2005;201:647-57.
36. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010;11:373-84.
37. Wilkinson K, El Khoury J. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer’s disease. Int J Alzheimers Dis 2012;2012:489456.
38. Pei Z, Pang H, Qian L, Yang S, Wang T, et al. MAC1 mediates LPS-induced production of superoxide by microglia: the role of pattern recognition receptors in dopaminergic neurotoxicity. Glia 2007;55:1362-73.
39. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010;10:826-37.
40. Shechter R, Schwartz M. CNS sterile injury: just another wound healing? Trends Mol Med 2013;19:135-43.
41. McPherson CA, Merrick BA, Harry GJ. In vivo molecular markers for pro-inflammatory cytokine M1 stage and resident microglia in trimethyltin-induced hippocampal injury. Neurotox Res 2014;25:45-56.
42. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 2008;8:279-89.
44. Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci 2007;30:596-602.
45. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 2007;10:1387-94.
46. Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature 2010;468:253-62.
47. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011;91:461-553.
48. Domercq M, Vázquez-Villoldo N, Matute C. Neurotransmitter signaling in the pathophysiology of microglia. Front Cell Neurosci 2013;7:49.
50. Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci 2007;30:527-35.
51. Garcia-Revilla J, Alonso-Bellido IM, Burguillos MA, Herrera AJ, Espinosa-Oliva AM. Reformulating pro-oxidant microglia in neurodegeneration. J Clin Med 2019;8:1719.
52. Lee JK, Chung J, Kannarkat GT, Tansey MG. Critical role of regulator G-protein signaling 10 (RGS10) in modulating macrophage M1/M2 activation. PLoS One 2013;8:e81785.
53. Seifert S, Pannell M, Uckert W, Färber K, Kettenmann H. Transmitter- and hormone-activated Ca(2+) responses in adult microglia/brain macrophages in situ recorded after viral transduction of a recombinant Ca(2+) sensor. Cell Calcium 2011;49:365-75.
54. Ajami B, Samusik N, Wieghofer P, Ho PP, Crotti A, et al. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat Neurosci 2018;21:541-51.
55. Pettersen JS, Fuentes-Duculan J, Suárez-Fariñas M, Pierson KC, Pitts-Kiefer A, et al. Tumor-associated macrophages in the cutaneous SCC microenvironment are heterogeneously activated. J Invest Dermatol 2011;131:1322-30.
56. Ziegler-Heitbrock L, Hofer TPJ. Toward a refined definition of monocyte subsets. Front Immunol 2013;4:23.
57. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008;8:958-69.
60. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010;11:889-96.
61. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012;122:787-95.
62. Hume DA, Freeman TC. Transcriptomic analysis of mononuclear phagocyte differentiation and activation. Immunol Rev 2014;262:74-84.
63. Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 2009;4:399-418.
64. Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol 2009;210:3-12.
65. Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 2013;39:3-18.
66. Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 2014;11:98.
67. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014;41:14-20.
68. Loane D, Kumar A. Microglia in the TBI Brain: the good, the bad, And the dysregulated. Exp Neurol 2016;275:316-27.
69. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 2016;173:649-65.
70. Gerrick KY, Gerrick ER, Gupta A, Wheelan SJ, Yegnasubramanian S, et al. Transcriptional profiling identifies novel regulators of macrophage polarization. PLoS One 2018;13:e0208602.
72. Stein M. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med 1992;176:287-92.
73. Doyle AG, Herbein G, Montaner LJ, Minty AJ, Caput D, et al. Interleukin-13 alters the activation state of murine macrophages in vitro: comparison with interleukin-4 and interferon-gamma. Eur J Immunol 1994;24:1441-5.
74. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 2014;6:1-13.
75. Mills EL, O’Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol 2016;46:13-21.
76. Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol 2006;6:379-86.
77. Bordt EA, Polster BM. NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: a bipartisan affair? Free Radic Biol Med 2014;76:34-46.
79. Liemburg-Apers DC, Schirris TJ, Russel FG, Willems PH, Koopman WJ. Mitoenergetic dysfunction triggers a rapid compensatory increase in steady-state glucose flux. Biophys J 2015;109:1372-86.
