
The immune regulation of PD-1/PDL-1 axis, a potential biomarker in multiple sclerosis

Abstract
Multiple sclerosis is an autoimmune disease characterised by a chronic inflammation within the central nervous system. In the last ten years, studies on multiple sclerosis have been concentrated on the discovery of new biomarkers of disease and potential therapeutic targets. In chronic infection or in cancer, the immune system response is faulty and maintained in a condition defined as T-cell exhaustion induced by expression of co-inhibitory receptors. The PD-1/PDL-1 pathway is demonstrated to be the main one responsible for promoting T-cell exhaustion, and immunotherapies targeting PD-1 or PDL-1 have shown beneficial clinical outcomes in several tumours and chronic diseases. Contrarily, transcriptional T-cell exhaustion signature and high expression of co-inhibitor receptor PD-1 are associated with favourable prognosis in multiple sclerosis and other autoimmune diseases. Several studies have clearly demonstrated PD-1 has a dual role in immune self-tolerance: to constrain autoreactive T cells in anergic condition and to protect the tissue from the damage caused by the activation of endogenous autoreactive T cells. Consequently, immune checkpoint inhibitor therapies that target inhibitory receptors in cancer cause an exacerbation of autoimmune diseases. This review describes the roles of the PD-1/ PDL-1 pathway in cancer and autoimmune diseases, especially in multiple sclerosis, and how manipulating PD-1 can be a therapeutic approach in multiple sclerosis.
Keywords
T-cell exhaustion
The word “exhaustion” originates from the Latin “exaurire” and was used for the first time to explain a mechanism for silencing antiviral T-cell response during a Lymphocytic Choriomeningitis Virus infection (LCMV)[1]. Antigen-specific CD8 T cells remove viral infection by killing infected cells and the production of antiviral cytokines such as interferon gamma (IFNγ). The damping of immune response in LCMV was associated with two mechanisms silencing the CD8 Cytotoxic T Lymphocytes (CTLs) response: the depletion of nucleoprotein-specific CD8 T cells and the persistence of exhausted glycoprotein-specific CD8 T cells unable to kill virus-infected cells and release antiviral cytokines[1]. Further investigations showed that the exhaustion process suppresses the CD8 antiviral activity by the hierarchical loss of T cell function[2]. Proliferation, release of IL-2 and cytolysis were lost at an early stage of exhaustion, followed by tumor necrosis factor alpha (TNFα) production and, at the severe late stage, IFNγ production. CD4 T helper cells (Th cells) drive the fate of CD8 T-cell responses in chronic viral infections. Mice with transient depletion of CD4 T cells before infection with chronic strains of LCMV develop CD8 T-cell exhaustion and high viral load compared with non-treated mice[3]. Th cells are necessary for the generation of stable and functional CD8 memory cells[4-6]. During a chronic infection, CD8 T cells develop an exhaustion phenotype that produces a state of immunosuppression in the absence of CD4 T cells[7]. The exhaustion process induces low levels of Th cells[8-10] and affects CD4 T cell functions with loss of proliferation and IL-2 and TNFα production[11]. Moreover, CD8 T cell and B cell response was restored when functional LCMV-specific CD4 T cells were transfected in LCMV chronically infected mice. PD-1 expression increased in LCMV-specific CD4 T cells by two weeks after transfer in chronically infected mice and programmed death 1 (PD-1) blockage improved the CD4 T-cell activity[12,13]. In addition, the rescue of CD8 T cell function in terms of proliferation and cytokine release was greater in mice receiving the combination of PD-1 blockade and Th cells compared with the mice receiving either treatment alone[12].
Inhibitory checkpoint pathways
Cytotoxic T Lymphocytes A-4 (CTLA-4), PD-1 and programmed death ligand 1 (PDL-1) are the first inhibitory checkpoint receptors to be discovered and targeted in cancer immunotherapy and chronic viral infection. The amplitude of T-cell response depends on the activation of co-stimulatory (CD28) or inhibitory receptors after the engagement of T-cell receptor (TCR) with the cognate-peptide-major histocompatibility complex. Co-inhibitory receptors show distinct patterns of expression and different mechanisms of action and signalling.
The knockout CTLA-4 mice has shown a lethal hyperactivation phenotype, confirming that CTLA-4 is a vital inhibitor checkpoint of the immune system. After TCR activation, CTLA-4 upregulates in the CD4 T cells and competes with the co-stimulatory receptor CD28 for its ligands CD80 and CD86, for which CTLA-4 has more binding affinity. The link of CTLA-4 to CD80 and CD86 inhibits T-cell activation. Because antigen-presenting cells and dendritic cells express CD80 and CD86, the suppression of anti-tumour immunity by CTLA-4 is thought to occur in the secondary lymphoid organs as well as in the tumour microenvironment.
PD-1 is an inhibitory receptor that belongs to the CD28 family. The receptor has been detected on activated T lymphocytes, B lymphocytes, dendritic cells, macrophages and natural killer cells after a transcriptional activation[14]. PDL-1 is the ligand of PD-1, belongs to B7 family and is present on B lymphocytes, antigen-presenting cells (APC) and tissue cells, including several types of cancer. PD-1 engagement activates the inhibitory phosphatase PP2A and SHP-2 by immune receptor tyrosine inhibitory motif and immune receptor tyrosine switch motif, inhibits T-cell activation and increases T-cell migration within tissues.
T-cell exhaustion in cancer and infectious diseases. T-cell exhaustion has been described in animal models of polyomavirus[15] and adenovirus[16], as well as in chronic human infections mediated by human immunodeficiency virus (HIV)[17] and hepatitis B and C virus (HBV, HCV)[18,19]. The loss of antiviral activity on CD8 T cells is associated with the upregulation of PD-1 in an animal model of LCMV followed by hierarchy suppression of cyto- and cytotoxic function[20]. The CD8 cytotoxic function against infected cells and antiviral cytokines production can be restored by blocking the PD-1/PDL-1 pathway, leading to clearance of infection in LCMV. The PD-1/PDL-1 pathway is the principal regulator of T-cell exhaustion in the animal model of LCMV. Studies on PD-1 in human chronic infections such as Human HIV and HCV have shown an increase of PD-1 on virus-specific CD8 T cells. PD-1 increases in HIV-specific CD4 and CD8 T cells and is directly correlated with the viral load and inversely with CD4 T cell counts. Furthermore, PD-1 increases in patients with HIV progression as compared with patients with long-term progression.
Moreover, T-cell exhaustion suppresses cancer immune-surveillance, leading to tumour spread. Restoring the immune surveillance by blocking PD-1/PDL-1 pathway has been an essential improvement in the cancer treatment. The function of PD-1 and its deregulations are summarised in Figure 1.
