
Microglial contributions to aberrant neurogenesis and pathophysiology of epilepsy

Abstract
Microglia are dynamic cells that constitute the brain’s innate immune system. Recently, research has demonstrated microglial roles beyond immunity, which include homeostatic roles in the central nervous system. The function of microglia is an active area of study, with insights into changes in neurogenesis and synaptic pruning being discovered in both health and disease. In epilepsy, activated microglia contribute to several changes that occur during epileptogenesis. In this review, we focus on the effects of microglia on neurogenesis and synaptic pruning, and discuss the current state of anti-seizure drugs and how they affect microglia during these processes. Our understanding of the role of microglia post-seizure is still limited and may be pivotal in recognizing new therapeutic targets for seizure intervention.
Keywords
Introduction
Epilepsy is a neurological disorder characterized by recurrent seizures. Microglia, the innate immune cells of the central nervous system (CNS), are increasingly recognized as mediators of seizures and contributors to the epileptogenic process. The progression to epilepsy is characterized by the presence of neuroinflammation, as well as structural and molecular alterations in the brain, that subsequently lead to increased neuronal hyperexcitability and a lasting disposition towards spontaneous recurrent seizures (SRS)[1]. Microglia regulate neuroinflammation and axonal sprouting and have been reported to modulate neurogenesis. Following seizures, microglia are activated, functioning as resident macrophages of the brain and respond quickly to injury while trying to maintain the physiological processes under its control[2]. Changes in neuronal homeostasis are also observed, highlighting the diverse ways in which microglia could be contributing to the development of epilepsy.
This review will discuss the roles of microglia in neuroinflammation and neurogenesis, and how these contributions are altered post-seizure. We will examine microglia in the context of epileptogenesis, the process by which “the previously normal brain is functionally altered and biased towards the generation of abnormal electrical activity that subserves chronic seizures”[3]. Additionally, we will explore studies of pharmacological reagents and their effects on microglia as a therapeutic target to mitigate the epileptogenic process that drives epilepsy.
Epilepsy
Epilepsy is a chronic brain disorder characterized by abnormal brain activity that causes seizures. The propensity to generate recurrent seizure events has neuropathological, cognitive, and social consequences[4]. Epileptic seizures are aberrant, excessive, or synchronous neuronal discharges and manifest in a variety of ways. According to the International League Against Epilepsy (ILAE), seizures are classified into three types based on their onset: generalized onset seizures do not have a determined area of origin and can affect both sides of the brain; focal onset seizures originate from one area of the brain; and unknown onset seizure when the onset is missed or obscured. Generalized onset seizures can present with a variety of manifestations that include non-motor and motor presentations: they range from absence seizures (that present with lapses in awareness, accompanied with staring into space, probably accompanied by rapid blinking and/or orofacial automatisms) to generalized tonic-clonic seizures with tonic and/or clonic spasms, and are always accompanied by loss of consciousness. Focal onset seizures may or may not be accompanied by a loss of awareness and their origin can be attributed to a specific area of the brain that causes motor or sensory changes, including taste or smell. Focal seizures may also result in a loss of awareness, manifested by a person who appears to be dazed, confused, and unable to respond to questions for several minutes. Focal seizures may become generalized if the original behavior, which was localized to one brain hemisphere, expands to behaviors that involve both sides of the brain[5]. The cause of epilepsy in many patients is not known, though acquired causes include stroke, traumatic brain injury (TBI), autoimmune disorders, infection, and tumors.
It is estimated that almost 10% of people will experience a seizure in their lifetime[6]. Epilepsy affects approximately 1.2% of the population in the United States alone[7]. Higher incidence rates have been reported in younger (early childhood and infancy) and older age groups (older than 55 years of age), while a lower prevalence is seen in the period between early adulthood and midlife[8]. The imbalance between excitatory and inhibitory neurotransmission (E/I imbalance), with a propensity towards increased excitation, is believed to be the underlying cause of seizures in epilepsy. Research demonstrates hyper-excitability during ictogenesis, when excitatory glutamatergic activity is increased while inhibitory gamma aminobutyric acid (GABA) ergic activity is dampened[9-11]. Currently, the treatment of epilepsy varies from patient to patient. Anti-seizure medications are typically the first choice of therapy for subsequent seizure prevention. When medication fails, surgery has been successful in significantly decreasing or making patients seizure free, though only a small number of patients with focal onset seizures would qualify for surgical options[12]. When surgery is not an option, patients are treated with antiepileptic drugs (AEDs). There have been > 30 medicines that have been approved by the United States Food and Drug Administration (FDA) or the European Medicines Agency (EMA). Even though many seizure medication options exist, nearly 33% of patients fail to respond to them[13]. Some patients with pharmacologically refractory epilepsy try to control seizures by exploring dietary changes, such as employing the ketogenic diet, a high fat/low carbohydrate diet which can be successful in reducing seizures in about 50% of adult patients[14]. Though originally believed to result in an increase in levels of GABA production[15], there may be multiple mechanisms that contribute to its success in seizure cessation[16]. Neurostimulatory devices, such as deep brain or vagus nerve stimulation therapies, have also been used with varying success, as they help to normalize the excitatory state of the brain[17].
Epileptogenesis
Epileptogenesis is the process by which structural and molecular changes occur in the brain and predispose towards epileptic seizures[18]. The epileptogenic process can be initiated by multiple underlying causes such as tumors, infections, stroke, and brain injuries. Epileptogenesis occurs prior to an unprovoked seizure and continues beyond the event. It is a dynamic process that can occur very quickly, after brain injury or stroke, or over an extended period of time (up to months in animal models, and years in humans)[18,19]. This window presents a temporal opportunity for treatment approaches, but also provides challenges for studying the process. Understanding the pathophysiological changes that occur during epileptogenesis is a pivotal part of developing new therapies.
Changes during epileptogenesis occur in both neuronal and glial cells, all of which contribute to the dysfunction of neuronal circuits. The mechanisms underlying epileptogenesis suggest that the pathophysiological and compensatory changes are connected. Animal models of epileptogenesis have displayed histologically-detectable changes, such as sprouting along the mossy fiber pathway, neurogenesis, and gliosis [Figure 1] alterations, all of which can contribute to the potential for hyperexcitability[20]. The condition most frequently associated with mossy fiber sprouting is temporal lobe epilepsy (TLE), the most common type of epilepsy in adults[21], but can occur in epilepsy patients without TLE[22]. Sprouting occurs when granule cell axons in the inner molecular layer (mossy fibers) project into the hilus of the dentate gyrus and CA3 region of the hippocampal formation, creating their own dendritic field. Mossy fibers synapse onto hilar mossy cells, CA3 pyramidal cells, and interneurons[23] to create de novo recurrent excitatory circuits. Aberrant sprouting in a model of TLE was reported to contribute to excitatory feedback loops of normal and ectopic granule cells[24]. Another study described aberrant mossy fibers that drive inhibitory basket cells to reduce neuronal excitability[25]. Mossy fiber sprouting is increased through the activation of several granule cell factors, such as neuromodulin and brain-derived neurotrophic factor (BDNF)[26], and involves the secretion and deposition of molecules of the extracellular matrix that facilitate aberrant growth[27-29]. The number of granule cells also affects mossy fiber sprouting. Hippocampal neurogenesis, which leads to the formation of new granule cells, is increased shortly after an epileptic seizure, but the increase is transient. The development of new granule cells, and their ectopic integration into neuronal networks contribute to aberrant mossy fiber sprouting that is evident post-seizure.
