
Defining activation states of microglia in human brain tissue: an unresolved issue for Alzheimer's disease

Abstract
The development of concepts concerning the role of microglia in different brain diseases has relied on studies of animal models or human brain tissue, which primarily use antibodies and immunohistochemistry techniques to make observations. Since initial studies defined increased expression of the major histocompatibility complex II protein human leukocyte antigen-DR as a means of identifying reactive, and therefore by implication, damage-causing microglia in Alzheimer’s disease (AD) or Parkinson’s disease (PD), understanding and describing their activation states has evolved to an unexpected complexity. It is still difficult to ascertain the specific functions of individual microglia, particularly those associated with pathological structures, using a narrow range of antigenic markers. As many approaches to developing treatments for AD or PD are focused on anti-inflammatory strategies, a more refined understanding of microglial function is needed. In recent years, gene expression studies of human and rodent microglia have attempted to add clarity to the issue by sub-classification of messenger RNA expression of cell-sorted microglia to identify disease-associated profiles from homeostatic functions. Ultimately all newly identified markers will need to be studied in situ in human brain tissue. This review will consider the gaps in knowledge between using traditional immunohistochemistry approaches with small groups of markers that can be defined with antibodies, and the findings from cell-sorted and single-cell RNA sequencing transcription profiles. There have been three approaches to studying microglia in tissue samples: using antigenic markers identified from studies of peripheral macrophages, studying proteins associated with altered genetic risk factors for disease, and studying microglial proteins identified from mRNA expression analyses from cell-sorting and gene profiling. The technical aspects of studying microglia in human brain samples, inherent issues of working with antibodies, and findings of a range of different functional microglial markers will be reviewed. In particular, we will consider markers of microglia with expression profiles that do not definitively fall into the pro-inflammatory or anti-inflammatory classification. These additional markers include triggering receptor expressed on myeloid cells-2, CD33 and progranulin, identified from genetic findings, colony stimulating factor-1 receptor, purinergic receptor P2RY12, CD68 and Toll-like receptors. Further directions will be considered for addressing crucial issues.
Keywords
Introduction
Alzheimer’s disease (AD) and Parkinson’s disease (PD) have become the most significant and feared brain diseases of elderly populations who are now enjoying longer lifespans due to more effective treatments for cancer, cardiovascular and metabolic diseases. AD is the most common cause of cognitive decline and dementia in elderly populations[1], while PD can lead to severe loss of mobility and independence, amongst other features[2]. Both diseases are significant causes of morbidity in elderly populations and share many common pathological features involving the accumulation of aggregated proteins. In AD, it is extracellular amyloid and neurofibrillary-associated phosphorylated tau neurites and tangles[3], while in PD, it is aggregated/phosphorylated alpha-synuclein accumulated into pathological inclusions[4]. These diseases are distinctive on account of the degenerative changes occurring in different brain regions; however, one common feature is the appearance of “activated” microglia within brain regions showing degenerative changes. Inflammation has become one of the targets being investigated as treatment strategies for these diseases, and the importance of studying microglia in relation to many different brain diseases is widely appreciated[5-7].
Initial antibody-based observations on “activated” microglia in postmortem brain tissues were made 30 years ago and gave rise to the inflammatory hypotheses for neurodegeneration. This is illustrated in Figure 1 and suggests that initial cell death or accumulation of aberrant/aggregated proteins [amyloid beta (Aβ), tau or alpha-synuclein (α-syn)] results in proinflammatory activation of microglia, causing the production of toxic and/or inflammatory cytokines. The resulting neurotoxicity would then accelerate further inflammation, thus exacerbating the neurodegenerative process. These concepts developed in the 80’s and 90’s might now be considered imprecise based on more recent findings. However, it was from this hypothesis that treatments for AD and PD with anti-inflammatory agents were developed and tested. This approach was supported by data from epidemiological studies that patients who had long-term usage of anti-inflammatory drugs for inflammatory conditions such as arthritis, had less dementia, which appeared to support the inflammatory hypothesis of AD[8]. However, although many anti-inflammatory compounds and strategies have proven effective in AD animal models, clinical trials of AD patients have generally shown no significant effect[9]. The purpose of this review is to consider the approaches used to define changes in microglial phenotypes and neuroinflammation in brain tissue, and discuss how the role of microglia in neurodegeneration should be considered in light of a wider range of markers identified from recent transcriptional profiling of microglia. The focus of this review will be on studies relating to AD, but many of the concepts might be applicable to PD, multiple sclerosis (MS) or stroke. The aim for this review is to bridge the gap between the studies that have analyzed transcription in large numbers of samples or isolated cells with in situ studies in human brain samples with antibodies to define the microenvironments of microglia in the specialized neuroanatomy of the human brain[10-12]. Ultimately, the best way to define the microglia responsible for damaging inflammation in brain samples will be with a single or small panel of markers that can be studied reliably in widely available types of pathological brain samples.
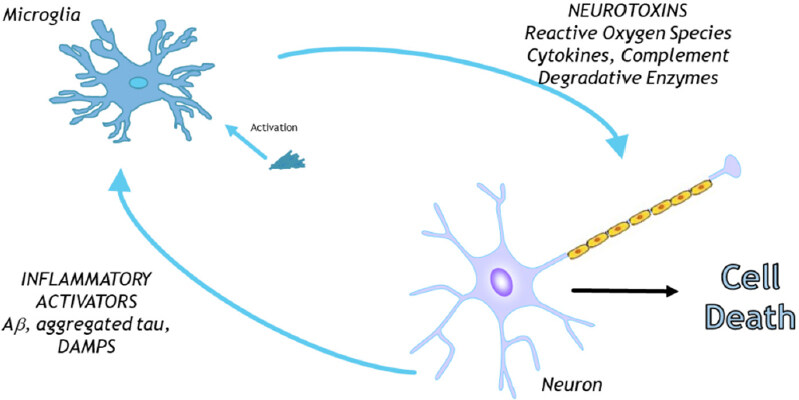
Figure 1. Hypothesis on the involvement of microglia in neurodegeneration
Methodology for investigating microglial phenotypes
Antibody-based methodology for in situ localization of microglial antigens
Pioneering observations on microglia by Rio-Hortega[13] used traditional metal-based histological stains to identify these cells. The fascinating history of their discovery has been reviewed by Tremblay et al.[14], but it was the use of specific antibodies in more recent times to sensitively identify microglia in human brain samples that re-launched this field of study. Studies by McGeer and colleagues employed an antibody to the major histocompatibility complex class II (MHCII) protein human leukocyte antigen-DR to identify what was described as “activated” or “reactive” microglia in AD and PD brain samples[15-18]. These types of microglia were enhanced around AD and PD pathological structures. Similar observations were made by Rogers and colleagues using the same marker[19]. Early studies highlighted recurring issues in the study of microglia in human autopsy tissue, namely with the antibodies used and the fixation methods of brain tissue samples for study[20]. Many microglial antigens, including human leukocyte antigen (HLA)-DR, are membrane-associated glycoproteins that are sensitive to tissue fixation with cross-linking fixatives such as paraformaldehyde/formalin and glutaraldehyde. The most widely available tissue samples for research are those taken for routine pathological examination and diagnosis at autopsy and usually involve long-term immersion fixation and paraffin-embedding using treatment with alcohols, xylene and similar solvents. Many validated monoclonal antibodies to macrophage/microglia antigens will not recognize them in tissue fixed in this manner, though a newer generation of antibodies, particularly monoclonal antibodies developed in rabbits, work more effectively when combined with different antigen-retrieval techniques[21]. Optimally-fixed tissue is tissue with short postmortem interval between death of donor subject and start of fixation, and a short period of fixation (48 h) of sliced brain coronal sections (not whole hemispheres) in buffered formalin/paraformaldehyde followed by preservation at 4 °C or -20 °C in an anti-freeze solution. Tissue preserved in this manner, which is then sectioned and processed for immunohistochemistry without paraffin embedding, has given optimal results for this investigator for detecting a number of different microglial proteins in situ[22,23].
Cell-sorting, nuclei-sorting, transcriptional profiling of inflammation and microglia
Expression profiling methods used to address this question have evolved rapidly over the last few years with RNA-sequencing becoming the predominant method of identification and quantification of genes expressed. There are now enormous amounts of data available online for carrying out analyses using various statistical criteria to identify the interactions of expressed microglial genes. For a more detailed understanding of these analytical approaches, the review of Chew and Petretto provides an overview of the different analytical approaches focusing on how the identification of transcriptional networks of microglia in AD can give insight into disease pathogenesis[10]. One observation by these authors was the lack of agreement between studies on which genes/markers should be the targets for tissue validation.
The findings from a number of RNA sequencing experiments of batch-sorted or single cell microglia isolated from AD or immune-stimulated animal models or human brain tissues will be considered. The results have been analyzed comparing AD animals with non-transgenic control animals, or between AD and controls from aged human tissues[24]. Although the focus of this article is on understanding how to define activation states in human brains, findings from rodent models in this context have to be considered. Microglia can be directly isolated from the mouse brain by Dounce homogenization to break up tissue, filtering through 70 μm mesh, separation by magnetic beads conjugated with anti-myelin antibody to remove myelin, and then selected using a fluorescence-activated cell sorter with appropriate labeled antibodies (e.g., CD11b, CD45) to isolate immune cells, including microglia[25]. This basic approach will isolate populations of cells that can be diluted to allow the isolation of single cells or analysis in bulk. Refinements to these techniques have allowed the sorting and RNA profiling of cellular nuclei from frozen human and animal tissue samples[11,26]. Different approaches for microglial profiling are illustrated in Figure 2.