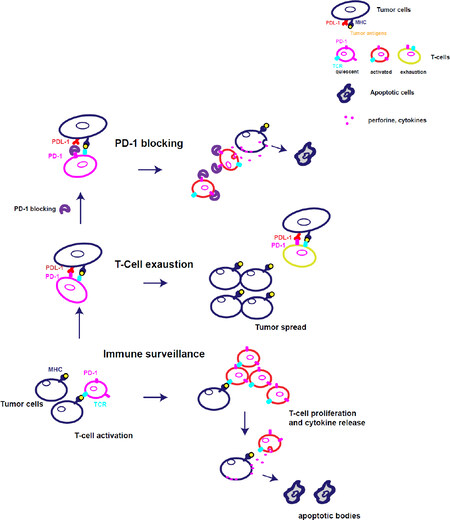
Figure 1. PD-1 inhibitor checkpoint regulates T cell activation during immune surveillance and induces T-cell exhaustion in cancer. (1) PD-1 is expressed on activated T cells and regulates the activity of late differentiate effector cells. The foreign antigens are presented to T cells by the TCR engagment with MHC expressed on dendritic cells, infected cells or tumor cells. Thus, T cells proliferate and differentiate in effector and memory cells. Effector cells kill the foreign antigens express on infected or tumor cells by releasing inflammatory cyokines and cytotoxic granules that induce target-cells to apoptosis. PD-1 is an inhibitory checkpoint able to regulate the T cell activation when the inflammation is resolved. (2) After activation, PD-1 increases on T cells and the link with the ligand PDL-1 reduces the T cell functionality. Thus, T cells are not able to kill the tumor or reduce the viral load and this condition called T-cell exaustion favours a persistent infection and tumor spread. (3) The blocking of PD-1 with monoclonal antibodies restores T cell function. T cells proliferate and differentiate in effector and memory cells contributing to resolve the infection or tumor. MHC: major histocompatibility complex
Nonetheless, some tumours develop resistance to PD-1 blocking, which is regulated by the tumour microenvironment where infiltrates of regulatory and immune suppressor cells (myeloid suppressor cells, regulatory T cells, immature dendritic cells and immune-suppressive macrophages) reduce the activity of cytotoxic CD8 T cells. Any treatment that induces changes in the levels of hormones and growth factors increases the vulnerability of cancer cells to cytotoxic drugs, which become sensitive to PD-1 treatment. Furthermore, short-term starvation (STS) has been described to reduce levels of insulin-like growth factor 1 (IGF-1) in the lung cancer microenvironment with an increase in the infiltration of immune cells and cytotoxic CD8 T cells[21]. The combining of PD-1 blockade treatment with STS boosts the immune system, reducing the tumour size significantly in a mouse model of KRAS-driven lung adenocarcinoma and Lewis lung carcinoma[21]. The combination of the two treatments induced in the mice an extended lasting memory response. The immunological study has shown an increase in tumour-infiltrating CD8 and natural killer cells by reducing the proportion of CD4 and B cells. CD8 and CD4 T cells showed a reduction in PD-1 expression. Depletion of CD8 T cells abrogated utterly the effect of the STS and anti-PD-1 treatment, confirming that STS sensitises the lung cancer to CD8 T cells reactivated by PD-1 blocking. The tumour-immune infiltrate treated with anti-PD-1 after STS was analysed with a flow cytometer and presented an increase in the frequency of tumour-specific IFN-γ-producing T cells as compared with mice treated with only one agent or vehicle[21].
A selective ablation of PD-1 on myeloid cells or T cells has essentially contributed to understanding the function of PD-1 in the cancer-immunity cycle. Mice with PD-1 ablated only on myeloid cells showed an increase of effector memory T cells and an enhanced response against the tumours. Ablation of PD-1 on myeloid cells changes the tumour microenvironment, skewing the myeloid cell fate toward differentiation of monocytes, macrophages and CD11c+MHCII+ dendritic cells (DC) rather than myeloid suppressor cells and granulocyte/macrophage progenitors. The reduction of myeloid suppressor cells due to PD-1 ablation contributes to restoring the functionality of effector memory T cells and, consequently, an immune response to the tumour[22].
PD-1/PDL-1 axis in autoimmune disease
Autoimmune thyroid diseases (AIDTs) are an organ-specific autoimmune disease that affects 50/100,000 people per year, with a prevalence in females[23]. Infiltrating lymphocytes generating follicle structures are described in the thyroid glands in Hashimoto thyroiditis and Grave’s disease (GD), the most common AIDTs[24]. Interferon signalling and increased expression of PD-1 and M2 macrophages markers were revealed in the transcriptomic analysis of GD glands[25]. Thyroid autoimmunity is one of the most common Immune-Related Adverse Events observed after immune checkpoint inhibitors (ICI) treatments in cancer[26]. The PD-1/PDL-1 axis has been investigated in the peripheral blood and the infiltrating lymphocytes in glands of patients with GD and compared with non-multinodular goitres as non-autoimmune controls and healthy controls (HC)[27]. A decrease of naïve as well as an increase of memory and effector subsets of CD4 T cells was observed in GD as compared with the healthy donors (HD)[27]. Besides, infiltrating lymphocytes in the gland of GD patients were predominantly effector and memory cells. PD-1 was found higher in GD than HD in CD4 T cells, and it increased in effector memory T cells re-expressing CD45RA (TEMRA), effector and central memory subsets. PD-1 expression increased in infiltrating CD4 and CD8 T cells in infiltrating lymphocytes with predominance on effector and memory subsets. The expression of PDL-1 but not programmed death ligand 2 (PDL-2) was observed in epithelial thyroid follicular cells in the thyroid tissue from GD patients but not in non-multinodular goitres patients[27].
Rheumatoid arthritis (RA) is a chronic progressive inflammatory disorder characterised by damage of articular cartilage and joint destruction[28-31]. Environmental, genetic, infectious and hormonal factors can contribute to the pathogenesis of the disease[32,33]. The overproduction of TNFα generates inflammation and damages the joints. The interaction of B and T lymphocytes[34] with synovial-like fibroblasts and macrophages causes the overproduction of TNFα that induces the production of several inflammatory cytokines, such as interleukin-6 (IL-6)[35,36]. Several animal and clinical studies revealed the presence of CD4 T cells in the perivascular cuff and infiltration of CD8 T cells into the tissue. Depletion of T cells or treatment of anti-cytokines that are involved in T-cell activation or promote antigen-presentation reduces inflammation. T helper 17 cells are the primary T cell subsets involved in inflammation and autoimmunity in RA[37]. PD-1-/- C57BL/6 mice developed arthritis. PD-1 polymorphisms have been reported to be associated with RA[38,39]. Expression of PD-1 was detected in synovial T cells and macrophages in patients with RA[40]. In the peripheral blood of RA patients, PD-1 was significantly decreased in CD4 T cells (P = 0.002) and CD8 T cells (P < 0.001) as compared with HC (P < 0.05)[41]. DAS28 score is a measure of disease activity in RA, and PD-1 expression was found inversely correlated with DAS28 scores in RA patients[41]. Besides, CRP is an indicator of inflammation and cases with positive CRP detection had a lower proportion of PD-1+CD4+ T cells than those with negative CRP[42].