80. Vazquez A, Liu J, Zhou Y, Oltvai ZN. Catabolic efficiency of aerobic glycolysis: the Warburg effect revisited. BMC Syst Biol 2010;4:58.
82. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029-33.
83. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015;42:406-17.
84. Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 2015;25:771-84.
85. Mehta MM, Weinberg SE, Chandel NS. Mitochondrial control of immunity: beyond ATP. Nat Rev Immunol 2017;17:608-20.
86. Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol 2019;10:1462.
89. Ryan DG, O’Neill LAJ. Krebs cycle reborn in macrophage immunometabolism. Annu Rev Immunol 2020. Epub ahead of print. doi: 10.1146/annurev-immunol-081619-104850
90. Bernhart E, Kollroser M, Rechberger G, Reicher H, Heinemann A. Lysophosphatidic acid receptor activation affects the C13NJ microglia cell line proteome leading to alterations in glycolysis, motility, and cytoskeletal architecture. Proteomics 2010;10:141-58.
91. Moss DW, Bates TE. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Eur J Neurosci 2001;13:529-38.
93. Lartigue L, Faustin B. Mitochondria: Metabolic regulators of innate immune responses to pathogens and cell stress. Int J Biochem Cell Biol 2013;45:2052-56.
94. Voloboueva LA, Emery JF, Sun X, Giffard RG. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett 2013;587:756-62.
95. Gimeno-Bayon J, Lopez-Lopez A, Rodriguez MJ, Mahy N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci Res 2014;92:723-31.
96. Chenais B, Morjani H, Drapier JC. Impact of endogenous nitric oxide on microglial cell energy metabolism and labile iron pool. J Neurochem 2002;81:615-23.
97. Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragones J, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 2010;185:605-14.
98. Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol 2011;6:275-97.
99. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol 2013;229:176-85.
100. Galvan-Pena S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol 2014;5:420.
101. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol 2017;18:488-98.
102. Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J 1986;239:121-5.
103. Newsholme P, Gordon S, Newsholme EA. Rates of utilization and fates of glucose, glutamine, pyruvate, fatty acids and ketone bodies by mouse macrophages. Biochem J 1987;242:631-6.
104. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab 2012;15:813-26.
105. Blagih J, Jones RG. Polarizing macrophages through reprogramming of glucose metabolism. Cell Metab 2012;15:793-5.
106. Bonora M, Patergnani S, Rimessi A, De Marchi E, Suski JM, et al. ATP synthesis and storage. Purinergic Signal 2012;8:343-57.
107. Bernier LP, Bohlen CJ, York EM, Choi HB, Kamyabi A, et al. Nanoscale surveillance of the brain by microglia via cAMP-regulated filopodia. Cell Reports 2019;27:2895-908.
108. Madry C, Kyrargyri V, Arancibia-Carcamo IL, Jolivet R, Kohsaka S, et al. Microglial ramification, surveillance, and interleukin-1beta release are regulated by the two-pore domain K(+) channel THIK-1. Neuron 2018;97:299-312.
109. Ghosh S, Castillo E, Frias ES, Swanson RA. Bioenergetic regulation of microglia. Glia 2018;66:1200-12.
110. Cherry JD, Olschowka JA, O’Banion MK. Are ‘resting’ microglia more ‘m2’? Front Immunol 2014;5:594.
111. Baik SH, Kang S, Lee W, Choi H, Chung S, et al. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab 2019;30:493-507.e6.
112. Holland R, McIntosh AL, Finucane OM, Mela V, Rubio-Araiz A, et al. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav Immun 2017;68:183-96.
113. McIntosh A, Mela V, Harty C, Minogue AM, Costello DA, et al. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol 2019;29:606-21.
114. Wang T, Liu H, Lian G, Zhang SY, Wang X, et al. HIF1 α -induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediators Inflamm 2017;2017:1-10.
115. Nair S, Sobotka KS, Joshi P, Gressens P, Fleiss B, et al. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 2019;67:1047-61.
116. Chénais B, Morjani H, Drapier JC. Impact of endogenous nitric oxide on microglial cell energy metabolism and labile iron pool. J Neurochem 2002;81:615-23.
117. Banati RB, Egensperger R, Maassen A, Hager G, Kreutzberg GW, et al. Mitochondria in activated microglia in vitro. J Neurocytol 2004;33:535-41.