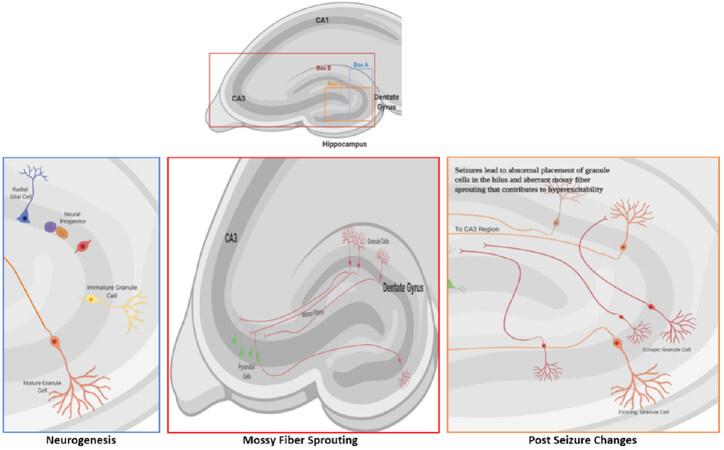
Figure 1. Granule cell neurogenesis and mossy fiber sprouting. A: Neurogenesis occurs in the dentate gyrus of the hippocampus. The cells proliferate in the subgranular zone and then migrate a short distance to the granule cell layer where they differentiate into mature granule cells; B: the axons of granule cells (mossy fibers) normally project to the cells in the CA3 region of the dentate gyrus; C: during seizures, several factors contribute to aberrant migration of granule cells that leads to their ectopic placement in the hilus. Ectopic granule cells (red cells) form functioning neural connections to the pyramidal neurons in the CA3 region and contribute to hyperexcitability and epileptogenesis through aberrant ‘sprouting’ along the mossy fiber pathway. Image created with BioRender.com
Reactive gliosis has also been identified as a contributor to epileptogenesis in genetic and chemically-induced animal models of epilepsy[30]. Activated astrocytes and microglia exhibit changes that promote network hyperexcitability[31,32]. Microglia can be activated by cytokines and monocytes circulating in blood[33], neurotransmitters released by activated or damaged neurons, or by molecules migrating across the blood brain barrier (BBB)[31]. Disruption of the BBB during status epilepticus (SE) leads to the transport of plasma proteins and immune cells into the brain. The combined effects on astrocytic functions, ion concentration changes, entry of infiltrating systemic components, and potential pathogens into the CNS may lead to neuronal dysfunction, neuroinflammation, and neurodegeneration[34]. The BBB plays a pivotal role in diseases associated with neuronal hyperexcitability such as epilepsy, TBI, and post-stroke seizure activity[35-37]. Microglia-neuron signaling had been shown initially by the release of the neuronal chemokine fractalkine, which activates the CXC-chemokine receptor 1 (CXCR1) on microglia. Neurogenesis, synaptic plasticity, and neuronal survival have all been reported to be affected by the CXCR1 signaling pathway[31]. Cytokine release of IL-1β and tumor necrosis factor-α (TNF-α) and other signals (such as HMGB1 and ATP) from activated astrocytes and microglia lead to hyperexcitability in neurons[38,39]. Precise targeting of reactive astrocytes and microglia for therapeutic intervention during epilepsy and epileptogenesis may be beneficial due to microglial involvement in the processes of neurogenesis, axonal sprouting, and neuroinflammation.
Models of epilepsy
The pursuit of AEDs has provided > 30 medications, with many that were developed in the 1980s[40]. Although several animal models of epilepsy exist, clinically validated models, ones that are validated to predict efficacy and tolerability, are limited and currently only consist of three models: the maximal electroshock (MES) seizure protocol, subcutaneous pentylenetetrazol (scPTZ) acute seizure tests, and the kindled rodent model of chronic hyperexcitability[41]. Though not validated, multiple other animal models have been developed that have contributed to the understanding of the premise of new therapeutic options[42]. Still, newer drugs continue to have similar adverse events or side effects without exhibiting greater efficacy[43]. Variation in seizure models can result in acute or chronic seizure paradigms, differences in severity, or the intervening time until seizures start[44]. Acute models lack persisting changes, like a decrease in seizure threshold or spontaneous seizures. Chronic seizure models of epilepsy accommodate a period during which epileptogenesis takes place and may better represent human epilepsy[45]. Newer models, such as the post-SE model, kindling[46], or genetic models, have become more extensively used due to their ability to result in spontaneous seizures. The kindling model, where repeated electrical stimulation leads to enhanced seizure susceptibility, is commonly utilized as it has been associated with seizure induced plasticity and provides a way to study such plasticity. Combining SRS with convulsive behavior or video-electroencephalogram (EEG) represents a more accurate epilepsy model, though it is not considered a clinically validated model for AED discovery.
The chemical induction of status epilepticus, usually by injection of kainic acid or pilocarpine[47,48], can result in animals exhibiting SRSs days to weeks after SE, and allows for the determination of post-seizure changes in the brain neuropil. Models using chemoconvulsants and kindling have provided researchers with a way to study changes in mossy fiber sprouting, neurogenesis, and neuroinflammation post-seizure.
Microglia
Microglia, which make up approximately 10% of the brain’s cells, are the central nervous system’s primary form of immune defense. Originally thought to only serve immune response functions, they are now widely recognized to perform important functions that contribute to the development and maintenance of a healthy brain. Microglia are dynamic cells that survey their environment for injury or infection. Ramified microglia rapidly and constantly extend and retract their processes to assess the environment[49]. By evaluating their surroundings, microglia can actively participate in neurogenesis[50,51], neurotrophic functions[52], neuronal phagocytosis[53], modulation of axonal processes[54], synapse formation and pruning[55-57]. It has also been proposed that microglia aid in neurotransmitter clearance, specifically glutamate[58], due to their upregulation of glutamate transporter GLT-1 in a cortical injury model[59]. Many of these functions however, are reported to be similarly performed by astrocytes.