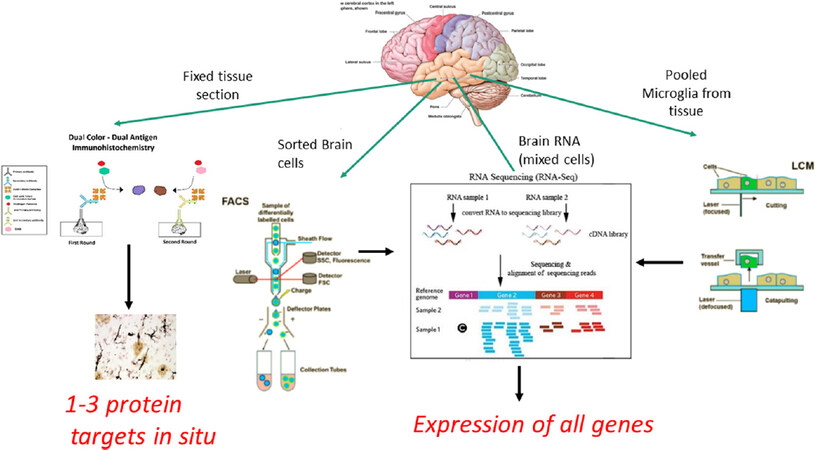
Figure 2. Technical approaches to characterizing microglia phenotypes in human brain tissue. Figure 2 illustrates different methodological approaches for defining microglia phenotypes using established immunohistochemistry methods compared to RNA gene expression profiling in sorted or single cell microglia populations, or microglia that had been laser micro-dissected from brain tissue sections
Using these approaches, it was shown that trans-membrane protein 119 (TMEM119) was a specific marker for microglia in mouse and human brains[25]. The isolation of microglia from human brains using the same methodology is possible but has some limitations. Human brain tissue is not usually amenable to Dounce homogenization and requires additional enzymatic digestion to dissociate tissue into single cells, and density gradient centrifugation to separate the myelin content from the cellular components[27]. Studies using these approaches aimed to define genes that are unique to microglia and not expressed, or expressed at low levels in blood monocytes/macrophages[28-31].
Recent important studies relating to inflammatory changes in AD brains identified a type of “disease-associated microglia” (DAM) that appear to be associated primarily with preventing inflammatory pathology rather than enhancing it[26,32,33]. Another key study involved the meta-analyses of multiple different gene profiling studies related to brain inflammation[24]. Amongst other findings, these studies confirmed TMEM119, purinergic receptor P2YR12 and fractalkine receptor CX3CR1 as markers with highly enriched expression in microglia compared to monocytes/macrophages.
DAM
The identification of DAM was primarily carried out using single cell microglia RNA sequencing in AD model mice (5xFAD) of different ages followed by validation in human tissue samples. The progressive changes in microglia phenotypes was from homeostatic (non-activated) to stage 1 DAM, which represents a state of proinflammatory activation, to stage 2 DAM, an altered phenotype that restricts neurodegenerative changes. Based on results that included the use of mice that are gene deficient (knockout) for the crucial triggering receptor expressed on myeloid cells (TREM2), it was shown that the transition from stage 1 to stage 2 DAM was dependent on Trem2 signaling. These data have implied that the activation of Trem2 signaling was protective rather than pathogenic[32]. [Gene identification primarily from studies using rodents will use the lower case abbreviation; genes primarily identified in human will use the upper-case abbreviation]. This study defined expression of genes Hexb (Beta-hexosaminidase subunit beta), Cst3 (Cystatin C), Cx3cr1 (Fractalkine receptor), Ctsd (Cathepsin D), Csf1r (Colony stimulating factor-1 receptor), Ctss (Cathepsin S), Sparc (Osteonectin), Tmsb4x (Thymosin beta-4), P2ry12 (Purinergic P2Y receptor 12), C1qa (Complement C1q subunit A), C1qb (Complement C1q subunit B) as features of homeostatic microglia. Activation and transition to stage 1 DAM involved downregulation of identified homeostatic genes Cx3cr1 (Fractalkine), P2ry12 (Purinergic 2Y receptor 12) and TMEM119 (Transmembrane Protein 119), along with P2ry13 (Purinergic P2Y receptor 13), Tgfbr1 (Transforming growth factor receptor beta 1), Txnip (Thioredoxin-interacting protein) and Glu1 (Glucoamylase 1), and upregulation of Tyrobp (TYRO protein tyrosine kinase binding protein - DAP12), Ctsb (Cathepsin B), Cstb (Cytstatin B), Ctsd (Cathepsin D) Apoe (Apolipoprotein E), B2m (Beta-2 microglobulin), Fth1 (Ferritin heavy chain-1), Timp2 (Tissue inhibitor of metalloprotease-2), H2-D1 (H2 class 1 histocompatability antigen) and Lyz2 (Lysozyme C-2). The expression of homeostatic genes C1qc (Complement C1q subunit C), C1qb, C1qa, Ctss, Hexb, Olfml3 (Olfactomedin-like 3), Csf1r and Cst3 remained unchanged between the different classes of microglia. Transition to stage 2 DAM involved upregulation of Trem2, Ankh (Progressive ankylosis protein), Cd9 (Tetraspanin), Cd63 (Tetraspanin-30), Serpine2 (Serine Protease inhibitor-2), Ctsz, Cd68 (macrosialin), Cadm1 (Cell-adhesion molecule-1), Spp1 (Secreted phosphoprotein-1), Cd52 (CD52), Ctsa, Clec7a (C-type lectin domain family 7 member A), Axl (AXL receptor tyrosine kinase), Ctsl, Lpl (Lipoprotein lipase), Ccl6 (C-C chemokine 6), Csf1 (Colony stimulating factor-1), Hif1a (Hypoxia inducible factor-1 alpha), Cusb (Cation efflux system protein CusB), Itgax (Integrin Subunit Alpha X) and Cst7 and downregulation of Cx3cr1, P2ry12, CD33 and TMEM119[32,33]. This study showed that Trem2 expression levels increased progressively upon activation from homeostatic to stage 1 and stage 2 DAM. This study chose Lpl (lipoprotein lipase), Timp2 (Tissue inhibitor of metalloprotease-2) and Itgax (CD11c) for antibody validation in human AD brain sections and showed strong expression in AD plaque-associated microglia. Lipoprotein lipase functions as a homodimer, and has the dual functions of triglyceride hydrolase and ligand/bridging factor for receptor-mediated lipoprotein uptake. Its expression is not specific for microglia/macrophage cells. Other stage 2 DAM markers that have been studied in human AD brains include TREM2 (see separate section below), CD68 and ITGAX (CD11c). There has been controversy about the significance of CD68 expression by microglia. This is a monocyte specific lysosomal-associated membrane protein that becomes upregulated with increased phagocytosis[34]. It has been considered an activation marker, but this does raise the question whether phagocytosis markers are genuine proinflammatory markers or whether increased phagocytosis is a reparative response. CD11c (a complement C3b integrin receptor CR4) is a cell adhesion and phagocytosis marker for dendritic cells. An earlier study had shown that CD11c was constitutively expressed by microglia in brain with some upregulation in reactive microglia in AD brains[35,36]. It has been considered an activation marker, but this does raise the Table 1. List of key genes associated with Disease-Associated Microglia.
List of key genes described associated with disease-associated microglia
| Gene | Gene | Gene |
|---|---|---|
| Homeostatic Microglia | Stage 1 DAM | Stage 2 DAM |
| aHexb | bCD33 | cTrem2 |
| aCst3 | bCx3cr1 | cAnkh |
| aCx3cr1 | bP2ry12 | cCd63 |
| Ctsd | bP2ry13 | cCd9 |
| Csf1r | bTgfbr1 | cSerpine2 |
| Ctss | bTxnip | cCtsz |
| Sparc | bGlu1 | cCd68 |
| Tmsb4x | bTmem119 | cCadm1 |
| Tmem119 | cTyrobp | cSpp1 |
| P2ry12 | cCtsb | cCd52 |
| C1qa | cCstb | cCtsa |
| C1qb | cCtsd | cClec7a |
| cApoe | cAxl | |
| cB2m | cCts1 | |
| cFth1 | cLpl | |
| cTimp2 | cCcl6 | |
| cH2-d1 | cCsf1 | |
| cLyz2 | cHif1a | |
| cCusb | ||
| cItgax |
The identification of ApoE as a microglial activation marker does not concur with previous immunohistochemistry results. Possession of the APOE allele e4 is the most significant risk factor for developing sporadic AD[37,38]. Subjects homozygous for APOE e4 have up to a 7-fold greater risk of developing AD but the mechanism(s) are still unclear. The possession of APOE e4 allele has also been associated with increased inflammation in the brain but immunohistochemistry studies have identified ApoE protein in neurofibrillary tangles, amyloid plaques and reactive astrocytes[39-42], not in microglia[43]. This is surprising as human microglia in culture and isolated brain microglia express high levels of APOE mRNA and protein. It is possible that the protein is rapidly secreted by microglia after synthesis, but evidence to date does not support ApoE as a marker for describing microglial activation states in human brain tissue. Complement C1q protein has also been studied in relation to AD pathogenesis. Antibodies to C1q have reactivity with amyloid plaques and neurofibrillary tangles in human brains[44,45]. Similar to ApoE, cultured brain-isolated microglia express high levels of complement C1q protein[46], but the reason microglia do not show immunoreactivity to ApoE or C1q in brain sections is unclear.