118. Ferger AI, Campanelli L, Reimer V, Muth KN, Merdian I, et al. Effects of mitochondrial dysfunction on the immunological properties of microglia. J Neuroinflamm 2010;7:45.
119. Finucane OM, Sugrue J, Rubio-Araiz A, Guillot-Sestier MV, Lynch MA. The NLRP3 inflammasome modulates glycolysis by increasing PFKFB3 in an IL-1beta dependent manner in macrophages. Sci Rep 2019;9:4034.
120. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017;356:513-9.
121. Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol 2011;89:557-63.
122. Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol Res 2012;53:11-24.
123. Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 2014;15:846-55.
124. Liu PS, Wang H, Li X, Chao T, Teav T, et al. Alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 2017;18:985-94.
125. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, et al. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab 2006;4:13-24.
126. Mills CD, Kincaid K, Alt JM, Hellman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol 2000;164:6166-73.
127. De Simone R, Ajmone-Cat MA, Pandolfi M, Bernardo A, De Nuccio C, et al. The mitochondrial uncoupling protein-2 is a master regulator of both M1 and M2 microglial responses. J Neurochem 2015;135:147-56.
128. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011;472:476-80.
129. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep 2016;17:684-96.
130. Poderoso JJ, Helfenberger K, Poderoso C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 2019;88:61-72.
131. Hu Y, Mai W, Chen L, Cao K, Zhang B, et al. mTOR-mediated metabolic reprogramming shapes distinct microglia functions in response to lipopolysaccharide and ATP. Glia 2020;68:1031-45.
132. Li D, Wang C, Yao Y, Chen L, Liu G, et al. mTORC1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from M1 type to M2 type. FASEB J 2016;30:3388-99.
133. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, et al. TREM2 maintains microglial metabolic fitness in alzheimer’s disease. Cell 2017;170:649-63.e13.
134. Zhao X, Liao Y, Morgan S, Mathur R, Feustel P, et al. Noninflammatory changes of microglia are sufficient to cause epilepsy. Cell Rep 2018;22:2080-93.
135. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245-313.
136. Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferon gamma. J Neurosci Res 2004;77:540-51.
137. Qin L, Liu Y, Wang T, Wei SJ, Block ML, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 2004;279:1415-21.
138. Loane DJ, Stoica BA, Pajoohesh-Ganji A, Byrnes KR, Faden AI. Activation of metabotropic glutamate receptor 5 modulates microglial reactivity and neurotoxicity by inhibiting NADPH oxidase. J Biol Chem 2009;284:15629-39.
139. Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J Neurochem 2012;120:292-301.
140. Eguchi H, Fujiwara N, Sakiyama H, Yoshihara D, Suzuki K. Hydrogen peroxide enhances LPS-induced nitric oxide production via the expression of interferon beta in BV-2 microglial cells. Neurosci Lett 2011;494:29-33.
141. Mander PK, Jekabsone A, Brown GC. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J Immunol 2006;176:1046-52.
142. Roy A, Jana A, Yatish K, Freidt MB, Fung YK, et al. Reactive oxygen species up-regulate CD11b in microglia via nitric oxide: implications for neurodegenerative diseases. Free Radic Biol Med 2008;45:686-99.
143. Taetzsch T, Levesque S, McGraw C, Brookins S, Luqa R, et al. Redox Regulation of NF-κB p50 and M1 Polarization in Microglia. Glia 2015;63:423-40.
144. Yuan YH, Sun JD, Wu MM, Hu JF, Peng SY, et al. Rotenone could activate microglia through NF-kB associated pathway. Neurochem Res 2013;38:1553-60.
145. Kletzien RF, Harris PK, Foellmi LA. Glucose-6-phosphate dehydrogenase: a “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J 1994;8:174-81.
146. Salvemini F, Franzé A, Iervolino A, Filosa S, Salzano S, et al. Enhanced glutathione levels and oxidoresistance mediated by increased glucose-6-phosphate dehydrogenase expression. J Biol Chem 1999;274:2750-7.
148. Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 2008;10:179-206.
149. Shen Y, Kapfhamer D, Minnella AM, Kim JE, Won SJ, et al. Bioenergetic state regulates innate inflammatory responses through the transcriptional co-repressor CtBP. Nat Commun 2017;8:624.