Microglial contribution to epileptogenesis
Models of epilepsy provide insight into neuronal and glial behavior post-seizure. Microglia sense the injury, and their activation cascade is initiated[60,61] as they migrate to the region of insult, where they then remain activated for about 4-5 weeks post-seizure[62], creating an inflammatory environment around the site of seizure onset. The extent and duration of microglial activation depends on the model used[63]. Most, though not all, chronic seizure models of epileptogenesis present a persistent inflammatory state in neural tissue[64]. After an inciting event, inflammatory cascades can either begin in the CNS, or be activated by molecules in the systemic circulation via breakdown of the BBB[65]. The seizure-induced activation of microglia can be visualized and followed non-invasively by positron emission tomography using 11C-PK11195, a radiolabeled TSPO (a selective translocator protein) that is expressed at low levels in the healthy CNS, but upregulated when neuroinflammation is initiated. Although TSPO does not distinguish between microglia and infiltrating macrophages[66], its upregulation provides clear proof of the neuroinflammatory state of post-seizure CNS. Acute neuroinflammation is thought to contribute to chronic neuroinflammation states or worsen a pre-existing state[67]. Understanding how and when microglia are activated after seizures, and how they contribute over time to neuroinflammation may provide a target for downregulating or attenuating epileptogenesis.
Cytokines are signaling molecules that modulate inflammatory responses and are produced by neurons and glial cells after seizures. Interleukin-1β (IL-1β), IL-2, and IL-6 are present in the brain at low concentrations, which increase post-seizure[68]. Following seizures, mRNA expression of IL-1β, IL-6, TNF-α, TGF-β, and vascular endothelial growth factor (VEGF) were all reported to be upregulated ithe hippocampus. IL-1β may induce seizures by upregulating N-methyl-D-aspartate (NMDA) receptors on post-synaptic cells[69]. Studies also suggest that uncontrolled levels of IL-1β impair synaptic plasticity and cause neuronal dysfunction[70]. Other studies have demonstrated that IL-1β decreased GABA-mediated neurotransmission, leading to neuronal hyperexcitability and seizures[71]. When IL-1β activity was blocked, acute or recurrent seizures were reduced in rodent models[38,72,73]. Anakinra, a recombinant IL-1 receptor antagonist, was successfully used in a clinical study to treat febrile infection-related epilepsy syndrome (FIRES), demonstrating that IL-1β may be a crucial target in controlling seizure recurrence[74]. TNF-α is released by microglia and astrocytes, when low levels of glutamate are detected, to maintain neuronal excitation levels by upregulating synapses[75]. TNF-α also increases microglial glutamate release through glutaminase and gap junction regulation[76] and regulates the adhesion molecule N-cadherin, which is involved in the organization of synapses[77]. Like IL-1β, TNF-α also affects GABA levels by increasing GABA receptor endocytosis, reducing its inhibitory action[78]. Another pro-inflammatory cytokine, IL-6, is upregulated by TNF-α and IL-1β. IL-6 has been reported to decrease hippocampal neurogenesis while increasing microgliosis, possibly contributing to epileptogenesis[79].
Changes in microglia post-seizure
The question of microglial activation status and its effects post-seizure have yet to be answered. Microglia modulate the severity of early seizures in a pilocarpine model with lipopolysaccharide (LPS) pre-conditioning[80]: ablation of microglia prior to seizure onset resulted in dramatic increases of seizure severity. Since no other cell types were affected by the method of microglia ablation[81,82], it is suggested that microglia may play a role early on in seizure induction to protect the CNS from exaggerated neuronal activity. The presence of microglia may thus be beneficial during seizure; however, evidence suggests that their activation may be detrimental post-seizure. Minocycline, a tetracyclic antibiotic that has anti-inflammatory properties, has been shown to act as an inhibitor of microglial proliferation/activation[83]. Studies that used minocycline have reported that it protects against neuronal cell death after seizures, thus indicating that microglia contribute to neurodegeneration following seizures[84]. Other studies demonstrated that a 2-week course of minocycline post-status epilepticus decreased the number, duration, and severity of spontaneous recurrent seizures, suggesting that microglia are involved in the propagation of these SRS[85-87]. It should also be noted, on the other hand, that there are studies that show only partial effectiveness by minocycline[88], or inability to reverse the increase of epileptogenesis[89,90].
Inflammatory cytokines increase neuronal excitability and are believed to contribute to epileptogenesis[91]. Though inflammatory cytokines are expressed by several cell types in the brain, microglia-specific pro-inflammatory cytokines, such as IL-1β, IL6 and TNF-α, showed increased expression three days after SE but had diminished by day 21[63]. Levels of anti-inflammatory cytokines, such as Arg1, IL-4 and IL-10, were also increased. These data contribute to the existing controversy on the role that microglia and cytokines play post-seizure. Additionally, Toll-like receptor (TLR) signaling has been implicated in the production of cytokines in seizure models. Studies have demonstrated that the downregulation of TLR3 and TLR4 activities reduces recurrent and acute seizures, respectively[92,93]. Another study showed that the activated TLR4 pathway (mediated by MyD88) was part of the molecular response contributing to a pro-inflammatory environment post-SE[94]. Matsuda et al.[95] reported that microglia secrete TNF-α to decrease the proliferation of neural progenitor cells (NPCs) in the subgranular zone (SGZ) and demonstrated that microglial activation is partly mediated through TLR9 post-SE. These studies emphasize the need for a better understanding of the role of cytokine signaling post-seizure.
Neurogenesis
Neurogenesis, the incorporation of new neurons into the hippocampus, is a controlled process that affects fundamental brain activities such as memory formation and learning. Neurogenesis, and the newborn cells generated, contribute to brain plasticity and can be followed through maturation using specific markers. The progression from newborn cells to mature neurons can be tracked using markers such as Nestin and Sox-2 for newborn cells, doublecortin and polysialylated neuronal cell adhesion molecule for immature neural progenitor cells, and NeuN for mature neurons[96]. In recent years, there has been an increased effort to determine some of the major regulators of the neurogenic process in the adult brain[97-99]. Neurogenesis, mediated by the activation and differentiation of adult neural stem cells (NSCs), has been documented to occur primarily in two regions of the adult CNS: the subventricular zone (SVZ) of the lateral ventricles, and within the SGZ of the dentate gyrus (DG) in the hippocampus[100,101]. Neurogenesis in the hippocampus will be the main focus of this section, as the hippocampal region has been intimately linked and affected by seizures and epilepsy.