Core transcriptional signature of human microglia
A different approach to address the question of an AD-specific microglial gene signature was carried out by analyzing 9 different datasets obtained from profiling either sorted cells or brain tissue using unbiased correction network analysis[12]. Despite the heterogeneity between the datasets, a consensus list of 249 genes was identified, and when used to compare AD vs. age-matched controls, 52 genes were identified. Key genes from this list were CD84 (Signaling Lymphocytic activation molecule-5), dedicator of cytokinesis 2 (DOCK2), hepatitis A virus cellular receptor 2 (HAVCR2), Fc fragment of IgG receptor IIa (FCGR2A), linker for activation of T cells family member 2 (LAT2), CD86 (B7-2), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma (PIK3CG), apoptosis-associated speck-like protein containing a CARD (PYCARD), tetraspanin-26 (CD37), myosin IF (MYO1F), leukocyte immunoglobulin like receptor A2 (LILRA2), protein tyrosine phosphatase receptor type C - CD45 (PTPRC), inositol polyphosphate-5-phosphatase D (INPP5D), CD33, Toll-like receptor-5 (TLR5), SH3 domain and nuclear localization signals 1 (SAMSN1), integrin alpha M chain - CD11b (ITGAM), dedicator of cytokinesis 8 - Zir3 (DOCK8), ribosomal protein S6 kinase A1 (RPS6KA1), colony stimulator factor-3 receptor (CSF3R), SLC7A7 (Y+L amino acid transporter 1), Oxidized low-density lipoprotein receptor 1 (OLR1), chemokine-like factor (CKLF), Parkin co-regulated gene protein (PARCG), lysozyme (LYZ), lymphocyte antigen 86 (LY86), arachidonate 5-lipoxygenase activating protein (ALOX5AP), Ras and Rab interactor 3 (RIN3), regulator of G-protein signaling 18 (RGS18), colony stimulating factor 2 receptor beta common subunit (CSF2RB), Rho GTPase activating protein 15 (ARHGAP15), Rho GTPase activating protein 45 (ARHGAP45), regulator of G-protein signaling 10 (RGS10), interleukin 10 receptor subunit A (IL10RA), macrophage scavenger receptor-1 (MSR1), bridging integrator-2 (BIN2), and cytokine-like 1 (CYTL1). Most of these have not been studied in AD brain tissues. Another study addressed the issue of microglial specific genes by performing meta-analysis of a number of different datasets[47]. This study only examined rodent gene datasets. Thirteen microglia-enriched genes were identified and 14 genes were differentially expressed in monocytes/macrophages.
Meta-analyses of multiple microglial-inflammation profiling studies
Complex analyses of a number of different gene expression profiling studies of different disease animal models, human diseased tissue and sorted murine and human cells identified multiple signatures (modules) for microglia associated with neurodegeneration. In human material, there appeared to be elevated expression of genes that were not observed in animal models. The microglia cluster of genes contain those more highly expressed in microglia compared to other myeloid cells, but these are not necessarily microglia-specific. The cluster contained Rho GTPase Activating Protein-5 (Arhgap5), C-C Chemokine receptor-5 (Ccr5), Sialomucin core protein 24 (Cd164), CUB and sushi multiple domains 3 (Csmd3), Cst3, Cx3cr1, Gcnt1 (Beta-1,3-galactosyl-O-glycosyl-glycoprotein beta-1,6-N-acetylglucosaminyltransferase), Golgi membrane protein-1 (Golm1), G protein-coupled receptor 155 (Gpr155), G protein-coupled receptor 34 (Gpr34), G protein-coupled receptor 56 (Gpr56), General Transcription Factor IIH Subunit 2 (Gtf2h2), LPS responsive beige-like anchor protein (LRBA), leucine-rich repeat-containing protein (Lrrc3), Geranyltranstransferase (FPPS), microfibril-associated glycoprotein 3 (Mfap3), P2ry12, P2ry13, Plexin domain-containing protein 1 (Plxdc1), prostate transmembrane protein, androgen induced 1 (Prmepa1), Ras-related protein Rab-39 (Rab39), Spalt-like transcription factor 1 (Sali1), Selectin-P ligand, CD162 (Selplg), Siglech (SIGLEC-H), Toll-like receptor-3 (Tlr3), and TMEM119. A panel of 134 neurodegeneration-related genes were defined (for complete list refer to supplementary data in reference[24]). It was noted that 75% of these genes were associated with plasma membrane or extracellular space proteins. It was concluded that this was due to changes in the way microglia interact with the degenerative environment. Other genes included transcription factors Bhihe40 (Clast-5), retinoid X receptor gamma (Rxrγ), Hif1a and melanocyte inducing transcription factor (MITF), and 10 lysosome-associated genes including cathepsins (Ctsb, Ctsl and Ctsz). In a number of the models analyzed, increased expression of ApoE was consistently detected, and microglial responses to Aβ were highly dependent on Trem2 signaling. The central role for TREM2 in microglial responses has been demonstrated in network analysis showing TYROBP (DAP12), the essential adaptor protein that mediates TREM2 signaling, is a central hub gene for many of the above-listed inflammatory genes[31,48].
A recent publication employing single-nucleus transcriptomics combined with proteomic validation studies comparing AD model mice and human AD materials[26] showed that there were large differences in the glial phenotypes between AD mice and human AD samples. These investigators confirmed that the “Disease Associated Microglia” signature was dependent on expression of TREM2. There was increased expression of a number of “homeostatic” genes, including P2RY12, TMEM119 and CX3CR1, which have been downregulated in AD mouse models. This study also identified an amyloid-driven oligodendrocyte signature showing disruption in myelination, possibly driven by enhanced white-matter inflammation. In addition to the previously mentioned TREM2, APOE, HLA-DRA, and Alpha-2 macroglobulin (A2M), this study identified a number of additional markers that could be characterized in AD brains. These include Suppressor of Cytokine Signaling-6 (SOCS6), ZFP36 ring finger protein like 2 (ZFP36L2), SELPLG (Selectin-P ligand, CD162), sortilin related receptor 1 (SORL1), and chitinase-3-like protein 1 (CHI3L1), which were upregulated, and SLC11A1 (natural resistance-associated macrophage protein-1), S100A8 (S100 calcium binding protein A8), HAMP (Hepcidin), FTH1, SLC2A3 (Glucose transporter-3), Interleukin-1 beta (IL1B), Interferon Induced Transmembrane Protein 2 (IFITM2), S100 Calcium Binding Protein A9 (S100A9), regulator of G-protein signaling 1 (RGS1) and SLC25A37.
Differences between old and middle-aged human microglia
Noticeable differences in gene expression profiles were identified in microglia isolated from middle aged (young-mean age 53) and old brains (aged-mean age 94)[49]. This study produced RNA sequencing profiles of aged brain microglia and compared the results with those in another published study to show that 1060 genes were significantly upregulated in aged microglia and 1174 were downregulated[50]. Many of the significantly upregulated genes included those with genetic associations to AD risk [Table 2].
Prominent microglia genes that are altered with age[50]
| Upregulated | Downregulated |
|---|---|
| CTSD | CD83 |
| GRN | FLT1 |
| LTBR | ILIB |
| TSPO | PTGS2 |
| CYBA | CCL4 |
| CD14 | CCL2 |
| C1QA | CCL3 |
| C1QC | TLR4 |
| IRF7 | PTGER1 |
| TGFBR2 | |
| MRC1 |
Prominent in the upregulated group were Cathepsin D (CTSD), Progranulin (GRN), Lymphotoxin beta receptor (LTBR), Translocator protein (TSPO), Cytochrome B245 alpha (CYBA), CD14 (LPS receptor), C1QA, C1QC and interferon regulatory factor-7 (IRF7), while prominent in the downregulated group were CD83, FLT1 (vascular endothelial growth factor receptor-1), nuclear factor kappa-light-chain-enhancer of activated B cells (NFKB), interleukin-1 beta (IL1B), cycloxygenase-2 (PTGS2), CCL4 (macrophage inflammatory protein-1 beta), CCL2 (monocyte chemoattractant protein-1), CCL3 (macrophage inflammatory protein-1 alpha), Toll-like receptor-4 (TLR4), prostaglandin E receptor-1 (PTGER1), transforming growth factor beta receptor 2 (TGFBR2) and mannose receptor C-type 1-CD206 (MRC1).
A similar approach was carried out comparing the RNA sequencing profiles of aged human microglia bulk isolated from the human parietal cortex in 39 autopsy cases. Data were compared with available datasets of human and rodent microglia[51]. The paper contained large datasets of differentially expressed microglia genes with aging but the key finding was the altered expression of genes associated with cell adhesion, cell motility and different cell surface receptors [Table 3].
Human microglia associated genes that change with aging[51]
| Cell adhesion/axonal guidance | Cell surface receptors | ||
|---|---|---|---|
| Upregulated | Downre gulated | Upregulated | Downregulated |
| CAECAM1 | ADGRE5 | CD163 | IFNGR1 |
| CDH3 | CDH12 | CLEC2B | IL6R |
| DOCK1/5 | CDH19 | CLEC5A | LPAR1 |
| NLGN2 | CHL1 | CXCR4 | LPAR5 |
| NRP1/2 | ICAM3 | IGF2R | MRC2 |
| PLXNC1 | ROBO2 | P2RX1 | MST1R |
| PCDHGA2/4-8 | SEMA3C | TNFRSF14 | NTRK2 |
| PDHGB2-4 | SEMA7A | IL15 | P2YR12 |
| PTK7 | CLEC17A | ||
| ROBO4 | TLR10 | ||
| SE MA4A | TREML4 | ||
The microglia genes related to cell adhesion and axonal guidance were altered in aged microglia. Upregulated genes were: carcinoembryonic antigen-related cell adhesion molecule 1-CD66a (CAECAM1), cadherin-3 (CDH3), dedicator of cytokinesis-1/-5 (DOCK1/5), neuroligin-2 (NLGN2), neuropilin-1/-2 (NRP1/2), Plexin C1 (PLXNC1), protocaderin gamma -2,-4, 8 (PCDHGA2/4/8), protocadherin beta-2, -4 (PCDHGB-2, -4), protein tyrosine kinase-7 (PTK7), roundabout guidance receptor 4 (ROBO4), and Semaphorin 4A (SEMA4A). Downregulated genes were: ADGRE5 (Adhesion G-protein-coupled receptor CD97), Cadherin-12 (CDH12), CHL1, intercellular adhesion molecule-3 (ICAM3), roundabout guidance receptor 2 (ROBO2), Semaphorin 3A (SEMA3C), and Semaphorin 7A (SEMA7A). Immune receptor-related genes were also altered in aged microglia. Upregulated genes were: CD163 (Hemoglobin scavenger receptor), C-type lectin domain family 2B (CLEC2B), C-type lectin domain family 5A (CLEC5A), Chemokine receptor type 4 (CXCR4), insulin growth factor 2 receptor (IGF2R), purinergic receptor X1 (P2RX1), tumor necrosis factor receptor superfamily14 (TNFRSF14), and interleukin-15 (IL15). Downregulated genes were: interferon gamma receptor 1 (IFNGR1), interleukin 6 receptor (IL6R), lysophosphatidic acid receptor 1 (LPAR1), lysophosphatidic acid receptor 5 (LPAR5), mannose receptor C type 2 (MRC2), macrophage stimulating 1 receptor (MST1R), neurotrophic receptor tyrosine kinase 2 (NTRK2), purinergic receptor Y12 (P2RY12), C-type lectin domain family 17A (CLEC17A), toll-like receptor 10 (TLR10) and triggering receptor expressed on myeloid cells like-4 (TREML4).