150. Wang L, Pavolu S, Du X, Bhuckory M, Xu H, et al. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol Neurodegener 2019;14:2.
151. Kalsbeek MJ, Mulder L, Yi CX. Microglia energy metabolism in metabolic disorder. Mol Cell Endocrinol 2016;438:27-35.
152. Payne J, Maher F, Simpson I, Mattice L, Davies P. Glucose transporter Glut 5 expression in microglial cells. Glia 1997;21:327-31.
153. Hsieh C, Liu C, Lee C, Yu LE, Wang JY. Acute glucose fluctuation impacts microglial activity, leading to inflammatory activation or self-degradation. Sci Rep 2019;9:840.
154. Chen J, Sun Z, Jin M, Tu Y, Wang S, et al. Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-κB pathway. J Neuroimmunol 2017;305:108-14.
155. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013;496:238-42.
156. O’Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med 2016;213:15-23.
157. Wang F, Zhang S, Jeon R, Vuckovic, Jiang X, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EbioMedicine 2018;30:303-16.
158. Choi SJ, Shin IJ, Je KH, Min EK, Kim EJ, et al. Hypoxia antagonizes glucose deprivation on interleukin 6 expression in an Akt dependent, but HIF-1/2α independent manner. PLoS One 2013;8:e58662.
159. Churchward MA, Tchir DR, Todd KG. Microglial function during glucose deprivation: inflammatory and neuropsychiatric implications. Mol Neurobiol 2018;55:1477-87.
160. Weng F, Zhang S, Vuckovic I, Ryounghoon J, Lerman A, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab 2018;28:463-75.
161. Venter G, Oerlemans FTJJ, Wijers M, Willemse M, Fransen JAM, et al. Glucose controls morphodynamics of LPS-stimulated macrophages. PLoS One 2014;9:e96786.
162. Meiser J, Krämer L, Sapcariu SC, Battello N, Ghelfi J, et al. Pro-inflammatory macrophages sustain pyruvate oxidation through pyruvate dehydrogenase for the synthesis of itaconate and to enable cytokine expression. J Biol Chem 2016;291:3932-46.
163. Shay JE, Celeste-Simon M. Hypoxia-inducible factors: crosstalk between inflammation and metabolism. Semin Cell Dev Biol 2012;23:389-94.
164. Liu PS, Wang H, Li X, Chao T, Teav T, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 2017;8:985-94.
165. Moon JS, Hisata S, Park MA, DeNicola GM, Ryter SW, et al. mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep 2015;12:102-15.
166. Xie M, Yu Y, Kang R, Zhu S, Yang L, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun 2016;7:13280.
167. Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 2013;39:432-41.
168. Adamczak SE, de Rivero Vaccari JP, Dale G, Brand FJ, Nonner D, et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J Cereb Blood Flow Metab 2014;34:621-29.
169. Kaushal V, Dye R, Pakavathkumar P. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ 2015;22:1676-86.
170. Yogarajah T, Ong KC, Perera D, Wong KT. AIM2 inflammasome-mediated pyroptosis in enterovirus A71-infected neuronal cells restricts viral replication. Sci Rep 2017;7:5845.
171. Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013;87:3655-67.
172. Freeman L, Guo H, David CN, Brickey WJ, Jha S, et al. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med 2017;214:1351-70.
173. de Rivero Vaccari JP, Dietrich WD, Keane RW. Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J Cereb Blood Flow Metab 2014;34:369-75.
174. Burm SM, Zuiderwijk-Sick EA, ‘t Jong AE, van der Putten C, Veth J, et al. Inflammasome-induced IL-1β secretion in microglia is characterized by delayed kinetics and is only partially dependent on inflammatory caspases. J Neurosci 2015;35:678-87.
175. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008;9:847-56.
176. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 2009;10:241-7.
177. Hanamsagar R, Torres W, Kielian T. Inflammasome activation and IL-1beta/IL-18 processing are influenced by distinct pathways in microglia. J Neurochem 2011;119:736-48.
178. Lunov O, Syrovets T, Loos C, Nienhaus GU, Mailander V, et al. Amino-Functionalized polystyrene nanoparticles activate the NLRP3 inflammasome in human macrophages. ACS Nano 2011;5:9648-57.