In rodent models of neurogenesis, radial glia-like NSCs located in the SGZ give rise to NPCs[102]. The neurogenic process involves five intricate stages, ultimately leading to the integration of newly mature granule cells in the hippocampus. During the first stage, NSCs proliferate and generate neural progenitors in the SGZ. Stage 2 is the continuous phase of survival, where NSC and progenitor cells are lost through apoptosis, in this early part of the process. During stage 3, progenitor cells undergo fate determination and differentiate into immature neurons. In stage 4, immature neurons migrate a short distance within the granule cell layer where they continue their maturation and integrate (Stage 5) into the hippocampal circuitry, receiving input from the entorhinal cortex, and projecting axons to the CA3 (mossy fibers) and hilar regions of the hippocampus[101,103-106], which further synapse with CA1 pyramidal cells[107].
In epilepsy, while the stimuli to trigger adult neurogenesis are activated, the orchestrated differentiation process is dysregulated at various steps. The newly formed granule neurons do not integrate appropriately into the dentate gyrus, thus forming aberrant connections with other neuronal cells, and contributes to epilepsy and associated cognitive decline[108-110].
The role of microglia in physiological neurogenesis
Variations in neurogenesis properties from the embryonic stages to adulthood have been studied and show that newborn neuron populations decrease with age[111], potentially due to a lowered ability of NSCs to regenerate[112], or changes in environmental cues in the hippocampus, including an activated state of microglia[113]. Microglia have been shown to participate in neurogenesis, during multiple stages of the process through the contribution of factors that affect the proliferation and survival of NSCs[114,115]. Cognitive decline has been correlated with decreased neurogenesis[116], and studies provide support to the idea that exercise or enriched environments result in an increase in neurogenesis[117-119], which may be modulated by microglial activation[120]. A pro-inflammatory environment has been demonstrated to inhibit adult neurogenesis, while anti-inflammatory treatments were able to rescue the phenotype[121,122]. All these findings demonstrate the need to understand the role of microglia in neurogenesis that takes place in the physiological and pathological CNS. The function of microglia is most likely influenced by the environmental signals in a particular setting, which will dictate the direction of their activation status.
Microglia constantly survey their environment and are in the proximity of all cell types during neurogenesis, including newborn neurons. They are also involved in the phagocytosis of NPCs and neuroblasts in a homeostatic role for maintaining neurogenic stem cells without releasing pro-inflammatory cytokines[51]. In concordance with these data, ablating microglia in the DG inhibited adult neurogenesis by diminishing neuroblast survival[123]. Although these effects are most likely mediated by the secretion of cytokines and by microglial-regulated phagocytosis, the influence of microglia on neurogenesis also extends beyond these molecular steps and events. There is a growing body of evidence demonstrating that microglial receptors can modulate their activity in neurogenesis. For example, microglial P2Y13 receptor was recently described to contribute to microglial structural integrity. When the P2Y13 receptor is knocked out, increases in proliferation of NPCs and new neurons are observed, and this may be another way to regulate neurogenesis[124]. CX3CR1 has also been demonstrated to be involved in the regulation of adult neurogenesis: microglia have been reported to activate NPCs through CX3CR1 pathways in the hippocampus[125], and CX3CR1 null (-/-) mice exhibited impaired connectivity and aberrant synapse formation[126]. This was further supported by genetic and pharmacological inhibition of CX3CR1 signaling, which also led to aberrant neurogenesis[127,128].
Abundant data show that microglia are critical in adult neurogenesis and regulate several stages of accurate incorporation of new neurons into the hippocampal circuitry. As several seizure disorders and models manifest predominantly in the hippocampus, the effects of epileptic activity on SGZ neurogenesis is starting to be uncovered.
Neurogenesis and the pathophysiology of epilepsy
Adult neurogenesis increases following SE in animal models, resulting in an increased number of granule cells[129,130]. These additional granule cells undergo aberrant differentiation, axonal sprouting, and ectopic displacement in the hilar region of the dentate gyrus[109,131,132]. Ectopic granule cells are thought to contribute to pro-epileptic activity[133-135]; studies show that axonal sprouting and aberrant placement of granule cells were reduced when newborn granule cells were eliminated[132]. Following SE, microglia regulate the number of new granule cells through selective phagocytosis to maintain homeostasis in the dentate gyrus circuitry[136] and are capable of engulfing viable neurons in the hippocampus as well[137]. It has been suggested that microglia modulate each step (proliferation, survival, and maturation) of adult neurogenesis in both homeostasis and epileptic states[138], though their exact role in the integration of new cells has not been elucidated. Microglia may also suppress aberrant neurogenesis through the secretion of TNF-α[95], potentially leading to anti-epileptic effects [Figure 2]. Recent studies depleting microglia from the SVZ suggested that they might not be necessary for NSC proliferation[139,140], although this has not been shown in the hippocampus.
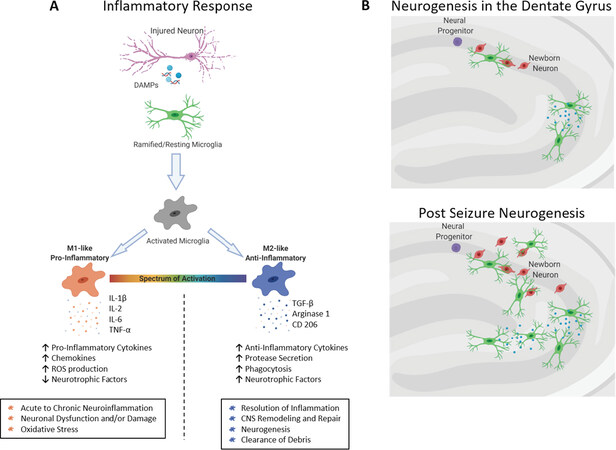
Figure 2. Microglial responses in fnflammation and neurogenesis. A: Microglia activate in response to damage associated molecular patterns (DAMPs) released by injured neurons post-seizure. Upon activation, microglial adopt one of two phenotypes: M1-like, which presents a pro-inflammatory profile that consists of decreased expression of neurotrophic factors and increased levels of pro-inflammatory chemokines and cytokines and reactive oxygen species, or M2-like, which is an anti-inflammatory response that includes the resolution of the inflammatory profile, neurogenesis and the clearance of debris; B: during neurogenesis in the hippocampus, unchallenged microglia clear cellular debris and control the number of newborn neuronal cells through phagocytosis. Post-seizure, the increased numbers of newborn cells may be cleared by microglia to reduce the potential for ectopic connections that contribute to pro-epileptic activity. Image created with Biorender.com
Conclusion
Investigation of inflammatory and neurogenic processes in epilepsy has revealed potential and critical roles of microglia in several facets of seizure generation. Epilepsy patients take AED with the aim of preventing seizures, yet studies looking at the anti-inflammatory and neurogenic effects of these drugs are sparse. Interrogating the literature for effects of AEDs in vivo on microglia, an important modulator of these processes, result in surprisingly few reports[141-143].