Role of classical pro-inflammatory cytokines in ad neuroinflammation
The early theories of the role of neuroinflammation in AD suggested the involvement of classical proinflammatory cytokines and chemokines such as interleukin (IL)-1α, IL-1β, IL-6, IL-8, tumor necrosis factor-α (TNF-α), interferon-gamma (IFN-γ) and CCL-2. Demonstration of cytokines IL-1α, IL-1β and TNF-α in AD brain microglia has been reported but these are not widely-used markers for describing microglia in tissue[52-55]. There appears to be technical difficulties in localizing these secreted cytokines in tissue, and it should be noted that these classical cytokines do not prominently feature in the microglial disease-associated gene signatures of recent studies[10,11,24,32]. In the paper of Friedman et al.[24], they provide supplementary data from gene expression profiles of two studies comparing control and AD samples; neither of these detected increased expression of these classical cytokines in AD samples (supplementary data file in reference[24]).
Discrepancies between gene profiling results and tissue studies of microglia
Apolipoprotein E and Complement C1q
Apolipoprotein E (APOE/Apoe) and the three complement C1q genes (C1QA/C1qa, C1QB/C1qb, C1QC/C1qc) have been identified as microglial markers by expression studies, but these proteins have not been identified in microglia in AD tissue sections. APOE and C1Q proteins can be detected and are associated with Aβ plaques in AD brains[42,44], with expression of C1Q protein being detectable in neurons[45,56]. Recent experimental studies with mouse models showed that the majority of C1QA proteins in mouse brain was derived from microglia[57], and that C1Q overexpression can have a neuroprotective rather than pathogenic role[58]. As the majority of microglial gene profiling studies used rodent AD models, it is possible that these discrepancies are due to species differences in gene expression of human compared to rodent cells.
TREM2
TREM2 has become the most widely studied inflammatory/microglia marker for studies linking inflammation and AD. This came about due to the identification of heterozygous single nucleotide polymorphisms (SNP)/mutations associated with increased risk of developing AD[59,60]; and studies of Nasu-Hakola disease (NHD), which is associated with homozygous mutations in TREM2 gene or TYROBP gene[61]. TYROBP gene encodes DAP12, the essential adaptor protein that mediates TREM2 signaling. Patients with NHD, also known as polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL), develop a type of dementia similar to frontotemporal dementia. This dementia appears to be directly caused by microglial dysfunction resulting from the loss of function of TREM2 signaling. The function of TREM2 has been extensively characterized as a receptor for lipids, lipoprotein[62-64], including Apoe[65], heat shock protein 60 (HSP60), and Aβ peptide[66]. TREM2 appears to be a pattern recognition type of receptor rather than being ligand sequence specific. It is still not clear whether TREM2 is recognizing Aβ in plaques, or one of the many different plaque-associated proteins or lipids that accumulate, including lipoproteins such as ApoE and ApoJ (clusterin)[67]. An interaction of TREM2 and APOE signaling pathways has been implicated in altering microglia to a more damaging phenotype[68]. However, there are still inconsistencies about whether TREM2 signaling functions in a pro-inflammatory (damaging) or anti-inflammatory (reparative) manner[32].
If one now considers the theme of this review as to whether TREM2 expression can be used to describe the phenotypes of microglia in human brain tissue, findings on this have been scarce and divergent. Studies have consistently shown that TREM2 mRNA is highly expressed in human and animal brains and in microglia, and many mechanistic studies of TREM2 associated with disease assume that all microglia express TREM2. However, few studies have successfully localized TREM2 expression to microglia in human or rodent brain tissue. An earlier study using aged Aβ plaque-developing mice showed TREM2 expression by microglia associated with plaques[69], but two studies that used hard-fixed paraffin embedded human brain tissue with antigen retrieval concluded that TREM2 was not expressed by human brain microglia[70,71]. One study that included characterization of a number of different commercial antibodies to TREM2 concluded that its expression was primarily in blood monocytes and possibly neurons[70]. Another study similarly concluded that TREM2 was expressed by monocytes or infiltrating macrophages and not by microglia[71]. These authors did not report neuronal staining[71]. The discrepancies found in these studies need to be resolved. Both of these studies carefully characterized the specificity of the TREM2 antibodies used and included positive control samples that demonstrated positive staining in blood or spleen monocytes[70,71]. By contrast, our laboratory was able to identify TREM2 immunoreactivity in different types of microglia, particularly those associated with plaques and tangles in AD brains. In our study, the expression of TREM2 by microglia was not extensive or robust, and small cells with shorter processes were identified, which is not typical of the features of tissue microglia identified with antibodies to HLA-DR or IBA-1[72].
These plaque-associated microglia might still represent infiltrating macrophages and further studies would be needed, but their localization in brain neuropil and morphology were consistent with microglia. Our study used a validated antibody, but the major difference between studies was our use of brain tissues that had not been hard-fixed or paraffin-embedded. These findings do illustrate the technical difficulties of showing TREM2-positive microglia in human brains. In further follow up studies, the demonstration of TREM2 immunolocalization using antibody AF1828 (R&D Systems, Minneapolis, MN, USA) was repeatable, but demonstration of positive TREM2 staining was not consistent, with some sections showing good microglial staining while others showing none (Figure 3: unpublished observations). Figure 3 demonstrates types of TREM2-positive microglia in an AD case (temporal cortex). In Figure 3B, the demonstration of colocalization of P2RY12 expression (brown) in TREM2 positive cells appears to define the phenotype of TREM2 expression as non-activated. P2RY12 expression has been defined to identify homeostatic microglia with this marker being downregulated with activation[28]. Our study showed increased levels of TREM2 protein in AD cases compared to control cases as measured by Western blotting[72], a finding repeated by others[73], though this latter paper did not demonstrate TREM2 immunoreactivity in microglia in tissue sections.
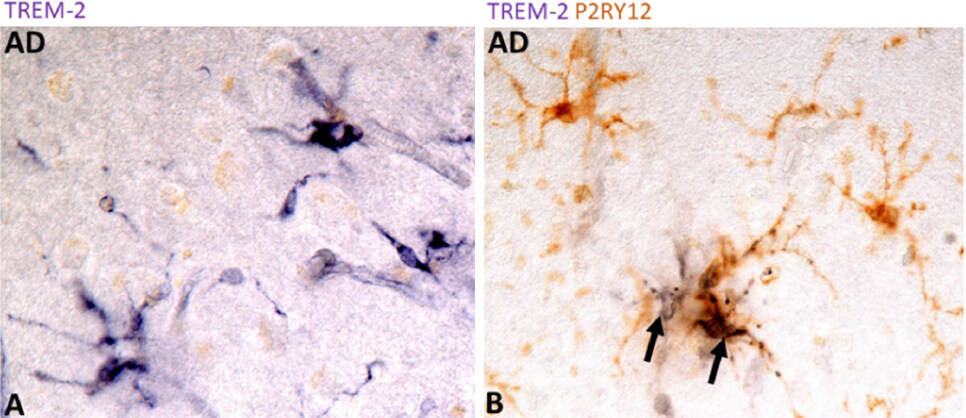
Figure 3. TREM2 expression by microglia in AD brain tissue sections. Immunohistochemical localization of TREM2 protein using antibody AF1828 (R&D systems). Sections from an AD case are shown: (A) single-stained section showing microglial-like structures immunoreactive for TREM2. Purple represents reaction with nickel-enhanced diaminobenzidine substrate; (B) double-staining (TREM2 - purple - P2RY12- brown) of AD section showing colocalization of TREM2 and P2RY12. Arrows indicate TREM2 positive microglia (purple) adjacent to P2RY12 microglia
A more recent paper suggested that there was a decrease in TREM2 protein levels in AD brains, and that neuronal and microglial TREM2 staining was also observable. These authors posited the hypothesis that brain TREM2 was derived from soluble forms of TREM2 from peripheral erythromyeloid cells and monocyte/macrophages that had trafficked into the brain[74]. These authors did not provide details on the type and fixation of the brain tissue samples employed. Replication of these findings by others would clarify the questions raised. Overall, we have demonstrated that TREM2 expression can be observed with appropriately fixed materials and a verified antibody, however, there are still inconsistencies that need to be addressed with optimal fixation methods and the availability of higher affinity antibodies. Due to the significance that experimental and gene expression studies have placed on TREM2 in AD inflammation, reliable methods to detect TREM2 in situ are needed to adequately define the phenotypes of TREM2-expressing microglia.