Systemic Lupus Erythematous (SLE) is an autoimmune disease generated by the production of antibodies against self-antigens and deposition of immune complexes in different tissues. Inflammation and multisystem disorders characterise the disease[43]. The disorder affects mainly women of reproductive age with an incidence of 20-70 cases per 100,000 individuals[44,45]. Environmental, genetic and hormonal factors are relevant in the pathogenesis of the disease[46-48]. Genetic variations in the immune checkpoint genes such as PD-1, T-cell immunoglobulin domain, mucin domain (TIM) and CTLA-4 increase the susceptibility to develop the autoimmune disease as a consequence of the breakdown of immune tolerance to self-antigens[49,50]. Several single-nucleotide polymorphisms (SNPs) have been identified to affect PD-1 function and to contribute to tumours and autoimmune disease[49,50]. The frequencies of PD-1 SNPs (PD1.1, PD1.3, PD1.5 and PD1.9) were analysed in SLE patients. The PD1.5 genotype frequency was increased in Iranian, Malaysian and European patients with SLE as compared with healthy donors[51-53]. The distribution of PD1.5 C/C, PD1.5 C/T and PD1.5 T/T genotypes versus other genotypes in patients with SLE differed from healthy controls[53]. In addition, there were significant differences in the PD1.5 genotypes between patients with renal involvement and neurological involvement and between neurological involvement and HC[53]. The allelic analysis revealed that there was a significant association between PD1.5 allele frequency and SLE susceptibility[53].
Type I diabetes (TID) is caused by autoreactive cells that destroy the insulin-producing beta cells in the pancreatic islet of Langerhans[54]. PD-1 and PDL-1 protect from TID. PD-1 deficiency accelerates the onset and the frequency of TID in NOD (non-obese diabetic) mice and infiltration of T cells into the islets. PD-1 or PDL-1 but not PDL-2 blockage rapidly induces diabetes in NOD mice with an expansion of activated glutamic acid decarboxylase (GAD)-reactive cells[55-59]. In addition, despite CTLA-4 blockage showing a negative regulation of autoimmune diabetes only in early stages of the life, the PD1-PDL-1 pathway regulated autoreactive T cells throughout the life span of the animal and appeared to be critical for progression of autoimmune diabetes[59]. Moreover, polymorphisms that reduce the function of PD-1 are associated with human TID[60]. PD-1 function was also investigated by using a model that mimics the naïve pre-immune repertoire. Fewer islet specific BDC2.5 transgenic naïve CD4 T cells were transferred into prediabetic NOD mice[61]. BDC2.5 CD4 T cells accumulated in the pancreas surrounding the islet (peri-insulitis)[61]. When BDC2.5 naïve T cells were preactivated in vitro and then transferred into NOD mice, the majority of them accumulated in the pancreas but within the islet (insulitis), developing severer TID[61]. The majority of BDC2.5 cells differentiate in IFNγ-producing cells. Anti-PDL-1 administration caused a conversion from peri-insulitis to destructive insulitis[61]. PD-1 on BDC2.5 naïve T cells regulate proliferation, C-X-C Motif Chemokine Receptor 3 (CXCR3) expression, infiltration of the pancreas, and release of inflammatory cytokines IFNγ, TNFα and IL-2. Moreover, PD-1 but not PDL-1 expressed by BDC2.5 cells is required to suppress proliferation and infiltration of the pancreas[61].
A fusion protein containing a single-chain variable fragment (scFv) of PD-1 antibody (aPD-1), an albumin-binding protein and Pseudomonas aeruginosa exotoxin A was used to select and kill PD-1+ cells[62]. The treatment was tried first in animal model of TID. Depletion of PD-1+ cells inhibited the development of TID in NOD mice, reducing the pancreatic infiltration of PD-1+ cells as compared with the controls. Contrarily, anti-PD-1 was observed to induce a TID progression in NOD mice, suggesting that PD-1 blocking restores the proliferation and effector function of autoreactive cells. The TID progression was reduced in mice pre-treated with PD-1 depletion before PD-1 blocking, confirming that PD1 is expressed in autoreactive cells[62].
Multiple Sclerosis (MS) is an autoimmune disease of the central nervous system (CNS). Disease genetic and cellular studies sustain that autoreactive T cells are responsible for CNS damage[63,64]. Post-mortem studies showed that T and B cells infiltrate the CNS and, in the long term, develop lymphoid follicles with a functional germinal centre in the meninges and this meningeal inflammation causes white matter demyelination[65]. Further investigations demonstrated that inflammatory cytokines and molecules involved in T and B cell development and lymphoid-neogenesis increased in the cerebrospinal fluid (CSF) from post-mortem MS cases with a high level of meningeal inflammation and Gray matter demyelination, as well as in the CSF of patients with MS[66] and Gray matter damage at diagnosis[67]. Moreover, infiltration of T cells enriched the brain lesions[68] and T- and B-depleted therapies reduced activity and progression in MS[69].
IFNγ and IL-17-producing CD4 T cells have been defined as the effector populations driving CNS damage. Adoptive transfer of Th1 cells inducing experimental autoimmune encephalomyelitis (EAE) and the cytokine profile of cells isolated from the CNS of mice with acute EAE have shown that Th1 cytokines are released from infiltrating CD4+ T cells and TNFα is predominantly transcribed by macrophages and microglia[70-72]. T-bet is the transcription factor regulating Th1 development and IFNγ production, and it is induced by interferon γ transducer and activator of transcription (STAT)-1 signalling pathway during T-cell activation. The role of Th1 in inducing EAE was confirmed in STAT-4 and STAT-6 deficient mice. STAT-4 pathway controls the Th1 differentiation and STAT-4-/- mice showed resistance to the development of AEA. Mice deficient in STAT-6, which regulates the differentiation of Th2 cells, develop severer AEA and have more Th1 phenotype[73]. The IFNγ-producing CD4+T cells generate in the cervical lymph nodes and Th1 migration happens 24 h before the onset of neurological signs of EAE[74]. Although Th1 cells contribute to EAE, IFNγ knockdown mice are predisposed to develop EAE and infiltrates of lymphocytes, macrophages and granulocytes were detected in the CNS[75-77]. The results from IFNγ knockdown and STAT-1 deficient mice established the contribution of other effector cells to the disease pathogenesis. Besides, the discovery of IL-23 rather than IL-12 being crucial for EAE development led to evaluating Th17 cells and their transcriptional factor RORgt in the EAE pathogenesis[78-80], Moreover, Th1, Th17 and Th9 were defined to induce EAE with a different disease phenotype[79,81]. In addition to effector cells in EAE[82], the mechanisms of immune regulations, including regulatory B and T cells, and expression of inhibitory receptors were investigated in EAE. CD4 T cells were observed to protect against spontaneous development of CNS autoimmunity in EAE[83], and CD4 regulatory T cells characterised by high expression of CD25 and transcription factor FOXp3 isolated from peripheral blood had a reduced effector suppression function in patients with MS as compared with healthy donors[84]. A defect in regulatory B cells was also described to induce EAE and autoimmunity in mice and patients[85,86].