In vitro studies on microglial cells as mediators of inflammation have demonstrated that topiramate, a second generation AED, decreased the release of IL-1β, IL-6 and TNF-α[144]. Other AEDs such as levetiracetam, gabapentin, and phenobarbital showed slight modification in cytokine production[145]. The first generation AED valproic acid, was shown to increase IL-6 and TNF-α production in LPS-induced microglial cells[145], which contrasts with in vivo results where TNF-α and IL-1β were decreased after valproic acid treatment[143]. It was also demonstrated that the AED levetiracetam suppressed neuroinflammation and phagocytosis in a pilocarpine induced SE model[143]. Itoh et al.[146] reported that levetiracetam lessened microglial activation, as demonstrated by lower numbers of Iba-1 positive microglia, higher ramified shape, and low expression of pro-inflammatory cytokines. While the results of in vitro studies may eventually be applicable to the clinic, they highlight the need for clarification of the effects of AEDs on inflammation in vivo.
Studies concerning AEDs and neurogenesis are also extremely limited. Pregabalin, a widely used AED with an unknown mechanism of action, has been shown to accelerate the maturation of granule cells in the dentate gyrus[147]. In rats, lamotrigine increased the number of newborn cells in the hippocampus[148] and increased neurogenesis[149]. Valproic acid also induced neurogenesis, but these effects were not induced by phenobarbital and topiramate[149].
Epileptogenic changes in the brain are provoked by inflammation and increased neurogenic levels post-seizure. To control this process, a greater understanding of microglial contributions is needed and could provide a mechanism and target for a new generation of AEDs.
Declarations
AcknowledgmentsWe would like to thank members of the Tsirka lab for helpful discussions and feedback. TRV is a recipient of a National Science Foundation Graduate Research Fellowship.
Authors’ contributionsCo-wrote review article, edited review, generated the images: Victor TR
Co-wrote review article, edited article, correspondence: sirka SE
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was partially supported by the National Science Foundation Graduate Research Fellowship under grant no. 1315232, and NIH T32GM127253.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Laxer KD, Trinka E, Hirsch LJ, Cendes F, Langfitt J, et al. The consequences of refractory epilepsy and its treatment. Epilepsy Behav 2014;37:59-70.
2. Jin X, Yamashita T. Microglia in central nervous system repair after injury. J Biochem 2016;159:491-6.
3. Goldberg EM, Coulter DA. Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci 2013;14:337-49.
4. Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014;55:475-82.
5. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:522-30.
6. Hauser WA, Beghi E. First seizure definitions and worldwide incidence and mortality. Epilepsia 2008;49 Suppl 1:8-12.
7. Zack MM, Kobau R. National and state estimates of the numbers of adults and children with active epilepsy-United States, 2015. MMWR Morb Mortal Wkly Rep 2017;66:821-5.
8. Fiest KM, Sauro KM, Wiebe S, Patten SB, Kwon CS, et al. Prevalence and incidence of epilepsy: a systematic review and meta-analysis of international studies. Neurology 2017;88:296-303.
9. Epsztein J, Milh M, Bihi RI, Jorquera I, Ben-Ari Y, et al. Ongoing epileptiform activity in the post-ischemic hippocampus is associated with a permanent shift of the excitatory-inhibitory synaptic balance in CA3 pyramidal neurons. J Neurosci 2006;26:7082-92.
10. Huberfeld G, de La Prida LM, Pallud J, Cohen I, Le Van Quyen M, et al. Glutamatergic pre-ictal discharges emerge at the transition to seizure in human epilepsy. Nat Neurosci 2011;14:627-34.
11. Derchansky M, Jahromi SS, Mamani M, Shin DS, Sik A, et al. Transition to seizures in the isolated immature mouse hippocampus: a switch from dominant phasic inhibition to dominant phasic excitation. J Physiol 2008;586:477-94.
12. Wiebe S, Blume WT, Girvin JP, Eliasziw M; Effectiveness and Efficiency of Surgery for Temporal Lobe Epilepsy Study Group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med 2001;345:311-8.
13. Chen Z, Brodie MJ, Liew D, Kwan P. Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs: a 30-year longitudinal cohort study. JAMA Neurol 2018;75:279-86.
14. Liu H, Yang Y, Wang Y, Tang H, Zhang F, et al. Ketogenic diet for treatment of intractable epilepsy in adults: a meta-analysis of observational studies. Epilepsia Open 2018;3:9-17.
15. Hartman AL, Gasior M, Vining EP, Rogawski MA. The neuropharmacology of the ketogenic diet. Pediatr Neurol 2007;36:281-92.
16. Masino SA, Rho JM. Metabolism and epilepsy: ketogenic diets as a homeostatic link. Brain Res 2019;1703:26-30.
17. Starnes K, Miller K, Wong-Kisiel L, Lundstrom BN. A review of neurostimulation for epilepsy in pediatrics. Brain Sci 2019;9:283.
18. Dudek FE, Staley KJ. The time course and circuit mechanisms of acquired epileptogenesis. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th ed. Bethesda (MD): National Center for Biotechnology Information (US); 2012.
19. Pitkänen A, Lukasiuk K, Dudek FE, Staley KJ. Epileptogenesis. Cold Spring Harb Perspect Med 2015;5:a022822.
20. Varvel NH, Jiang J, Dingledine R. Candidate drug targets for prevention or modification of epilepsy. Annu Rev Pharmacol Toxicol 2015;55:229-47.
21. Engel Jr J. Mesial temporal lobe epilepsy. Epilepsy: a comprehensive textbook 1997;3:2417-26.
22. Thom M, Martinian L, Catarino C, Yogarajah M, Koepp M, et al. Bilateral reorganization of the dentate gyrus in hippocampal sclerosis: a postmortem study. Neurology 2009;73:1033-40.
23. Koyama R, Ikegaya Y. The molecular and cellular mechanisms of axon guidance in mossy fiber sprouting. Front Neurol 2018;9:382.
24. Buckmaster PS. Mossy Fiber Sprouting in the Dentate Gyrus. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th ed. Bethesda (MD): National Center for Biotechnology Information (US); 2012.
25. Sloviter RS, Zappone CA, Harvey BD, Frotscher M. Kainic acid-induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyper-inhibition in chronically epileptic rats. J Comp Neurol 2006;494:944-60.
26. Heck N, Garwood J, Loeffler JP, Larmet Y, Faissner A. Differential upregulation of extracellular matrix molecules associated with the appearance of granule cell dispersion and mossy fiber sprouting during epileptogenesis in a murine model of temporal lobe epilepsy. Neuroscience 2004;129:309-24.