CD33
CD33, also known as Sialic acid-binding Ig-like lectin 3 (Siglec-3) is a myeloid specific cell-surface protein that is activated by binding with sialic acid-modified proteins or lipoproteins. The rs3865444 SNP resulting in a substitution of A for C in the 5’ untranslated region of CD33 mRNA was found to be protective for developing AD[75]. Based on animal studies, it was concluded that high levels of microglial expression of CD33 inhibited phagocytosis of Aβ, while lower levels of CD33 present in subjects with the minor A variant of rs3865444[76] was associated with increased phagocytosis of Aβ[77]. We demonstrated by immunohistochemistry of brain sections that increased microglial expression of CD33 was evident in AD cases, particularly in plaque-associated microglia. Similar plaque-associated CD33-positive microglia were demonstrated in another study[78]. CD33 expression does not fit into the established parameters of pro- or anti-inflammatory phenotypic markers as in vitro activation of cultured microglia resulted in significant down-regulation of expression[76]. In addition, CD33 functions as an inhibitory receptor resulting in activation of its immunoreceptor tyrosine-based inhibitory motif (ITIM) that results in downregulation of inflammatory signaling. CD33 is not itself an Aβ phagocytic receptor, but activation could inhibit functioning of other known Aβ binding/phagocytic receptors. It has been suggested that increased expression of CD33 could be pathogenic in AD, but there is evidence that the opposite should be considered. Studies of CD33 expression in macrophages from diabetics, where expression is downregulated, showed lower CD33 expression correlated with increased levels of proinflammatory cytokine TNFα[79]. Increased levels of CD33 in AD brains might function to restrict inflammatory activation as well as inhibiting phagocytic receptors, but blocking CD33 activation could result in enhanced neuroinflammation. A clear demonstration of the interaction of CD33 and TREM2 signaling occurred in experimental mouse models[80]. The loss of CD33 in these models resulted in increased levels of key proinflammatory cytokines. Significant information on whether these proteins interact in AD brains could be obtained if high-resolution immunohistochemistry could show microglia staining for both proteins in vivo. The phenotypes of CD33 or TREM2 positive microglia have not been rigorously investigated to determine if they primarily express pro-inflammatory or reparative/homeostatic markers.
Progranulin
Increased expression of progranulin by microglia has also been observed in AD brains[81-83]. This protein has multiple functions including neurotrophic, anti-inflammatory and lysosomal function regulation[84]. Progranulin expression is not restricted to myeloid cells with abundant neuronal expression having been characterized. Mutations in GRN (progranulin gene) can cause some forms of frontotemporal dementia (FTD), a neurodegenerative disease associated with neurodegeneration in the frontal and temporal cortex. The mechanism for this degeneration has been associated with enhanced microglial inflammation caused by partial loss of progranulin protein and its associated activity[85]. This protein appears to be present in most brain microglia, colocalizing with lysosomal proteins in brain sections, with increased levels in plaque-associated microglia[83]. As increased levels of progranulin can be protective, upregulated expression in AD would be suggestive of a reparative stress-associated response to neurodegenerative changes. As a marker to define microglial phenotypes, progranulin has limited utility, but its continued expression by different types of microglia would suggest it is having an anti-inflammatory effect in AD affected tissue. A recent gene profiling study of middle aged compared to old brain-derived microglia showed that GRN mRNA expression was significantly higher in older microglia[49].
Toll-like receptors
Toll-like receptors (TLR) are a class of ten pattern recognition receptors associated with identifying ligands from bacteria, viruses and fungi. However, they have also been identified to have a large range of cellular ligands. Due to their demonstrated interactions with aggregated Aβ and α-synuclein, TLR2 and TLR4 have been implicated in AD and PD, though immunohistochemistry for TLR4 has demonstrated neuronal, not microglial, localization[86-88]. Activation of TLR9 by its ligand unmethylated double-stranded DNA caused increased microglial phagocytosis of Aβ in experimental AD models[89]; however, there have been no demonstrations of its localization in microglia in human brains. In a recent study focused on TLR3, native ligand double-stranded RNA and the neuronal protein stathmin[90], we demonstrated distinct microglial expression of TLR3 in human brains with increased expression in plaque-associated microglia, and in endothelial cells, but not neurons or astrocytes[91]. These results were different from previous studies and dependent on the antibody used for immunohistochemistry. TLR3 had previously been defined as a specific marker for dendritic myeloid cells including microglia[92].
Colony stimulating factor-1 receptor
The survival and proliferation of microglia is primarily dependent on the action of colony stimulating factor-1 (CSF1) and IL34, ligands for CSF1R[93]. Binding of these growth factors to CSF1R results in microglial proliferation. These growth factors have distinct structures and though their expression is regulated differently, they have overlapping properties. CSF1R expression in the brain is mainly restricted to microglia. Studies using different CSF1R antagonist administered to mice resulted in knockout of microglia from tissue with a variety of mostly therapeutic effects, although it can also be detrimental in some circumstances depending on the disease model[94-99]. These studies have defined the significance of CSF1R to microglial function. There has only been a single definitive study demonstrating localization of CSF1R in microglia in human brains[100]. This study showed that all microglia constitutively expressed CSF1R and this was increased in pathology-associated microglia in AD brains[100]. This original finding of CSF1R expression by microglia is now 25 years old and requires replication using modern microscopic techniques to define the phenotypes of expressing microglia. We recently demonstrated increased expression of CSF1R and CSF1 mRNA in AD brains that correlated with severity of pathology, but reduced expression of IL34 mRNA in these samples[101]. One study has shown that CSF1R can be expressed in neurons in lesioned rat brains, but this finding has not been replicated[102]. Further characterization of CSF1R in AD tissue would be helpful to map the sites of microglial proliferation in tissues. These studies could be combined with markers and transcription factors associated with microglial proliferation (e.g., PU.1, Ki67).
CD14-Lipopolysaccharide receptor
A number of blood macrophage markers that have been defined are expressed at increased levels in activated cells (reviewed[103]). These defining studies have generally utilized strong activation agents such as bacterial proinflammogens including lipopolysaccharide (LPS) and IFN-γ that induce strong inflammatory responses for killing invading microorganisms. One of the activation receptors is CD14, along with Toll-like receptor (TLR)-4, one of the components of the LPS receptor[104]. CD14 has been considered a constitutive marker for macrophages and microglia, but it is noticeable that its expression in human AD brains has only been described in a single study[105]. This study identified increased CD14 expression of plaque-associated microglia. In Figure 4, we show examples of CD14 staining of microglia, but only in AD cases. Studies characterizing the expression of CD14 in microglia freshly isolated from human brains showed low levels of constitutive expression that increased significantly when isolated microglia were cultured for 2-4 days in vitro[106,107]. This culture-associated increase in CD14 mRNA expression was not observed for TLR4, but culture-associated decreases of a number of genes (P2RY12, CX3CR1, TNFα, TGFβ HLA-DRA, CD11b, FCγR3) were detected[107]. Increased expression of CD14 was detected by immunohistochemistry in microglia and infiltrating monocytes in sections from cases with traumatic brain injury[108].
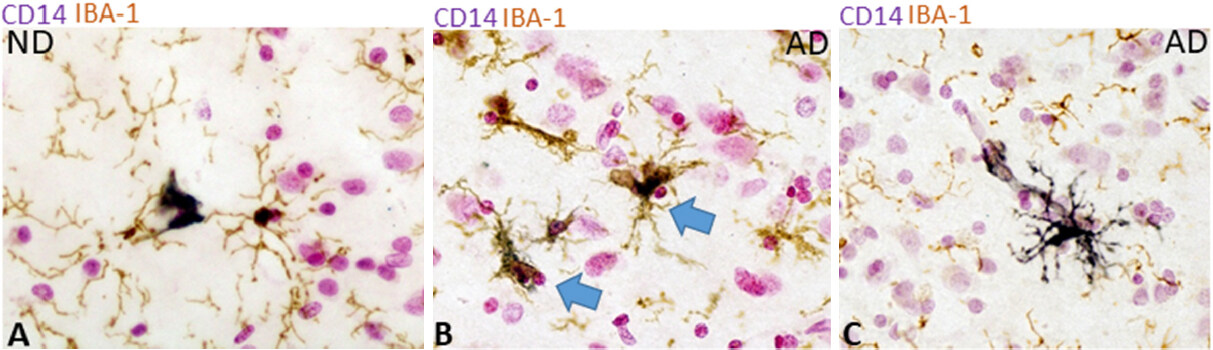
Figure 4. CD14 expression by microglia in ND and AD brain tissue sections. Immunohistochemical localization of CD14 protein using CD14 antibody (Clone 18D11, Biolegend # 812401). Sections from an ND AD case are shown: (A) Double-stained section showing strong CD14 immunoreactivity of blood monocyte (purple) but not IBA-1 positive microglia (brown). Purple represents reaction with nickel-enhanced diaminobenzidine substrate; (B, C) Double-staining (CD14 - purple - IBA-1- brown) of AD sections showing localization of CD14 and IBA-1; (B) Colocalization of CD14 and IBA-1 in microglia cells in AD case (blue arrows); (C) Strong CD14 staining of a perivascular microglia in AD section
The immunohistochemistry results presented here that follow up the earlier findings[105] suggest that using CD14 as a marker to define pro-inflammatory activation phenotypes in human brain should be reassessed. As had been encountered for other markers, successful CD14 staining was dependent on the antibody used, and also the use of antigen-retrieval methods (pH 8.0, 80 °C, 30 min). Other CD14 antibodies tested did not produce this staining of microglia under the same conditions.