Single-cell transcriptomics of blood and CSF cells isolated from patients with MS and healthy donors revealed that different mechanisms operate in the two compartments[87]. Analysis of the data showed that MS affects the cellular composition of the CSF and the transcriptional phenotype of blood cells[87]. Blood cells exhibited several transcriptional changes, including induction of activation markers (ICOS), cytokine receptors (IL17RA) and trafficking molecules (PECAM1/CD31 and ITGA5/a5 integrin) in T cells[87].
Contrarily, an enrichment of CD4 T cells with T helper 1 and T follicular helper (Tfh) profiles, regulatory T cells, myeloid lineage cells and late-stage B lineage cells were detected in the CSF[87]. Furthermore, Tfh cells expressing PD-1 were observed to correlate with the proportion of plasma cells and showed cytotoxicity and co-inhibitory function. Follicular T helper (Tfh) cells, a subset of T helper cells, are necessary for B cell differentiation and antibody production[87,88]. These cells express CXCR5, CD40 ligand and IL-21 as well as high levels of inducible T-cell constimulator (ICOS) and PD-1. They were described to migrate in the germinal centre and to activate B cells. An elevated frequency of circulating Tfh and B cells was identified in MS patients undergoing relapse and Tfh-like cells upregulated during the course of EAE progression[88]. In addition, an adoptive cell transfer experiment showed that myelin oligodendrocytes glycoprotein (MOG)-reactive Tfh-like cells induced a worsening of the disease, delaying the remission of EAE in vivo[88]. Despite the use of PD-1 to identify Tfh cells, the role of PD-1 signalling on Tfh cells is only beginning to be investigated. When PD-1 is engaged by its ligand PDL-1 on follicular B cells, a bystander mechanism is activated and PD-1-expressing Tfh cells are recruited into a special niche inside the GC, even though Tfh PD-1neg migrates to the follicle outside of the germinal centre[89]. Moreover, both ICOS and PD-1 are requested for maintaining the stringency of affinity-based selection between Tfh cells and antigen-specific cells[89].
The transcriptional signature of CD8 T-cell exhaustion predicted better prognosis in multiple autoimmune diseases[90]. Transcriptomes of CD4 and CD8 T cells isolated from a group of patients with active autoimmune diseases were analysed to identify modules of genes with a strong correlation with relapse rate. Modules corresponding to CD4 T-cell co-stimulation were found to correlate with clinical outcomes. In detail, CD4 co-stimulatory receptors, CD2, KAT2B and other surrogate markers were described to increase in MS patients with active autoimmune disease.
The immune regulatory role of PD-1 in MS was suggested by experiments in EAE, a mouse model of MS[59,91,92]. Mice in which PD-1 was deleted or the PD-1 pathway was inhibited by blocking the link between PD-1 and its ligand PDL-1 develop a worsening EAE with an increase of infiltrating immune cells, especially CD8 T cells into the CNS[59,92,93]. The deterioration of disease in PD-1-/- and PDL-1-/- mice was related to over production of inflammatory cytokines IFNg, TNFa, IL-6 and IL-17 released by draining lymph node cells during re-stimulation in vitro with different concentrations of MOG[93]. PDL-1 is rarely expressed in the brains of controls[94]. Contrarily, PDL-1 was detected in the majority of lesions expressed from astrocytes and microglia/macrophages with low expression of PD-1 on infiltrating T cells in post-mortem MS brain tissues[94]. A recent publication shows that the PDL-1 in dendritic cells improves EAE in mice. The authors used a hypomethylating agent 5-aza-2’-deoxycytidine, which reduces methylation in a “CPG” island located near the transcription site of Cd274 gene. The reduced methylation favours the gene transcription and upregulation of PDL-1 in dendritic cells. This effect was observed in DC isolated from EAE mice pre-treated with hypomethylating agent and in vitro treatment of bone marrow dendritic cells with the hypomethylating agent. Furthermore, DC isolated from EAE mice pre-treated with 5-aza or bone marrow dendritic cells treated in vitro with 5-aza suppressed proliferation and release of inflammatory cytokines such as IL-17 and TNFα when the DC were co-cultured with CD4 T cells isolated from EAE mice. In addition, an inhibition of EAE was observed in mice pre-treated with 5-aza before EAE induction. An increase of PDL-1 and PDL-2 was detected in DC isolated from EAE mice in accordance with the in vitro results. Moreover, blocking of PDL-1 but not PDL-2 exacerbated the EAE symptoms, confirming that the link of PDL-1 but not PDL-2 with PD-1 is relevant in the suppression of T cell function by DC[95].
PD-1 depletion was also applied in mice immunised with a peptide of myelin oligodendrocyte glycoprotein as adjuvant to develop EAE[62]. After PD-1 depletion, the mice recovered from EAE with a clinical score of one at the end of the experiments compared with the control mice that showed a score four without recovery. Depletion of PD-1 reduced the fractions of PD-1+CD4+ and PD-1+CD8+ T cells but not B cells in the CNS as compared with the controls. Besides, PD-1 depletion did not alter the ability of the treated mice to mount an immune response. The baseline number of PD-1+ cells in the blood and peripheral lymphoid organs was low, confirming that PD-1 is expressed on autoreactive cells infiltrating target organs[62].
In view of publications giving PD-1 a crucial role in protection against autoimmunity in human and animal models, Jang et al.[96] investigated how PD-1 controls the activation and accumulation of autoreactive T cells, by constraining them in anergic state. PD-1 is one of the checkpoint inhibitors investigated in self-tolerance and discussed previously in CD8 T cells. Self-reactive cells were deleted during thymic development[97] and, of those that survived thymic deletion, only 10%-25% preferentially differentiated into immune suppressive regulatory T cells (Tregs)[98,99]. Jiang et al.[96] demonstrated that PD-1 is required in culling endogenous peripheral high-affinity autoreactive CD4 T cells and protect against autoimmunity[96]. The tracking of endogenous autoreactive CD4 T cells showed that more than 90% of autoreactive CD4 T cells remained FOXp3- effectors and were not regulatory T cell precursors, despite the high TCR affinity[96]. Instead, self-reactive CD4 T cells acquired cell-intrinsic tolerance through the expression of the immune checkpoint molecule PD-1[96]. Monitoring the progeny of individual autoreactive CD4 T cell clones showed that the clones with the greatest expansion burst size and highest TCR affinity expressed high levels of PD-1 and the affinity for the self-antigen induces the expression of PD-1[96] and the absence of PD-1 converts this signal when priming with consequent cell activation. A similar mechanism was described to induce the peripheral CD8 T tolerance in vivo. The peripheral CD8 T tolerance is induced by resting dendritic cells and depends on activation of PD-1 and CTLA-4 pathways[100].