27. Wu YP, Siao CJ, Lu W, Sung TC, Frohman MA, et al. The tissue plasminogen activator (tPA)/plasmin extracellular proteolytic system regulates seizure-induced hippocampal mossy fiber outgrowth through a proteoglycan substrate. J Cell Biol 2000;148:1295-304.
28. Zhang Y, Huang P, Du G, Kanaho Y, Frohman MA, et al. Increased expression of two phospholipase D isoforms during experimentally induced hippocampal mossy fiber outgrowth. Glia 2004;46:74-83.
29. Zhang Y, Kanaho Y, Frohman MA, Tsirka SE. Phospholipase D1-promoted release of tissue plasminogen activator facilitates neurite outgrowth. J Neurosci 2005;25:1797-805.
30. Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci 2013;36:174-84.
32. Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, et al. Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci 2015;35:3330-45.
33. Takahashi Y, Yu Z, Sakai M, Tomita H. Linking activation of microglia and peripheral monocytic cells to the pathophysiology of psychiatric disorders. Front Cell Neurosci 2016;10:144.
34. Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med 2013;19:1584-96.
35. Friedman A, Heinemann U. Role of blood–brain barrier injury in epileptogenesis. Epilepsia 2010;51:34-34.
36. Christensen J, Pedersen MG, Pedersen CB, Sidenius P, Olsen J, et al. Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet 2009;373:1105-10.
37. Schoknecht K, Prager O, Vazana U, Kamintsky L, Harhausen D, et al. Monitoring stroke progression: in vivo imaging of cortical perfusion, blood-brain barrier permeability and cellular damage in the rat photothrombosis model. J Cereb Blood Flow Metab 2014;34:1791-801.
38. Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun 2011;25:1281-9.
39. Aronica E, Bauer S, Bozzi Y, Caleo M, Dingledine R, et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia 2017;58 Suppl 3:27-38.
40. French JA, Gazzola DM. New generation antiepileptic drugs: what do they offer in terms of improved tolerability and safety?. Ther Adv Drug Saf 2011;2:141-58.
41. Barker-Haliski M, Steve White H. Validated animal models for antiseizure drug (ASD) discovery: advantages and potential pitfalls in ASD screening. Neuropharmacology 2020;167:107750.
42. Löscher W. Animal models of seizures and epilepsy: past, present, and future role for the discovery of antiseizure drugs. Neurochem Res 2017;42:1873-88.
43. Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 2011;52:657-78.
44. Welzel L, Schidlitzki A, Twele F, Anjum M, Löscher W. A face-to-face comparison of the intra-amygdala and intrahippocampal kainate mouse models of mesial temporal lobe epilepsy and their utility for testing novel therapies. Epilepsia 2020;61:157-70.
45. Simonato M, Brooks-Kayal AR, Engel J Jr, Galanopoulou AS, Jensen FE, et al. The challenge and promise of anti-epileptic therapy development in animal models. Lancet Neurol 2014;13:949-60.
46. Kandratavicius L, Balista PA, Lopes-Aguiar C, Ruggiero RN, Umeoka EH, et al. Animal models of epilepsy: use and limitations. Neuropsychiatr Dis Treat 2014;10:1693-705.
47. Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 1985;14:375-403.
48. Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse 1989;3:154-71.
49. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005;308:1314-8.
50. Cunningham CL, Martínez-Cerdeño V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci 2013;33:4216-33.
51. Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010;7:483-5.
52. Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci 2013;16:543-551.
53. Peri F, Nüsslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 2008;133:916-27.
54. Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, et al. Microglia modulate wiring of the embryonic forebrain. Cell Rep 2014;8:1271-9.
55. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011;333:1456-8.
56. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012;74:691-705.
57. Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun 2016;7:12540.
58. Augusto-Oliveira M, Arrifano GP, Lopes-Araújo A, Santos-Sacramento L, Takeda PY, et al. What do microglia really do in healthy adult brain?. Cells 2019;8:1293.
59. van Landeghem FK, Stover JF, Bechmann I, Bruck W, Unterberg A, et al. Early expression of glutamate transporter proteins in ramified microglia after controlled cortical impact injury in the rat. Glia 2001;35:167-79.
60. Wyatt-Johnson SK, Herr SA, Brewster AL. Status epilepticus triggers time-dependent alterations in microglia abundance and morphological phenotypes in the hippocampus. Front Neurol 2017;8:700.
61. Tsirka SE, Gualandris A, Amaral DG, Strickland S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature 1995;377:340-4.
62. Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, et al. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol 2003;182:21-34.
63. Benson MJ, Manzanero S, Borges K. Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia 2015;56:895-905.
64. Alyu F, Dikmen M. Inflammatory aspects of epileptogenesis: contribution of molecular inflammatory mechanisms. Acta Neuropsychiatr 2017;29:1-16.
66. Politis M, Su P, Piccini P. Imaging of microglia in patients with neurodegenerative disorders. Front Pharmacol 2012;3:96.
67. Butler T, Li Y, Tsui W, Friedman D, Maoz A, et al. Transient and chronic seizure-induced inflammation in human focal epilepsy. Epilepsia 2016;57:e191-4.
68. Scorza CA, Marques MJG, Gomes da Silva S, Naffah-Mazzacoratti MDG, Scorza FA, et al. Status epilepticus does not induce acute brain inflammatory response in the Amazon rodent Proechimys, an animal model resistant to epileptogenesis. Neurosci Lett 2018;668:169-73.
69. Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 2003;23:8692-700.
70. Han T, Qin Y, Mou C, Wang M, Jiang M, et al. Seizure induced synaptic plasticity alteration in hippocampus is mediated by IL-1β receptor through PI3K/Akt pathway. Am J Transl Res 2016;8:4499-509.
71. Roseti C, van Vliet EA, Cifelli P, Ruffolo G, Baayen JC, et al. GABAA currents are decreased by IL-1β in epileptogenic tissue of patients with temporal lobe epilepsy: implications for ictogenesis. Neurobiol Dis 2015;82:311-20.
72. Maroso M, Balosso S, Ravizza T, Iori V, Wright CI, et al. Interleukin-1β biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics 2011;8:304-15.
73. Ravizza T, Vezzani A. Pharmacological targeting of brain inflammation in epilepsy: therapeutic perspectives from experimental and clinical studies. Epilepsia Open 2018;3:133-42.
74. Dilena R, Mauri E, Aronica E, Bernasconi P, Bana C, et al. Therapeutic effect of Anakinra in the relapsing chronic phase of febrile infection-related epilepsy syndrome. Epilepsia Open 2019;4:344-50.
76. Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 2006;281:21362-8.
77. Kubota K, Inoue K, Hashimoto R, Kumamoto N, Kosuga A, et al. Tumor necrosis factor receptor-associated protein 1 regulates cell adhesion and synaptic morphology via modulation of N-cadherin expression. J Neurochem 2009;110:496-508.
78. Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 2005;25:3219-28.
79. Levin SG, Godukhin OV. Modulating Effect of Cytokines on Mechanisms of Synaptic Plasticity in the Brain. Biochemistry (Mosc) 2017;82:264-74.
80. Mirrione MM, Konomos DK, Gravanis I, Dewey SL, Aguzzi A, et al. Microglial ablation and lipopolysaccharide preconditioning affects pilocarpine-induced seizures in mice. Neurobiol Dis 2010;39:85-97.
81. Torres L, Danver J, Ji K, Miyauchi JT, Chen D, et al. Dynamic microglial modulation of spatial learning and social behavior. Brain Behav Immun 2016;55:6-16.
82. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014;82:380-97.
83. Homsi S, Piaggio T, Croci N, Noble F, Plotkine M, et al. Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J Neurotrauma 2010;27:911-21.
84. Heo K, Cho YJ, Cho KJ, Kim HW, Kim HJ, et al. Minocycline inhibits caspase-dependent and -independent cell death pathways and is neuroprotective against hippocampal damage after treatment with kainic acid in mice. Neurosci Lett 2006;398:195-200.
85. Wang N, Mi X, Gao B, Gu J, Wang W, et al. Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience 2015;287:144-56.
86. Barker-Haliski ML, Heck TD, Dahle EJ, Vanegas F, Pruess TH, et al. Acute treatment with minocycline, but not valproic acid, improves long-term behavioral outcomes in the Theiler’s virus model of temporal lobe epilepsy. Epilepsia 2016;57:1958-67.
87. Wolf BJ, Brackhan M, Bascunana P, Leiter I, Langer BLN, et al. TSPO PET identifies different anti-inflammatory minocycline treatment response in two rodent models of epileptogenesis. Neurotherapeutics 2020;10.
88. Wang DD, Englot DJ, Garcia PA, Lawton MT, Young WL. Minocycline-and tetracycline-class antibiotics are protective against partial seizures in vivo. Epilepsy Behav 2012;24:314-8.
89. Russmann V, Goc J, Boes K, Ongerth T, Salvamoser JD, et al. Minocycline fails to exert antiepileptogenic effects in a rat status epilepticus model. Eur J Pharmacol 2016;771:29-39.
90. Dupuis N, Mazarati A, Desnous B, Chhor V, Fleiss B, et al. Pro-epileptogenic effects of viral-like inflammation in both mature and immature brains. J Neuroinflammation 2016;13:307.
91. Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Exp Neurol 2013;244:11-21.
92. Gross A, Benninger F, Madar R, Illouz T, Griffioen K, et al. Toll-like receptor 3 deficiency decreases epileptogenesis in a pilocarpine model of SE-induced epilepsy in mice. Epilepsia 2017;58:586-96.
93. Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 2010;16:413-9.
94. Liu JT, Wu SX, Zhang H, Kuang F. Inhibition of MyD88 signaling skews microglia/macrophage polarization and attenuates neuronal apoptosis in the hippocampus after status epilepticus in mice. Neurotherapeutics 2018;15:1093-111.
95. Matsuda T, Murao N, Katano Y, Juliandi B, Kohyama J, et al. TLR9 signalling in microglia attenuates seizure-induced aberrant neurogenesis in the adult hippocampus. Nat Commun 2015;6:6514.
96. Zhang J, Jiao J. Molecular biomarkers for embryonic and adult neural stem cell and neurogenesis. Biomed Res Int 2015;2015:727542.
97. Kempermann G. Environmental enrichment, new neurons and the neurobiology of individuality. Nat Rev Neurosci 2019;20:235-45.
98. Yao B, Christian KM, He C, Jin P, Ming GL, et al. Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci 2016;17:537-49.
99. Voss MW, Soto C, Yoo S, Sodoma M, Vivar C, et al. Exercise and hippocampal memory systems. Trends Cogn Sci 2019;23:318-33.
100. Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, et al. Neurogenesis in the adult human hippocampus. Nat Med 1998;4:1313-7.
101. Kempermann G, Jessberger S, Steiner B, Kronenberg G. Milestones of neuronal development in the adult hippocampus. Trends Neurosci 2004;27:447-52.
102. Dhaliwal J, Lagace DC. Visualization and genetic manipulation of adult neurogenesis using transgenic mice. Eur J Neurosci 2011;33:1025-36.
103. Gage FH, Coates PW, Palmer TD, Kuhn HG, Fisher LJ, et al. Survival and differentiation of adult neuronal progenitor cells transplanted to the adult brain. Proc Natl Acad Sci U S A 1995;92:11879-83.
104. Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 2011;70:687-702.
105. van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, et al. Functional neurogenesis in the adult hippocampus. Nature 2002;415:1030-4.
106. de Miranda AS, Zhang CJ, Katsumoto A, Teixeira AL. Hippocampal adult neurogenesis: Does the immune system matter?. J Neurol Sci 2017;372:482-95.
107. Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory?. Nat Rev Neurosci 2010;11:339-50.
108. Cho KO, Lybrand ZR, Ito N, Brulet R, Tafacory F, et al. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat Commun 2015;6:6606.
109. Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, Lowenstein DH. Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann Neurol 2006;59:81-91.
110. Jessberger S, Parent JM. Epilepsy and adult neurogenesis. Cold Spring Harb Perspect Biol 2015;7:a020677.
111. Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci 1996;16:2027-33.
112. Encinas JM, Michurina TV, Peunova N, Park JH, Tordo J, et al. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell 2011;8:566-79.
113. Mosher KI, Schaffer DV. Influence of hippocampal niche signals on neural stem cell functions during aging. Cell Tissue Res 2018;371:115-24.
114. Walton NM, Sutter BM, Laywell ED, Levkoff LH, Kearns SM, et al. Microglia instruct subventricular zone neurogenesis. Glia 2006;54:815-25.
115. Matsui TK, Mori E. Microglia support neural stem cell maintenance and growth. Biochem Biophys Res Commun 2018;503:1880-4.
116. Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011;477:90-4.
117. van Praag H, Shubert T, Zhao C, Gage FH. Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci 2005;25:8680-5.
118. van Praag H, Christie BR, Sejnowski TJ, Gage FH. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci U S A 1999;9:13427-31.
119. Kempermann G, Kuhn HG, Gage FH. Experience-induced neurogenesis in the senescent dentate gyrus. J Neurosci 1998;18:3206-12.