CD68
There has been some controversy in the literature about the status of CD68 (designated macrosialin for rodents) as an activation or functional phagocytic marker. CD68 is a myeloid cell-specific lysosomal associated membrane protein (LAMP) whose expression is increased in cells associated with elevated phagocytic and degradative activity. Under appropriate conditions, by employing antigen retrieval methods on free-floating sections, we can demonstrate CD68 positive structures in most microglia. We have recently demonstrated that the majority of progranulin-positive, P2RY12-positive and TLR3-positive microglia were CD68 positive to some extent, irrespective of whether the sections being examined were from non-demented control or AD cases[83,91,109]. Without antigen retrieval, there was a noticeable decrease in sensitivity of detection for CD68 with many microglia showing no reactivity (unpublished observations). It is clear that being able to optimize the sensitivity of detection of CD68 (and other antigens) has a significant effect on identifying the phenotypes of cells expressing this marker.
This is a good context to discuss semi-quantitative histological findings on the expression of a series of microglial markers, including CD68, carried out in a large series of cases by Boche and colleagues[34,110,111]. These investigators were able to examine the expression of microglial markers in samples from subjects who had received the Aβ vaccine as treatment to produce circulating levels of Aβ antibodies[110]. There were significantly decreased numbers of microglia positive for CD68, CD64, CD32 and MSR-1, but not IBA-1 in sections from immunized cases compared to control non-immunized subjects. The immunized cases had significantly reduced levels of Aβ[110]. In a follow-up study employing samples from 130 non-demented cases and 83 cases with AD pathology, the expression of markers CD68 (as an indicator of phagocytosis), HLA-DR, CD64, MSR1 and IBA-1 were quantified as a percentage of microglial load, and correlated with pathological and clinical indices. There were positive correlations between dementia status and microglial load for CD68, MSR-1, CD64, and a negative correlation with IBA-1 load; there was no correlation between HLA-DR load and dementia status[34]. It should be pointed out that the difference in mean load for CD68 between non-dementia and dementia with AD pathology was only 10%, confirming widespread expression of CD68 even in non-dementia brains. A similar study that focused on microglia expressing CD68, HLA-DR and IBA-1 in sections of white matter and gray matter from MS and AD cases showed stronger CD68 staining in normal white matter compared to gray matter with an increase in white matter showing MS-related demyelination[112]. Due to its intracellular location, these authors concluded that CD68 was not a good marker to describe morphological features of microglial activation.
Defining microglia by expression of homeostatic markers (TMEM119, P2RY12)
Transmembrane protein 119 (TMEM119)
TMEM119 has been identified to have a function in bone formation by promoting the differentiation of myeloblasts to osteoclasts. Its function in microglia has not been defined, but this marker was repeatedly shown to be expressed at much higher levels by microglia than macrophages, making it a good marker for specifically identifying microglia[25,28,47,113,114]. The only published report of TMEM119 expression in AD and non-demented brains demonstrated increased expression of TMEM119 mRNA in AD cases and no significant difference in the density of TMEM119 immunoreactive microglia or total protein levels in AD cases. There appeared to be an increased expression of TMEM119 in AD plaque-associated microglia[113]. This finding is contrary to the expected change in expression of TMEM119 mRNA, identified from gene expression profiling studies reported above, where microglial TMEM119/Tmem119 expression was decreased upon activation. A different result for TMEM119 was demonstrated in sections from multiple sclerosis cases with active and chronic white matter lesions. In these areas, there was a noticeable overall decrease in microglial immunoreactivity for TMEM119, but not in areas defined as pre-active white matter lesions[115].
Purinergic receptor P2RY12
P2RY12 is a receptor for adenosine triphosphate (ATP) and adenosine diphosphate (ADP). This receptor is primarily restricted to platelets and microglia. The activation of P2RY12 by ligands induces microglial chemotaxis to sources that can include necrotic and apoptotic neurons. P2RY12 has also been identified by most microglia gene profiling studies as being a specific marker for microglia. Its expression is highest in homeostatic microglia with significant downregulation upon inflammatory activation[30]. Recent studies of P2RY12 expression by microglia in multiple sclerosis tissue sections showed downregulation in areas of active white matter lesions, similar to TMEM119, but less in areas with gray matter lesions[28,115,116]. In addition, it was observed that microglia around many Aβ plaques had reduced or no P2RY12 immunoreactivity[116]. However, our recent study further characterized P2RY12 expression by microglia in AD brains, and observed that the pathological environment (diffuse or mature plaques) had an effect on microglial P2RY12 expression[109]. Our results were noticeably different from others. We have concluded, based on these observations of increased P2RY12 expressing microglia with activation morphologies, particularly in AD brains, that this marker could be identifying other classes of microglia in addition to homeostatic/resting microglia[109]. We observed that most P2RY12 positive microglia expressed CD68 and progranulin. It was observed that highly activated appearing microglia, but also expressing P2RY12, were frequently interacting with Aβ plaques. Although our results agreed that there was an overall decrease in P2RY12 immunoreactivity, the positive cells suggest an additional function besides as a marker for homeostatic microglia.
“Alternative activation” markers (CD200 receptor, CD206, CD163)
Additional markers that have been studied for defining microglial phenotypes are CD200 receptor (CD200R), CD206 and CD163. These antigens have been defined as markers for alternative activation. CD200R was shown to be induced in cultured microglia by IL-4 and IL-13, defining it as an alternative activation marker[117,118]. CD200R has been the focus of a number of mechanistic and experimental studies because activation of this microglia/monocyte by its ligand CD200 results in downregulation of inflammatory activation signaling[119]. Activation of this receptor has significant neuroprotective effects. We showed that there was significantly decreased expression of CD200R mRNA in AD brains[117]. Despite having validated antibodies to CD200R, we could not detect CD200R protein in microglia in any brain tissue sections examined[117]. A study employing tissue from multiple sclerosis cases also could not demonstrate microglial staining of CD200R[120]. It appears then that freshly isolated microglia from human brains have very low levels of CD200R expression[120,121].
CD206 (macrophage mannose receptor C) is another widely used marker for alternative activation. Increased levels of this receptor are associated with phagocytic activity. Preliminary studies of ND and AD tissue sections with a validated antibody have failed to identify CD206-positive microglia. In our results, we can detect strong staining of vascular macrophages and perivascular macrophages [Figure 5] but not in microglia. These findings would suggest a deficit in levels of IL-4 in brain parenchyma.
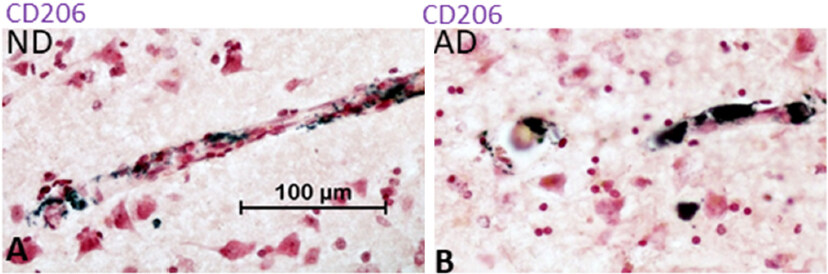
Figure 5. CD206 immunoreactivity by macrophages but not microglia in ND and AD brain tissue sections. Immunohistochemical localization of CD206 protein using antibody AF2534 (R&D systems). Sections from an ND and AD case are shown: (A) Single-stained section showing strong CD206 immunoreactivity of blood monocyte (purple). Purple represents reaction with nickel-enhanced diaminobenzidine substrate; (B) Single staining of AD section showing immunoreactivity of vascular and perivascular macrophages for CD206. No cells with morphologies of microglia were observed in sections examined. Similar findings observed by other investigators[122]
However, the localization of another alternative activation marker, the phagocytic receptor CD163 has been observed in microglia in AD and PD brains, and brains affected by human immunodeficiency virus (HIV)[113,122,123]. CD163 is a high-affinity scavenger receptor for hemogloblin-haptoglobin and a low-affinity receptor for hemoglobin alone. CD163 has been widely defined as a marker of macrophages rather than microglia, particularly those that infiltrate brain tissue following stroke. It can be highly expressed by macrophages but its role in neurodegeneration is unclear. Microglia strongly expressing CD163 were shown to be plaque-associated, and CD163 immunoreactivity was present in CD68-positive microglia, suggesting a phagocytic role, and also in TMEM119-positive microglia, a homeostatic role[122]. It should be mentioned that the description of microglia based on morphology is unreliable and so, CD163 immunoreactive microglia might be infiltrating macrophages.
Summary
At present, it is unresolved from studies of human brain tissues whether “activated’ microglia, defined in many studies based on morphology, have a predominantly pro-inflammatory phenotype or an alternative activation reparative phenotype. This remains an important issue for defining neuroinflammation in AD or other neurodegenerative diseases. Moving forward, investigators of the issues raised in this review need to consider using modern immunohistochemistry techniques that can localize multiple antigen markers to properly phenotype microglia associated with neuropathology (examples[124-126]).
Conclusion
Over thirty years of studies of tissue microglia in human brains and animal models of diseases have shown the increasingly complex behavior of microglial function in tissue, suggesting that classification into M1 or M2 schemes, or classical and alternative activation, is too simplistic to reflect this complexity in disease processes[127].
Recent gene expression profiling studies have shown (not unexpectedly) that there are significant differences between human and rodent microglia. This is particularly applicable when comparing microglia in diseased human brains, which have taken decades to develop a disease-phenotype, while microglia in mice brains develop disease phenotypes over weeks. Caution is thus needed in the interpretation of results from rodent models with aged humans.
Gene profiling technologies have now been applied to isolated microglia and these studies have challenged the hypothesis that there is an acute-type (microbial driven) of inflammation in human brains causing accelerated proinflammatory damage in AD. These studies have shown that many of the microglia genes expressed at increased levels reflect responses to restore homeostasis and limit inflammatory damage.
To fully understand the large amount of data from gene profiling technologies, ultimately there is the need for antibody-based studies to determine where a particular microglial marker is being expressed in the brain in relation to characteristic plaque and tangle pathology. Gene profiling studies have now identified a large number of new microglial antigenic markers that can be combined with established markers for phenotyping pathology-associated microglia.