Several studies have analysed the gene expression and protein levels of PD-1 and PDL-1 in MS, focusing on delineating any correlation with disease susceptibility or risk of progression in MS. PD-1 gene polymorphism has been investigated in MS, and the PD 1.3 SNP has been reported to correlate with progression of the disease, demonstrating that human polymorphisms that reduce PD-1 activity increase the risk of disease. Furthermore, a significant reduction in PD-1 expression was observed in patients with mutation as compared with donors with wild-type phenotype. Furthermore, patients bearing the mutual allele showed a lower suppression of IFNγ-producing CD4 T cells after aCD3-PD-1-microbead stimulation compared with healthy donors[101]. In addition, PD-1 and PDL-1 expression in peripheral blood mononuclear cells reduced in a cohort of patients with MS as compared with healthy donors[102]. The association of three PD-1 SNPs, namely PD-1.3, PD-1.5 and PD-1.9, with MS and disease outcome were investigated in a cohort of 203 patients with a diagnosis of relapsing-remitting and secondary-progressive MS showing any association with MS risk[103]. The expression of inhibitory receptor genes, including CTL-4, PD-1 and TIM-3, decreased in patients with MS as compared with healthy controls[104]. PD-1 is usually the most downregulated gene among the investigated inhibitors[104]. PD-1 was analysed on cytotoxic CD8+CD57+T cells in the peripheral blood of patients with relapsing–remitting MS and in T cells infiltrating the brain tissue in post-mortem MS cases. PD-1 increased in CD8+CD57+T cells in patients with stable disease and decreased in active-relapsing MS compared with healthy donors[105]. PD-1 was also found to increase in CD4 and CD8 T cells in MS patients early after autologous hematopoietic stem cell transplant[106]. A study of long-term immune reconstitution in MS patients after autologous hematopoietic stem cell transplant demonstrated that an early expansion of CD8+PD-1+T cells and CD19+PD-1+B cells is associated with favourable neurological outcomes[106]. PDL-1 was also investigated in post-mortem MS brain tissue. In MS lesions, glial cells with elevated PDL-1 and PD-1 expression were found absent in many infiltrating CD8 T cells[94]. Moreover, PDL-2 but not PDL-1 is expressed in human brain endothelial cells under basal culture conditions whilst both are upregulated under inflammatory condition[107]. PDL-1 or PDL-2 blockade lessens CD8 and CD4 T cell transmigration and CD8 T cells response[94]. Furthermore, PDL-1 is undetectable in the brain endothelium in normal tissues and MS lesions, even though PDL-2 is detectable in all blood vessels in normal brain tissue and in 50% of MS lesions[94].
Multiple sclerosis diagnosis after cancer immunotherapy
ICI treatments are immunotherapies engaged in restoring the immune response to tumour or viral infection by blocking the inhibitory pathways mediated by CTLA-4 and PD-1. ICI treatments have induced neurological immune-related adverse events. Patients with an MS history developed relapses after ICI treatment for melanoma[108,109] and a biopsy of the lesions revealed acute/inflammatory demyelination without any evidence of tumour cells[109]. A comparative functional profiling of myelin-reactive T cells of patients after ICI- treatment and 14 age/sex-matched patients with MS and healthy controls was performed. Myelin-reactive T cells isolated from ipilimumab-treated patients and MS patients showed a similar autoimmune response to myelin antigen but distinct from healthy controls. That confirmed that ICI treatment causes a reactivation of self-antigen cells in MS patients. The lack of outcomes to the ICI treatment in tumour is associated with an effect on the neurological condition. A case reported maintaining stable MS during ipilimumab treatment for melanoma that did not respond to the therapy, and the patient died from metastatic melanoma[110].
Furthermore, a 29-year-old man with metastatic melanoma underwent two cycles of ipilimumab before developing MS. The TCR repertoires of tumour-infiltrating T cells isolated from the primary melanoma and those of T cells isolated in two CSF samples, five and thirteen months after the second course of ipilimumab therapy, were analysed and compared. Distinct clonotypes of CD4 and CD8 T cells in the melanoma and the CSF were identified, demonstrating that the protective antitumor response and the anti-CNS response target different antigens[111]. Outcomes of MS relapse after ICI treatment were reported in a meta-analysis study including the published literature, the analysis of food and the drug administration adverse event reporting system database and a detailed case[112]. Fourteen cases were identified with MS, of which eight had a reported history of MS. All patients presented rapid disease progression, and two of them died from severe MS after ICI treatment[112]. The median age of MS diagnosis was 52.5 years, and ICI treatment was used as immunotherapy in several types of cancer: melanoma, non-small cell lung carcinoma, pleural mesothelioma, renal cell carcinoma and colorectal cancer[112]. ICI treatments such as nivolumab, ipilimumab, pembrolizumab and atezolizumab have caused MS relapse. In addition, Isitan and Wesley[113] described the case of a 49-year-old woman with a history of relapsing–remitting MS reported to develop a severe progressive MS after atezolizumab (monoclonal antibody targeting PD-L1) therapy for metastatic colonic adenocarcinoma. The women died after her first dose of atezolizumab. Although ICI therapy has given beneficial outcomes in cancer and infectious diseases, this treatment has shown neurological side effects in patients with MS history, inducing a rapid worsening of neurological conditions. The examined cases showed a worsening of the conditions associated with the activation of T cells recognising self-antigen in the CNS. The relevance of PD-1 blocking and depletion of PD-1+ cells in the pathogenesis of multiple sclerosis is shown in Figure 2.
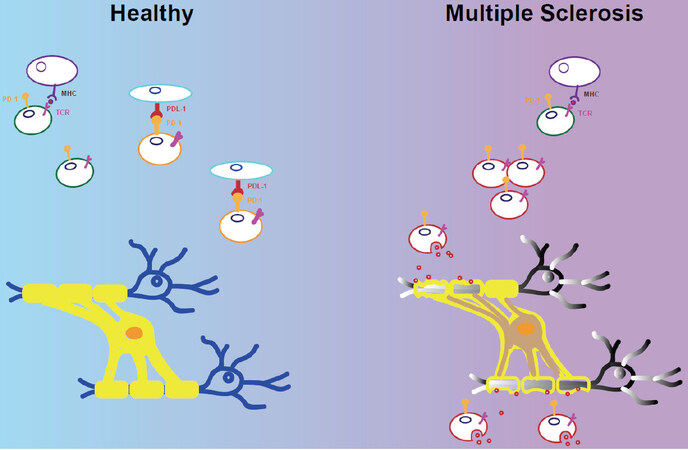
Figure 2. Immune checkpoints inhibitor treatment induces reactivation of multiple Sclerosis. PD-1 has been described on autoreactive T cells and TFH cells in multiple sclerosis. The link of PD-1 with the ligand PDL-1 controls T cell activation mantaining tollerance (Healthy). The blocking of PD-1 breaks the tollerance by reactivation of antigen-specific cells and causing damage of the target-cells with persistent inflammation in the CNS (Multiple Sclerosis). Contrary, depletion of PD-1+ T cells eliminates activated antigen-specific cells reducing the T cell infiltrates and inflammation in the target-organ (PD-1 blocking). TFH: T Follicular helper cells
Conclusion
PD-1 is an immune checkpoint inhibitor demonstrated to reduce the immune system response in cancer and chronic infection. Recent investigations have highlighted the dual role of PD-1 in immune tolerance, and the loss of PD-1 causes autoimmune diseases. Depletion of PD-1+ T cells has given beneficial effect in autoimmune disease, slowing down the inflammation and disease progression. A decrease of PD-1 is predisposed to autoimmunity, as described in experiments of PD-1 blocking or knockout in mice. PD-1 could be a target of immunotherapies in MS, although further investigations are required to define the role of PD-1 in MS. The majority of information has been derived from animal models and sporadic studies in humans. To this purpose, expression and levels of PD-1 and PDL-1 in the peripheral blood, CSF and post-mortem MS brain tissues and the correlation of their levels with risk of MS disease and inflammation could make relevant contributions for considering PD-1 a target in MS immunotherapies.