179. Schnaars M, Beckert H, Halle A. Assessing β-amyloid-induced NLRP3 inflammasome activation in primary microglia. Methods Mol Biol 2013;1040:1-8.
181. Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, et al. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol 2013;191:5230-8.
182. Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, et al. NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 2015;19:10:e0130624.
183. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation - the negative regulation of NF-kB and the NLRP3 inflammasome. Nat Immunol 2017;18:861-9.
184. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev 2015;265:35-52.
185. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016;16:407-20.
186. Zambetti LP, Laudisi F, Licandro G, Ricciardi-Castagnoli P, Mortellaro A. The rhapsody of NLRPs: master players of inflammation...and a lot more. Immunol Res 2012;53:78-90.
187. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009;27:519-50.
188. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, et al. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 2018;48:35-44.e6.
189. Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, et al. The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur J Immunol 2018;48:584-92.
190. Lee HM, Kang J, Lee SJ, Jo EK. Microglial activation of the NLRP3 inflammasome by the priming signals derived from macrophages infected with mycobacteria. Glia 2013;61:441-52.
191. Viganò E, Mortellaro A. Caspase-11: the driving factor for noncanonical inflammasomes. Eur J Immunol 2013;43:2240-5.
192. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 2014;15:727-37.
193. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017;552:355-61.
194. Gurung P, Lukens JR, Kanneganti TD. Mitochondria: diversity in the regulation of NLRP3 inflammasome. Trends Mol Med 2015;3:193-201.
195. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012;36:401-14.
196. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011;12:222-30.
197. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018;560:198-203.
198. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011;469:221-5.
199. Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, et al. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 2011;187:613-7.
200. Elliott EI, Miller A, Banoth B, Iyer SS, Stotland A, et al. Cutting edge: mitochondrial assembly of the NLRP3 inflammasome complex is initiated at priming. J Immunol 2018;200:3047-52.
201. Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, et al. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS One 2013;8:e55375.
202. Parajuli B, Sonobe Y, Horiuchi H, Takeuchi H, Mizuno T, et al. Oligomeric amyloid beta induces IL-1beta processing via production of ROS: implication in Alzheimer’s disease. Cell Death Dis 2013;4:e975.
203. Bellezza I, Grottelli S, Costanzi E, Scarpelli P, Pigna E, et al. Peroxynitrite activates the NLRP3 inflammasome cascade in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Mol Neurobiol 2018;55:2350-61.
204. Venegas C, Heneka MT. Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J 2019;33:13075-84.
205. Deora V, Lee JD, Albornoz EA, McAlary L, Jagaraj CJ, et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 2020;68:407-21.
206. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014;40:274-88.
207. Healy LM, Yaqubi M, Ludwin S, Antel JP. Species differences in immune-mediated CNS tissue injury and repair: a (neuro)inflammatory topic. Glia 2020;68:811-29.
208. Stoger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 2012;225:461-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Harry GJ, Childers G, Giridharan S, Hernandes IL. An association between mitochondria and microglia effector function: what do we think we know?. Neurosciences 2020;7:150-65. http://dx.doi.org/10.20517/2347-8659.2020.07
AMA Style
Harry GJ, Childers G, Giridharan S, Hernandes IL. An association between mitochondria and microglia effector function: what do we think we know?. Neuroimmunology and Neuroinflammation. 2020; 7(2): 150-65. http://dx.doi.org/10.20517/2347-8659.2020.07
Chicago/Turabian Style
Harry, Gaylia Jean, Gabrielle Childers, Sahana Giridharan, Irisyunuel Lopez Hernandes. 2020. "An association between mitochondria and microglia effector function: what do we think we know?" Neuroimmunology and Neuroinflammation. 7, no.2: 150-65. http://dx.doi.org/10.20517/2347-8659.2020.07
ACS Style
Harry, GJ.; Childers G.; Giridharan S.; Hernandes IL. An association between mitochondria and microglia effector function: what do we think we know?. Neurosciences. 2020, 7, 150-65. http://dx.doi.org/10.20517/2347-8659.2020.07
About This Article
Copyright

Data & Comments
Data


 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.