To successfully accomplish immunohistochemistry in human brains, greater appreciation is needed for differences in the specificity and sensitivity of antibodies being used and the consequences of differences in tissue being examined (fixation, cause of death, postmortem autolysis).
To obtain consistency between laboratories in human tissue studies of microglia, some established protocols are needed to ensure that results do not simply reflect technical differences in tissue fixation and preparation, quality of antibodies being used, and sensitivity in detection of antigenic signals.
Declarations
AcknowledgmentsThe author thanks the support from the Banner Sun Health Research Institute (BSHRI) and their Brain and Body Donation Program (BBDP) for providing high quality brain samples for studies on human brain microglia over the last 20 years that have allowed him to make these comments and observations.
Authors’ contributionsConceived and wrote this article, took responsibility for the ideas presented: Walker DG
Availability of data and materialsNot applicable
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateThe author’s publications that are referenced in this review article utilized human brain tissue samples provided by the Brain Bank, BSHRI. Most of the studies referenced refer to anonymous tissue studies carried out in the USA, which are considered non-human subject research under U.S. federal regulations. Some of these studies were completed in Japan using the same tissue samples that had been transferred to Japan with the author. Tissue studies carried out in Japan were approved by the Shiga University of Medical Science Ethical Committee (Certificate No. 29-114).
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Ulep MG, Saraon SK, McLea S. Alzheimer disease. J Nurse Pract 2018;14:129-35.
2. Simon DK, Tanner CM, Brundin P. Parkinson disease epidemiology, Pathology, genetics, and pathophysiology. Clin Geriatr Med 2020;36:1-12.
3. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 2019;14:32.
4. Kim WS, Kagedal K, Halliday GM. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res Ther 2014;6:73.
5. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 2015;16:358-72.
6. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med 2017;23:1018-27.
7. Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener 2015;4:19.
8. in t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med 2001;345:1515-21.
9. Wang J, Tan L, Wang HF, Tan CC, Meng XF, et al. Anti-inflammatory drugs and risk of Alzheimer’s disease: an updated systematic review and meta-analysis. J Alzheimers Dis 2015;44:385-96.
10. Chew G, Petretto E. Transcriptional networks of microglia in Alzheimer’s disease and insights into pathogenesis. Genes (Basel) 2019;10.
11. Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep 2017;21:366-80.
12. Patir A, Shih B, McColl BW, Freeman TC. A core transcriptional signature of human microglia: derivation and utility in describing region-dependent alterations associated with Alzheimer’s disease. Glia 2019;67:1240-53.
13. Rio-Hortega P. Del the Microglia. Lancet 1939;233:1023-6.
14. Tremblay ME, Lecours C, Samson L, Sanchez-Zafra V, Sierra A. From the cajal alumni achucarro and Rio-Hortega to the rediscovery of never-resting microglia. Front Neuroanat 2015;9:45.
15. McGeer PL, Itagaki S, McGeer EG. Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol 1988;76:550-7.
16. McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 1987;79:195-200.
17. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988;38:1285-91.
18. Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 1989;24:173-82.
19. Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiol Aging 1988;9:339-49.
20. Mattiace LA, Davies P, Dickson DW. Detection of HLA-DR on microglia in the human brain is a function of both clinical and technical factors. Am J Pathol 1990;136:1101-14.
21. Nakagawa T, Ohnishi K, Kosaki Y, Saito Y, Horlad H, et al. Optimum immunohistochemical procedures for analysis of macrophages in human and mouse formalin fixed paraffin-embedded tissue samples. J Clin Exp Hematop 2017;57:31-6.
22. Beach TG, Adler CH, Sue LI, Serrano G, Shill HA, Walker DG, et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 2015;35:354-89.
23. Beach TG, Sue LI, Walker DG, Roher AE, Lue L, et al. The sun health research institute brain donation program: description and experience, 1987-2007. Cell Tissue Bank 2008;9:229-45.
24. Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep 2018;22:83247.
25. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 2016;113:E1738-46.
26. Zhou Y, Song WM, Andhey PS, Swain A, Levy T, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 2020;26:131-42.
27. van der Poel M, Ulas T, Mizee MR, Hsiao CC, Miedema SSM, et al. Transcriptional profiling of human microglia reveals grey-white matter heterogeneity and multiple sclerosis-associated changes. Nat Commun 2019;10:1139.
28. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, et al. Loss of “homeostatic” microglia and patterns of their activation in active multiple sclerosis. Brain 2017;140:1900-13.
29. Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 2019;101:207-23.e10.
30. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 2014;17:131-43.
31. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 2013;16:1896-905.
32. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017;169:1276-90.e17.
33. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, et al. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 2018;173:1073-81.
34. Minett T, Classey J, Matthews FE, Fahrenhold M, Taga M, et al. Microglial immunophenotype in dementia with Alzheimer’s pathology. J Neuroinflammation 2016;13:135.
35. Akiyama H, McGeer PL. Brain microglia constitutively express beta-2 integrins. J Neuroimmunol 1990;30:81-93.
36. McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, et al. Microglia in degenerative neurological disease. Glia 1993;7:84-92.
37. Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, et al. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A 1994;91:11183-6.
38. Saunders AM, Schmader K, Breitner JC, Benson MD, Brown WT, et al. Apolipoprotein E epsilon 4 allele distributions in late-onset Alzheimer’s disease and in other amyloid-forming diseases. Lancet (London, England) 1993;342:710-1.
39. Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, et al. Apolipoprotein E associates with beta amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J Clin Invest 1994;94:860-9.
40. Yamaguchi H, Ishiguro K, Sugihara S, Nakazato Y, Kawarabayashi T, et al. Presence of apolipoprotein E on extracellular neurofibrillary tangles and on meningeal blood vessels precedes the Alzheimer beta-amyloid deposition. Acta Neuropathol 1994;88:413-9.
41. Diedrich JF, Minnigan H, Carp RI, Whitaker JN, Race R, et al. Neuropathological changes in scrapie and Alzheimer’s disease are associated with increased expression of apolipoprotein E and cathepsin D in astrocytes. J Virol 1991;65:4759-68.
42. Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res 1991;541:163-6.
43. Uchihara T, Duyckaerts C, He Y, Kobayashi K, Seilhean D, et al. ApoE immunoreactivity and microglial cells in Alzheimer’s disease brain. Neurosci Lett 1995;195:5-8.
44. McGeer PL, Akiyama H, Itagaki S, McGeer EG. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 1989;107:341-6.
45. Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ. Localization and cell association of C1q in Alzheimer’s disease brain. Exp Neurol 1996;138:22-32.
46. Walker DG, Kim SU, McGeer PL. Complement and cytokine gene expression in cultured microglial derived from postmortem human brains. J Neurosci Res 1995;40:478-93.
47. Haage V, Semtner M, Vidal RO, Hernandez DP, Pong WW, et al. Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol Commun 2019;7:20.
48. Mukherjee S, Klaus C, Pricop-Jeckstadt M, Miller JA, Struebing FL. A microglial signature directing human aging and neurodegeneration-related gene networks. Front Neurosci 2019;13:2.
49. Olah M, Patrick E, Villani AC, Xu J, White CC, et al. A transcriptomic atlas of aged human microglia. Nat Commun 2018;9:539.
50. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 2016;89:37-53.
51. Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci 2017;20:1162-71.
52. Sheng JG, Mrak RE, Griffin WS. Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol 1997;56:285-90.
53. Sheng JG, Griffin WS, Royston MC, Mrak RE. Distribution of interleukin-1-immunoreactive microglia in cerebral cortical layers: implications for neuritic plaque formation in Alzheimer’s disease. Neuropathol Appl Neurobiol 1998;24:278-83.
54. Li Y, Liu L, Barger SW, Griffin WST. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci 2003;23:1605-11.
55. Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 1993;7:75-83.
56. Terai K, Walker DG, McGeer EG, McGeer PL. Neurons express proteins of the classical complement pathway in Alzheimer disease. Brain Res 1997;769:385-90.
57. Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, et al. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation 2017;14:48.
58. Benoit ME, Tenner AJ. Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. J Neurosci 2011;31:3459-69.
59. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 2013;368:107-16.
60. Korvatska O, Leverenz JB, Jayadev S, McMillan P, Kurtz I, et al. R47H variant of TREM2 associated with alzheimer disease in a large late-onset family: clinical, genetic, and neuropathological study. JAMA Neurol 2015;72:920-7.
61. Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet 2002;71:656-62.
62. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 Binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 2016;91:328-40.
63. Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem 2015;290:26043-50.
64. Kosack L, Gawish R, Lercher A, Vilagos B, Hladik A, et al. The lipid-sensor TREM2 aggravates disease in a model of LCMV-induced hepatitis. Sci Rep 2017;7:11289.
65. Bailey CC, DeVaux LB, Farzan M. The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J Biol Chem 2015;290:26033-42.
66. Zhao Y, Wu X, Li X, Jiang LL, Gui X, et al. TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron 2018;97:1023-31.e7.
67. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015;160:1061-71.
68. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017;47:566-81.e9.
69. Frank S, Burbach GJ, Bonin M, Walter M, Streit W, et al. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 2008;56:1438-47.
70. Satoh JI, Kawana N, Yamamoto Y, Ishida T, Saito Y, et al. A survey of TREM2 antibodies reveals neuronal but not microglial staining in formalin-fixed paraffin-embedded postmortem Alzheimer’s brain tissues. Alzheimers Res Ther 2013;5:30.
71. Fahrenhold M, Rakic S, Classey J, Brayne C, Ince PG, et al. TREM2 expression in the human brain: a marker of monocyte recruitment? Brain Pathol 2017; doi: 10.1111/bpa.12564.