Declarations
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by Fondazione Italiana Sclerosi Multipla (ref. 2015/R/16 to PM), by Elena Pecci research project and Fondazione Careggi Onlus.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 1998;188:2205-13.
2. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 2003;77:4911-27.
3. Battegay M, Moskophidis D, Rahemtulla A, Hengartner H, Mak TW, et al. Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J Virol 1994;68:4700-4.
4. Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol 2004;5:927-33.
5. Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, et al. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003;421:852-6.
6. Day CL, Walker BD. Progress in defining CD4 helper cell responses in chronic viral infections. J Exp Med 2003;198:1773-7.
7. Urbani S, Amadei B, Fisicaro P, Tola D, Orlandini A, et al. Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology 2006;44:126-39.
8. Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 1994;68:8056-63.
9. Fuller MJ, Zajac AJ. Ablation of CD8 and CD4 T cell responses by high viral loads. J Immunol 2003;170:477-86.
10. Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993;362:758-61.
11. Iyasere C, Tilton JC, Johnson AJ, Younes S, Yassine-Diab B, et al. Diminished proliferation of human immunodeficiency virus-specific CD4+ T cells is associated with diminished interleukin-2 (IL-2) production and is recovered by exogenous IL-2. J Virol 2003;77:10900-9.
12. Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, et al. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci U S A 2011;108:21182-7.
13. West EE, Youngblood B, Tan WG, Jin HT, Araki K, et al. Tight regulation of memory CD8(+) T cells limits their effectiveness during sustained high viral load. Immunity 2011;35:285-98.
14. do Canto LM, Farias TD, Medeiros MD, Coêlho CC, Sereia AF, et al. Association of PDCD1 polymorphism to systemic lupus erythematosus and rheumatoid arthritis susceptibility. Rev Bras Reumatol Engl Ed 2016;56:483-9.
15. Moser JM, Altman JD, Lukacher AE. Antiviral CD8+ T cell responses in neonatal mice: susceptibility to polyoma virus-induced tumors is associated with lack of cytotoxic function by viral antigen-specific T cells. J Exp Med 2001;193:595-606.
16. Krebs P, Scandella E, Odermatt B, Ludewig B. Rapid functional exhaustion and deletion of CTL following immunization with recombinant adenovirus. J Immunol 2005;174:4559-66.
17. Kostense S, Vandenberghe K, Joling J, Van Baarle D, Nanlohy N, et al. Persistent numbers of tetramer+ CD8(+) T cells, but loss of interferon-gamma+ HIV-specific T cells during progression to AIDS. Blood 2002;99:2505-11.
18. Gruener NH, Lechner F, Jung MC, Diepolder H, Gerlach T, et al. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol 2001;75:5550-8.
19. Urbani S, Boni C, Missale G, Elia G, Cavallo C, et al. Virus-specific CD8+ lymphocytes share the same effector-memory phenotype but exhibit functional differences in acute hepatitis B and C. J Virol 2002;76:12423-34.
20. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006;439:682-7.
21. Ajona D, Ortiz-Espinosa S, Lozano T, Exposito F, Calvo A, et al. Short-term starvation reduces IGF-1 levels to sensitize lung tumors to PD-1 immune checkpoint blockade. Nat Cancer 2020;1:75-85.
22. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol 2020;5:eaay1863.
23. McLeod DS, Cooper DS. The incidence and prevalence of thyroid autoimmunity. Endocrine 2012;42:252-65.
24. Armengol MP, Juan M, Lucas-Martín A, Fernández-Figueras MT, Jaraquemada D, et al. Thyroid autoimmune disease: demonstration of thyroid antigen-specific B cells and recombination-activating gene expression in chemokine-containing active intrathyroidal germinal centers. Am J Pathol 2001;159:861-73.
25. Ruiz-Riol M, Barnils Mdel P, Colobran Oriol R, Pla AS, Borràs Serres FE, et al. Analysis of the cumulative changes in Graves’ disease thyroid glands points to IFN signature, plasmacytoid DCs and alternatively activated macrophages as chronicity determining factors. J Autoimmun 2011;36:189-200.
26. Chang LS, Barroso-Sousa R, Tolaney SM, Hodi FS, Kaiser UB, et al. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev 2019;40:17-65.
27. Álvarez-Sierra D, Marín-Sánchez A, Ruiz-Blázquez P, de Jesús Gil C, Iglesias-Felip C, et al. Analysis of the PD-1/PD-L1 axis in human autoimmune thyroid disease: insights into pathogenesis and clues to immunotherapy associated thyroid autoimmunity. J Autoimmun 2019;103:102285.
28. Gravallese EM, Harada Y, Wang JT, Gorn AH, Thornhill TS, et al. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol 1998;152:943-51.
29. Gravallese EM, Manning C, Tsay A, Naito A, Pan C, et al. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum 2000;43:250-8.
30. Shigeyama Y, Pap T, Kunzler P, Simmen BR, Gay RE, et al. Expression of osteoclast differentiation factor in rheumatoid arthritis. Arthritis Rheum 2000;43:2523-30.
32. van der Woude D, Houwing-Duistermaat JJ, Toes RE, Huizinga TW, Thomson W, et al. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum 2009;60:916-23.
33. Carlens C, Hergens MP, Grunewald J, Ekbom A, Eklund A, et al. Smoking, use of moist snuff, and risk of chronic inflammatory diseases. Am J Respir Crit Care Med 2010;181:1217-22.
34. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 2017;542:110-4.
36. Choy EH, Isenberg DA, Garrood T, Farrow S, Ioannou Y, et al. Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: a randomized, double-blind, placebo-controlled, dose-escalation trial. Arthritis Rheum 2002;46:3143-50.
37. VanderBorght A, Geusens P, Raus J, Stinissen P. The autoimmune pathogenesis of rheumatoid arthritis: role of autoreactive T cells and new immunotherapies. Semin Arthritis Rheum 2001;31:160-75.
38. Kong EK, Prokunina-Olsson L, Wong WH, Lau CS, Chan TM, et al. A new haplotype of PDCD1 is associated with rheumatoid arthritis in Hong Kong Chinese. Arthritis Rheum 2005;52:1058-62.
39. Prokunina L, Padyukov L, Bennet A, de Faire U, Wiman B, et al. Association of the PD-1.3A allele of the PDCD1 gene in patients with rheumatoid arthritis negative for rheumatoid factor and the shared epitope. Arthritis Rheum 2004;50:1770-3.