120. Ziv Y, Avidan H, Pluchino S, Martino G, Schwartz M. Synergy between immune cells and adult neural stem/progenitor cells promotes functional recovery from spinal cord injury. Proc Natl Acad Sci U S A 2006;103:13174-9.
121. Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A 2003;100:13632-7.
122. Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003;302:1760-5.
123. Kreisel T, Wolf B, Keshet E, Licht T. Unique role for dentate gyrus microglia in neuroblast survival and in VEGF-induced activation. Glia 2019;67:594-618.
124. Stefani J, Tschesnokowa O, Parrilla M, Robaye B, Boeynaems JM, et al. Disruption of the microglial ADP receptor P2Y13 enhances adult hippocampal neurogenesis. Front Cell Neurosci 2018;12:134.
125. Vukovic J, Colditz MJ, Blackmore DG, Ruitenberg MJ, Bartlett PF. Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J Neurosci 2012;32:6435-43.
126. Bolos M, Perea JR, Terreros-Roncal J, Pallas-Bazarra N, Jurado-Arjona J, et al. Absence of microglial CX3CR1 impairs the synaptic integration of adult-born hippocampal granule neurons. Brain Behav Immun 2018;68:76-89.
127. Bachstetter AD, Morganti JM, Jernberg J, Schlunk A, Mitchell SH, et al. Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol Aging 2011;32:2030-44.
128. Maggi L, Scianni M, Branchi I, D’Andrea I, Lauro C, et al. CX(3)CR1 deficiency alters hippocampal-dependent plasticity phenomena blunting the effects of enriched environment. Front Cell Neurosci 2011;5:22.
129. Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, et al. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 1997;17:3727-38.
130. Jessberger S, Römer B, Babu H, Kempermann G. Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp Neurol 2005;196:342-51.
131. Jessberger S, Zhao C, Toni N, Clemenson GD Jr, Li Y, et al. Seizure-associated, aberrant neurogenesis in adult rats characterized with retrovirus-mediated cell labeling. J Neurosci 2007;27:9400-7.
132. Kron MM, Zhang H, Parent JM. The developmental stage of dentate granule cells dictates their contribution to seizure-induced plasticity. J Neurosci 2010;30:2051-59.
133. Scharfman HE, Goodman JH, Sollas AL. Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J Neurosci 2000;20:6144-58.
134. Zhan RZ, Timofeeva O, Nadler JV. High ratio of synaptic excitation to synaptic inhibition in hilar ectopic granule cells of pilocarpine-treated rats. J Neurophysiol 2010;104:3293-304.
135. Pun RY, Rolle IJ, Lasarge CL, Hosford BE, Rosen JM, et al. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 2012;75:1022-34.
136. Luo C, Koyama R, Ikegaya Y. Microglia engulf viable newborn cells in the epileptic dentate gyrus. Glia 2016;64:1508-17.
137. Abiega O, Beccari S, Diaz-Aparicio I, Nadjar A, Laye S, et al. Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol 2016;14:e1002466.
138. Luo C, Ikegaya Y, Koyama R. Microglia and neurogenesis in the epileptic dentate gyrus. Neurogenesis (Austin) 2016;3:e1235525.
139. Kyle J, Wu M, Gourzi S, Tsirka SE. Proliferation and differentiation in the adult subventricular zone are not affected by CSF1R inhibition. Front Cell Neurosci 2019;13:97.
140. Ribeiro Xavier AL, Kress BT, Goldman SA, Lacerda de Menezes JR, Nedergaard M. A distinct population of microglia supports adult neurogenesis in the subventricular zone. J Neurosci 2015;35:11848-61.
141. Gibbons HM, Smith AM, Teoh HH, Bergin PM, Mee EW, et al. Valproic acid induces microglial dysfunction, not apoptosis, in human glial cultures. Neurobiol Dis 2011;41:96-103.
142. Su W, Xie M, Li Y, Gong X, Li J. Topiramate reverses physiological and behavioral alterations by postoperative cognitive dysfunction in rat model through inhibiting TNF signaling pathway. Neuromolecular Med 2020;22:227-38.
143. Itoh K, Taniguchi R, Matsuo T, Oguro A, Vogel CF, et al. Suppressive effects of levetiracetam on neuroinflammation and phagocytic microglia: A comparative study of levetiracetam, valproate and carbamazepine. Neurosci Lett 2019;708:134363.
144. Andrzejczak D, Woldan-Tambor A, Bednarska K, Zawilska JB. The effects of topiramate on lipopolysaccharide (LPS)-induced proinflammatory cytokine release from primary rat microglial cell cultures. Epilepsy Res 2016;127:352-7.
145. Shima T, Sakuma H, Suzuki T, Kohyama K, Matsuoka T, et al. Effects of antiepileptic drugs on microglial properties. Epilepsy & Seizure 2018;10:22-32.
146. Itoh K, Ishihara Y, Komori R, Nochi H, Taniguchi R, et al. Levetiracetam treatment influences blood-brain barrier failure associated with angiogenesis and inflammatory responses in the acute phase of epileptogenesis in post-status epilepticus mice. Brain Res 2016;1652:1-13.
147. Lempel AA, Coll L, Schinder AF, Uchitel OD, Piriz J. Chronic pregabalin treatment decreases excitability of dentate gyrus and accelerates maturation of adult-born granule cells. J Neurochem 2017;140:257-67.
148. Kondziella D, Strandberg J, Lindquist C, Asztely F. Lamotrigine increases the number of BrdU-labeled cells in the rat hippocampus. Neuroreport 2011;22:97-100.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Victor TR, Tsirka SE. Microglial contributions to aberrant neurogenesis and pathophysiology of epilepsy. Neurosciences 2020;7:234-47. http://dx.doi.org/10.20517/2347-8659.2020.02
AMA Style
Victor TR, Tsirka SE. Microglial contributions to aberrant neurogenesis and pathophysiology of epilepsy. Neuroimmunology and Neuroinflammation. 2020; 7(3): 234-47. http://dx.doi.org/10.20517/2347-8659.2020.02
Chicago/Turabian Style
Victor, Tanya R., Stella E. Tsirka. 2020. "Microglial contributions to aberrant neurogenesis and pathophysiology of epilepsy" Neuroimmunology and Neuroinflammation. 7, no.3: 234-47. http://dx.doi.org/10.20517/2347-8659.2020.02
ACS Style
Victor, TR.; Tsirka SE. Microglial contributions to aberrant neurogenesis and pathophysiology of epilepsy. Neurosciences. 2020, 7, 234-47. http://dx.doi.org/10.20517/2347-8659.2020.02
About This Article
Copyright

Data & Comments
Data


 Cite This Article 15 clicks
Cite This Article 15 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.