72. Lue LF, Schmitz CT, Serrano G, Sue LI, Beach TG, et al. .
73. Perez SE, Nadeem M, He B, Miguel JC, Malek-Ahmadi MH, et al. Neocortical and hippocampal TREM2 protein levels during the progression of Alzheimer’s disease. Neurobiol Aging 2017;54:133-43.
74. Raha-Chowdhury R, Henderson JW, Raha AA, Stott SRW, Vuono R, et al. Erythromyeloid-derived TREM2: a major determinant of Alzheimer’s disease pathology in down syndrome. J Alzheimers Dis 2018;61:1143-62.
75. Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 2011;43:436-41.
76. Walker DG, Whetzel AM, Serrano G, Sue LI, Beach TG, et al. Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol Aging 2015;36:571-82.
77. Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013;78:631-43.
78. Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci 2013;16:848-50.
79. Gonzalez Y, Herrera MT, Soldevila G, Garcia-Garcia L, Fabian G, et al. High glucose concentrations induce TNF-alpha production through the down-regulation of CD33 in primary human monocytes. BMC Immunol 2012;13:19.
80. Griciuc A, Patel S, Federico AN, Choi SH, Innes BJ, et al. TREM2 Acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron 2019;103:820-35.e7.
81. Pereson S, Wils H, Kleinberger G, McGowan E, Vandewoestyne M, et al. Progranulin expression correlates with dense-core amyloid plaque burden in Alzheimer disease mouse models. J Pathol 2009;219:173-81.
82. Minami SS, Min SW, Krabbe G, Wang C, Zhou Y, et al. Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nat Med 2014;20:1157-64.
83. Mendsaikhan A, Tooyama I, Bellier JP, Serrano GE, Sue LI, et al. Characterization of lysosomal proteins progranulin and prosaposin and their interactions in Alzheimer’s disease and aged brains: increased levels correlate with neuropathology. Acta Neuropathol Commun 2019;7:215.
84. Mendsaikhan A, Tooyama I, Walker DG. Microglial progranulin: involvement in Alzheimer’s disease and neurodegenerative diseases. Cells 2019;8.
85. Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell 2016;165:921-35.
86. Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci 2009;29:11982-92.
87. Chen K, Iribarren P, Hu J, Chen J, Gong W, et al. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem 2006;281:3651-9.
88. Tang SC, Lathia JD, Selvaraj PK, Jo DG, Mughal MR, et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol 2008;213:114-21.
89. Scholtzova H, Chianchiano P, Pan J, Sun Y, Goni F, et al. Amyloid beta and Tau Alzheimer’s disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol Commun 2014;2:101.
90. Bsibsi M, Bajramovic JJ, Vogt MHJ, van Duijvenvoorden E, Baghat A, et al. The microtubule regulator stathmin is an endogenous protein agonist for TLR3. J Immunol 2010;184:6929-37.
91. Walker DG, Tang TM, Lue LF. Increased expression of toll-like receptor 3, an anti-viral signaling molecule, and related genes in Alzheimer’s disease brains. Exp Neurol 2018;309:91-106.
92. Muzio M, Bosisio D, Polentarutti N, D’amico G, Stoppacciaro A, et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol 2000;164:5998-6004.
93. Baghdadi M, Umeyama Y, Hama N, Kobayashi T, Han N, et al. Interleukin-34, a comprehensive review. J Leukoc Biol 2018;104:931-51.
94. Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014;82:380-97.
95. Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MRP, et al. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain 2016;139:1265-81.
96. Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun 2019;10:3758.
97. Nissen JC, Thompson KK, West BL, Tsirka SE. Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp Neurol 2018;307:24-36.
98. Bennett RE, Bryant A, Hu M, Robbins AB, Hopp SC, et al. Partial reduction of microglia does not affect tau pathology in aged mice. J Neuroinflammation 2018;15:311.
99. Olmos-Alonso A, Schetters STT, Sri S, Askew K, Mancuso R, et al. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 2016;139:891-907.
100. Akiyama H, Nishimura T, Kondo H, Ikeda K, Hayashi Y, et al. Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Res 1994;639:171-4.
101. Walker DG, Tang TM, Lue LF. Studies on colony stimulating factor receptor-1 and ligands colony stimulating factor-1 and Interleukin-34 in Alzheimer’s disease brains and human microglia. Front Aging Neurosci 2017;9:244.
102. Luo J, Elwood F, Britschgi M, Villeda S, Zhang H, et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J Exp Med 2013;210:157-72.
103. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 2009;27:451-83.
104. Jiang Q, Akashi S, Miyake K, Petty HR. Lipopolysaccharide induces physical proximity between CD14 and toll-like receptor 4 (TLR4) prior to nuclear translocation of NF-kappa B. J Immunol 2000;165:3541-4.
105. Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain 2005;128:1778-89.
106. Melief J, Sneeboer MAM, Litjens M, Ormel PR, Palmen SJMC, et al. Characterizing primary human microglia: a comparative study with myeloid subsets and culture models. Glia 2016;64:1857-68.
107. Mizee MR, Miedema SSM, van der Poel M, Adelia Schuurman KG, van Strien ME, et al. Isolation of primary microglia from the human post-mortem brain: effects of ante- and post-mortem variables. Acta Neuropathol Commun 2017;5:16.
108. Beschorner R, Nguyen TD, Gozalan F, Pedal I, Mattern R, et al. CD14 expression by activated parenchymal microglia/macrophages and infiltrating monocytes following human traumatic brain injury. Acta Neuropathol 2002;103:541-9.
109. Walker DG, Tang TM, Mendsaikhan A, Tooyama I, Serrano GE, et al. Patterns of expression of purinergic receptor P2RY12, a putative marker for non-activated microglia, in aged and Alzheimer’s disease brains. Int J Mol Sci 2020;21.
110. Zotova E, Bharambe V, Cheaveau M, Morgan W, Holmes C, et al. Inflammatory components in human Alzheimer’s disease and after active amyloid-beta42 immunization. Brain 2013;136:2677-96.
111. Gomez-Nicola D, Boche D. Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease. Alzheimers Res Ther 2015;7:42.
112. Hendrickx DAE, van Eden CG, Schuurman KG, Hamann J, Huitinga I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J Neuroimmunol 2017;309:12-22.
113. Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016;36:39-49.
114. Gonzalez Ibanez F, Picard K, Bordelau M, Sharma K, Bisht K, et al. Immunofluorescence staining using IBA1 and TMEM119 for microglial density, morphology and peripheral myeloid cell infiltration analysis in mouse brain. J Vis Exp 2019; doi: 10.3791/60510.
115. van Wageningen TA, Vlaar E, Kooij G, Jongenelen CAM, Geurts JJG, et al. Regulation of microglial TMEM119 and P2RY12 immunoreactivity in multiple sclerosis white and grey matter lesions is dependent on their inflammatory environment. Acta Neuropathol Commun 2019;7:206.
116. Mildner A, Huang H, Radke J, Stenzel W, Priller J. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 2017;65:375-87.
117. Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue LF. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammation. Exp Neurol 2009;215:5-19.
118. Koning N, van Eijk M, Pouwels W, Brouwer MSM, Voehringer D, et al. Expression of the inhibitory CD200 receptor is associated with alternative macrophage activation. J Innate Immun 2010;2:195-200.
119. Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KHG, et al. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci 2007;27:8309-13.
120. Koning N, Swaab DF, Hoek RM, Huitinga I. Distribution of the immune inhibitory molecules CD200 and CD200R in the normal central nervous system and multiple sclerosis lesions suggests neuron-glia and glia-glia interactions. J Neuropathol Exp Neurol 2009;68:159-67.
121. Melief J, Koning N, Schuurman KG, Van De Garde MDB, Smolders J, et al. Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia 2012;60:1506-17.
122. Pey P, Pearce RKB, Kalaitzakis ME, Griffin WST, Gentleman SM. Phenotypic profile of alternative activation marker CD163 is different in Alzheimer’s and Parkinson’s disease. Acta Neuropathol Commun 2014;2:21.
123. Roberts ES, Masliah E, Fox HS. CD163 identifies a unique population of ramified microglia in HIV encephalitis (HIVE). J Neuropathol Exp Neurol 2004;63:1255-64.
124. Darmanis S, Gallant CJ, Marinescu VD, Niklasson M, Segerman A, et al. Simultaneous multiplexed measurement of RNA and proteins in single cells. Cell Rep 2016;14:380-9.
125. Genshaft AS, Li S, Gallant CJ, Darmanis S, Prakadan SM, et al. Multiplexed, targeted profiling of single-cell proteomes and transcriptomes in a single reaction. Genome Biol 2016;17:188.
126. Morrison LE, Lefever MR, Behman LJ, Leibold T, Roberts EA, et al. Brightfield multiplex immunohistochemistry with multispectral imaging. Lab Invest 2020; doi: 10.1038/s41374-020-0429-0.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Walker DG. Defining activation states of microglia in human brain tissue: an unresolved issue for Alzheimer's disease. Neurosciences 2020;7:194-214. http://dx.doi.org/10.20517/2347-8659.2020.09
AMA Style
Walker DG. Defining activation states of microglia in human brain tissue: an unresolved issue for Alzheimer's disease. Neuroimmunology and Neuroinflammation. 2020; 7(3): 194-214. http://dx.doi.org/10.20517/2347-8659.2020.09
Chicago/Turabian Style
Walker, Douglas G.. 2020. "Defining activation states of microglia in human brain tissue: an unresolved issue for Alzheimer's disease" Neuroimmunology and Neuroinflammation. 7, no.3: 194-214. http://dx.doi.org/10.20517/2347-8659.2020.09
ACS Style
Walker, DG. Defining activation states of microglia in human brain tissue: an unresolved issue for Alzheimer's disease. Neurosciences. 2020, 7, 194-214. http://dx.doi.org/10.20517/2347-8659.2020.09
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 61 clicks
Cite This Article 61 clicks

 Like This Article 18
likes
Like This Article 18
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.