40. Wan B, Nie H, Liu A, Feng G, He D, et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J Immunol 2006;177:8844-50.
41. Li S, Liao W, Chen M, Shan S, Song Y, et al. Expression of programmed death-1 (PD-1) on CD4+ and CD8+ T cells in rheumatoid arthritis. Inflammation 2014;37:116-21.
42. Shivaleela C, Suresh BS, Kumar GV, Lakshmiprabha S. Morphological study of the supracondylar process of the humerus and its clinical implications. J Clin Diagn Res 2014;8:1-3.
43. Xu J, He Y, Wang J, Li X, Huang L, et al. Influence of the TNFSF4 rs1234315 polymorphism in the susceptibility to systemic lupus erythematosus and rheumatoid arthritis. Hum Immunol 2019;80:270-5.
44. Gambino CM, Aiello A, Accardi G, Caruso C, Candore G. Autoimmune diseases and 8.1 ancestral haplotype: An update. HLA 2018;92:137-43.
45. Gao J, Gai N, Wang L, Liu K, Liu XH, et al. Meta-analysis of programmed cell death 1 polymorphisms with systemic lupus erythematosus risk. Oncotarget 2017;8:36885-97.
46. Al-Motwee S, Jawdat D, Jehani GS, Anazi H, Shubaili A, et al. Association of HLA-DRB1*15 and HLADQB1*06 with SLE in Saudis. Ann Saudi Med 2013;33:229-34.
47. Gao Z, Li X, Peng T, Hu Z, Liu J, et al. Systemic lupus erythematosus with Guillian-Barre syndrome: a case report and literature review. Medicine (Baltimore) 2018;97:e11160.
48. Mahmoudi M, Rezaiemanesh A, Salmaninejad A, Harsini S, Poursani S, et al. PDCD1 single nucleotide genes polymorphisms confer susceptibility to juvenile-onset systemic lupus erythematosus. Autoimmunity 2015;48:488-93.
49. Ferreiros-Vidal I, Gomez-Reino JJ, Barros F, Carracedo A, Carreira P, et al. Association of PDCD1 with susceptibility to systemic lupus erythematosus: evidence of population-specific effects. Arthritis Rheum 2004;50:2590-7.
50. Rocha MC, Santos LM, Bagatin E, Cohen Tervaert JW, Damoiseaux JG, et al. Genetic polymorphisms and surface expression of CTLA-4 and PD-1 on T cells of silica-exposed workers. Int J Hyg Environ Health 2012;215:562-9.
51. Chua KH, Lian LH, Sim XJ, Cheah TE, Lau TP. Association between PDCD1 gene polymorphisms and risk of systemic lupus erythematosus in three main ethnic groups of the malaysian population. Int J Mol Sci 2015;16:9794-803.
52. Lee YH, Woo JH, Choi SJ, Ji JD, Song GG. Association of programmed cell death 1 polymorphisms and systemic lupus erythematosus: a meta-analysis. Lupus 2009;18:9-15.
53. Fathi F, Sadeghi E, Lotfi N, Hafezi H, Ahmadi M, et al. Effects of the programmed cell death 1 (PDCD1) polymorphisms in susceptibility to systemic lupus erythematosus. Int J Immunogenet 2020;47:57-64.
54. Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr Rev 1994;15:516-42.
55. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med 2006;203:883-95.
56. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, et al. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A 2005;102:11823-8.
57. Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med 2003;198:63-9.
58. Fife BT, Pauken KE. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann N Y Acad Sci 2011;1217:45-59.
59. Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med 2003;198:71-8.
60. Nielsen C, Hansen D, Husby S, Jacobsen BB, Lillevang ST. Association of a putative regulatory polymorphism in the PD-1 gene with susceptibility to type 1 diabetes. Tissue Antigens 2003;62:492-7.
61. Pauken KE, Jenkins MK, Azuma M, Fife BT. PD-1, but not PD-L1, expressed by islet-reactive CD4+ T cells suppresses infiltration of the pancreas during type 1 diabetes. Diabetes 2013;62:2859-69.
62. Zhao P, Wang P, Dong S, Zhou Z, Cao Y, et al. Depletion of PD-1-positive cells ameliorates autoimmune disease. Nat Biomed Eng 2019;3:292-305.
64. Gourraud PA, Harbo HF, Hauser SL, Baranzini SE. The genetics of multiple sclerosis: an up-to-date review. Immunol Rev 2012;248:87-103.
65. Choi SR, Howell OW, Carassiti D, Magliozzi R, Gveric D, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 2012;135:2925-37.
66. Komori M, Blake A, Greenwood M, Lin YC, Kosa P, et al. Cerebrospinal fluid markers reveal intrathecal inflammation in progressive multiple sclerosis. Ann Neurol 2015;78:3-20.
67. Magliozzi R, Marastoni D, Rossi S, Castellaro M, Mazziotti V, et al. Increase of CSF inflammatory profile in a case of highly active multiple sclerosis. BMC Neurol 2019;19:231.
68. Machado-Santos J, Saji E, Tröscher AR, Paunovic M, Liblau R, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 2018;141:2066-82.
69. Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, et al; AFFIRM Investigators. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006;354:899-910.
70. Ben-Nun A, Cohen IR. Vaccination against autoimmune encephalomyelitis (EAE): attenuated autoimmune T lymphocytes confer resistance to induction of active EAE but not to EAE mediated by the intact T lymphocyte line. Eur J Immunol 1981;11:949-52.
71. Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2- T lymphocytes. J Immunol 1981;127:1420-3.
72. Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J Immunol 1995;154:944-53.
73. Chitnis T, Najafian N, Abdallah KA, Dong V, Yagita H, et al. CD28-independent induction of experimental autoimmune encephalomyelitis. J Clin Invest 2001;107:575-83.
74. Furtado GC, Marcondes MC, Latkowski JA, Tsai J, Wensky A, et al. Swift entry of myelin-specific T lymphocytes into the central nervous system in spontaneous autoimmune encephalomyelitis. J Immunol 2008;181:4648-55.
75. Ellmerich S, Mycko M, Takacs K, Waldner H, Wahid FN, et al. High incidence of spontaneous disease in an HLA-DR15 and TCR transgenic multiple sclerosis model. J Immunol 2005;174:1938-46.
76. Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol 1996;156:5-7.
77. Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol 1996;157:3223-7.
78. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006;126:1121-33.
79. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005;201:233-40.
80. Veldhoen M, Stockinger B. TGFbeta1, a “Jack of all trades”: the link with pro-inflammatory IL-17-producing T cells. Trends Immunol 2006;27:358-61.
81. Jäger A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol 2009;183:7169-77.
82. Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, et al. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol 2005;237:123-30.
83. Olivares-Villagómez D, Wang Y, Lafaille JJ. Regulatory CD4(+) T cells expressing endogenous T cell receptor chains protect myelin basic protein-specific transgenic mice from spontaneous autoimmune encephalomyelitis. J Exp Med 1998;188:1883-94.
84. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 2004;199:971-9.
85. Blair PA, Chavez-Rueda KA, Evans JG, Shlomchik MJ, Eddaoudi A, et al. Selective targeting of B cells with agonistic anti-CD40 is an efficacious strategy for the generation of induced regulatory T2-like B cells and for the suppression of lupus in MRL/lpr mice. J Immunol 2009;182:3492-502.
86. Mauri C, Blair PA. Regulatory B cells in autoimmunity: developments and controversies. Nat Rev Rheumatol 2010;6:636-43.
87. Schafflick D, Xu CA, Hartlehnert M, Cole M, Schulte-Mecklenbeck A, et al. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat Commun 2020;11:247.
88. Guo J, Zhao C, Wu F, Tao L, Zhang C, et al. T follicular helper-like cells are involved in the pathogenesis of experimental autoimmune encephalomyelitis. Front Immunol 2018;9:944.
89. Shi J, Hou S, Fang Q, Liu X, Liu X, et al. PD-1 controls follicular T helper cell positioning and function. Immunity 2018;49:264-74.e4.
90. McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015;523:612-6.
91. Zhu B, Guleria I, Khosroshahi A, Chitnis T, Imitola J, et al. Differential role of programmed death-ligand 1 [corrected] and programmed death-ligand 2 [corrected] in regulating the susceptibility and chronic progression of experimental autoimmune encephalomyelitis. J Immunol 2006;176:3480-9.
92. Kroner A, Schwab N, Ip CW, Ortler S, Göbel K, et al. Accelerated course of experimental autoimmune encephalomyelitis in PD-1-deficient central nervous system myelin mutants. Am J Pathol 2009;174:2290-9.
93. Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, et al. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol 2007;182:124-34.
94. Pittet CL, Newcombe J, Antel JP, Arbour N. The majority of infiltrating CD8 T lymphocytes in multiple sclerosis lesions is insensitive to enhanced PD-L1 levels on CNS cells. Glia 2011;59:841-56.
95. Chang CB, Lee SP, Chen WM, Wang CM, Song YC, et al. Dendritic cell upregulation of programmed death ligand-1 via DNA demethylation inhibits experimental autoimmune encephalomyelitis. J Autoimmun 2019:102362.
96. Jiang TT, Martinov T, Xin L, Kinder JM, Spanier JA, et al. Programmed death-1 culls peripheral accumulation of high-affinity autoreactive CD4 T cells to protect against autoimmunity. Cell Rep 2016;17:1783-94.
98. Bautista JL, Lio CW, Lathrop SK, Forbush K, Liang Y, et al. Intraclonal competition limits the fate determination of regulatory T cells in the thymus. Nat Immunol 2009;10:610-7.
99. Leung MW, Shen S, Lafaille JJ. TCR-dependent differentiation of thymic Foxp3+ cells is limited to small clonal sizes. J Exp Med 2009;206:2121-30.
100. Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol 2005;6:280-6.
101. Kroner A, Mehling M, Hemmer B, Rieckmann P, Toyka KV, et al. A PD-1 polymorphism is associated with disease progression in multiple sclerosis. Ann Neurol 2005;58:50-7.
102. Javan MR, Aslani S, Zamani MR, Rostamnejad J, Asadi M, et al. Downregulation of immunosuppressive molecules, PD-1 and PD-L1 but not PD-L2, in the patients with multiple sclerosis. Iran J Allergy Asthma Immunol 2016;15:296-302.
103. Pawlak-Adamska E, Nowak O, Karabon L, Pokryszko-Dragan A, Partyka A, et al. PD-1 gene polymorphic variation is linked with first symptom of disease and severity of relapsing-remitting form of MS. J Neuroimmunol 2017;305:115-27.
104. Mohammadzadeh A, Rad IA, Ahmadi-Salmasi B. CTLA-4, PD-1 and TIM-3 expression predominantly downregulated in MS patients. J Neuroimmunol 2018;323:105-8.
105. Cencioni MT, Magliozzi R, Nicholas R, Ali R, Malik O, et al. Programmed death 1 is highly expressed on CD8+ CD57+ T cells in patients with stable multiple sclerosis and inhibits their cytotoxic response to Epstein-Barr virus. Immunology 2017;152:660-76.
106. Arruda LCM, de Azevedo JTC, de Oliveira GLV, Scortegagna GT, Rodrigues ES, et al. Immunological correlates of favorable long-term clinical outcome in multiple sclerosis patients after autologous hematopoietic stem cell transplantation. Clin Immunol 2016;169:47-57.
107. Pittet CL, Newcombe J, Prat A, Arbour N. Human brain endothelial cells endeavor to immunoregulate CD8 T cells via PD-1 ligand expression in multiple sclerosis. J Neuroinflammation 2011;8:155.
108. Gettings EJ, Hackett CT, Scott TF. Severe relapse in a multiple sclerosis patient associated with ipilimumab treatment of melanoma. Mult Scler 2015;21:670.
109. Cao Y, Nylander A, Ramanan S, Goods BA, Ponath G, et al. CNS demyelination and enhanced myelin-reactive responses after ipilimumab treatment. Neurology 2016;86:1553-6.
110. Kyi C, Carvajal RD, Wolchok JD, Postow MA. Ipilimumab in patients with melanoma and autoimmune disease. J Immunother Cancer 2014;2:35.
111. Gerdes LA, Held K, Beltrán E, Berking C, Prinz JC, et al. CTLA4 as immunological checkpoint in the development of multiple sclerosis. Ann Neurol 2016;80:294-300.
112. Garcia CR, Jayswal R, Adams V, Anthony LB, Villano JL. Multiple Sclerosis outcomes after cancer immunotherapy. Clin Transl Oncol 2019. Epub 2019 Feb 20
113. Isitan C, Wesley S. Safety of checkpoint inhibitors for cancer treatment in patients with multiple sclerosis: a case report (P1.7-006). Neurology 2019;92 (15 Supplement):P1.7-006.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Cencioni MT. The immune regulation of PD-1/PDL-1 axis, a potential biomarker in multiple sclerosis. Neurosciences 2020;7:277-90. http://dx.doi.org/10.20517/2347-8659.2020.18
AMA Style
Cencioni MT. The immune regulation of PD-1/PDL-1 axis, a potential biomarker in multiple sclerosis. Neuroimmunology and Neuroinflammation. 2020; 7(3): 277-90. http://dx.doi.org/10.20517/2347-8659.2020.18
Chicago/Turabian Style
Cencioni, Maria Teresa. 2020. "The immune regulation of PD-1/PDL-1 axis, a potential biomarker in multiple sclerosis" Neuroimmunology and Neuroinflammation. 7, no.3: 277-90. http://dx.doi.org/10.20517/2347-8659.2020.18
ACS Style
Cencioni, MT. The immune regulation of PD-1/PDL-1 axis, a potential biomarker in multiple sclerosis. Neurosciences. 2020, 7, 277-90. http://dx.doi.org/10.20517/2347-8659.2020.18
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.