
β-funaltrexamine differentially modulates chemokine and cytokine expression in normal human astrocytes and C20 human microglial cells
 0
0Abstract
Aim: Emerging evidence implicates astrocyte/microglia dysregulation in a range of brain disorders, thereby making glial cells potential therapeutic targets. The novel anti-inflammatory actions of beta-funaltrexamine (β-FNA) are of particular interest. β-FNA is a derivative of naltrexone, and recognized as a selective, irreversible antagonist at the mu-opioid receptor (MOR). However, we discovered that β-FNA has novel anti-inflammatory actions that seem to be mediated through a MOR-independent mechanism. Thus far, we have focused on the acute effects of β-FNA on inflammatory signaling.
Methods: The effect of β-FNA treatment on interleukin-1β (IL-1β)-induced inflammatory signaling in normal human astrocytes (NHA) and C20 human microglial cells. Cytokine/chemokine expression was measured using ELISA, and nuclear factor-kappaB (NF-κB) p65 activation was evaluated by immunoblot.
Results: IL-1β-induced interferon-γ inducible protein-10 (CXCL10) production in NHA was more sensitive to chronic (3 day) β-FNA as indicated by an approximately 3-fold lower EC50 compared to that observed in acutely treated cells. Chronic β-FNA did not affect IL-1β-induced monocyte chemoattractant protein-1 (CCL2) or IL-6 production in NHA. β-FNA inhibited phosphorylation of NF-κB p65, suggesting that the inhibitory effects may be due in part to reduced NF-κB activation. We showed for the first time that C20 human microglial cells were insensitive to the anti-inflammatory actions of acute β-FNA.
Conclusion: β-FNA differentially affects inflammatory cytokine/chemokine expression in human astrocytes and microglia. These findings warrant further investigation into the novel anti-inflammatory actions of β-FNA, with a particular focus on astrocytes. These insights should contribute to the development of strategies to treat brain disorders that involve neuroinflammation.
Keywords
Introduction
Neuroinflammation is present in brain infection, trauma, neurodegenerative diseases, and psychiatric disorders as well[1-5]. Glial cells, including astrocytes and microglia are instrumental in neuroinflammation. The therapeutic effectiveness of certain antidepressant and antipsychotic drugs reportedly results, in part, from anti-inflammatory actions[6-9]. Astrocytes and microglia have integral roles in metabolic/neurotrophic support, ion and neurotransmitter homeostasis, synaptic plasticity, and modulation of neuronal excitability; and they respond to infection and cellular insults associated with neuropathology[10-13]. Astrocytes have a fundamental role in neuropathogenesis, in part, through the release of neurotoxic/neuroinflammatory factors including cytokines [i.e., interleukin (IL)-1β and IL-6] and chemokines [interferon-γ inducible protein-10 (CXCL10) and monocyte chemoattractant protein-1 (CCL2)]. These secreted factors can be directly neurotoxic or can induce further glial activation/dysregulation[14-17]. Microglia are resident CNS macrophages and thus play an important role in neuroinflammation, in part, through phagocytosis of cellular debris and release of inflammatory mediators including cytokines/chemokines. Importantly, emerging evidence highlights the importance of astrocyte/microglia dysregulation in a wide range of CNS disorders, thus implicating glial cells as potential therapeutic targets[18-21].
We, and others, are therefore interested in identifying novel, anti-inflammatory agents that are therapeutically effective in the treatment of neurological disorders. We are interested in the previously identified, novel anti-inflammatory actions of beta-funaltrexamine (β-FNA). As a fumaramate methyl ester derivative of naltrexone, β-FNA is most notably recognized as a selective, irreversible antagonist at the mu-opioid receptor (MOR)[22,23]. In both behavioral and in vitro assays, β-FNA acts initially as a reversible kappa-opioid receptor agonist, and then later results in MOR antagonism[24,25]. As an alkylating agent, β-FNA irreversibly antagonizes MOR by covalently binding at Lys233 on the receptor[23]. However, we discovered that β-FNA also has novel anti-inflammatory actions which seem to be mediated through MOR-independent actions[26-28]. For instance, neither naltrexone (a nonselective opioid receptor antagonist) nor D-Phe-Cys-Tyr-D-Trp-D-Arg-Pen-Thr-NH2 (CTAP) inhibits pro-inflammatory-induced CXCL10 expression in human astroglial cells[27,28]. Additionally, we predicted that if the anti-inflammatory actions of β-FNA were due to alkylation, this covalent modification should then remain after washout. Indeed, pre-treatment of astroglial cells with β-FNA for 60 min, followed by drug washout prior to stimulating with IL-1β (or tumor necrosis factor α), resulted in inhibition similar to 24 h co-exposure (cytokine stimulus + β-FNA). These findings suggested that β-FNA-induced modifications (i.e., alkylation) are persistent and lead to the disruption of signal transduction. Notably, our in vitro findings also showed that β-FNA reduces inflammatory signaling in astroglia, regardless of whether the stimulus is tumor necrosis factor α, IL-1β or bacterial lipopolysaccharide (LPS). We also determined that β-FNA inhibits LPS-induced pro-inflammatory cytokine expression in mouse brain (but not in plasma)[29]. Furthermore, treatment with β-FNA reduced LPS-induced sickness behavior in mice suggesting important translational implications[29].
The primary goal of the present study was to determine the effect of chronic β-FNA treatment on inflammatory signaling in NHA. Additionally, we assessed for the first time the effects of β-FNA on inflammatory signaling in human microglial cells.
Methods
Cells
Normal human astrocytes (NHA, cat# HA1800; ScienCell Research Laboratories, Carlsbad, CA, USA) were cultured as previously reported[28]. Briefly, cultures were maintained in growth medium containing Astrocyte Medium (cat# 1801), 1% Astrocyte Growth Supplement (cat# 1852), 1% penicillin/streptomycin solution (cat# 0503), and 2% fetal bovine serum (FBS). Culture medium was replaced every 48 h. Experimental cultures were seeded in either 24-well plates (1.15 × 104 cells/well) or 100-mm dishes (2.5 × 105 cells/dish) and grown until 90% confluent (15 days).
C20 human microglial cells were obtained from David Alvarez-Carbonell, PhD (Case Western Reserve University), and the details regarding the generation of this cell line have been reported[30]. The C20 cells that we received were evaluated by the Human Identity Testing Laboratory at Oklahoma State University Center for Health Sciences and confirmed to be of human origin as previously described (Davis et al.[31], 2018). Cultures were maintained in growth medium [Dulbecco’s Modified Eagle Medium/Ham’s F-12 50/50 mix supplemented with 2.5 mmol/L L-glutamine (Corning 10-090-CV), 10% FBS (Atlanta Biologicals S11550), and 1% penicillin/streptomycin (Lonza 17603E)] as previously reported[31].
Treatment of cells
To determine the dose-dependent effect of chronic (72 h) β-FNA (NIDA reagent supply program; Bethesda, MD, USA) on CXCL10 expression in NHA, cells were initially cultured in growth medium containing 0.04-10 µmol/L β-FNA for 24 h. The medium was then replaced with serum-free medium (SFM) containing β-FNA for an additional 48 h; IL-1β (3 ng/mL; Peprotech, Rocky Hill, NJ, USA) was added to cultures for the final 24 h of the 72 h exposure period. To determine the differential effects of β-FNA on chemokine/cytokine expression, NHA were treated as described above; however, only a single concentration of β-FNA was used (3 µmol/L; EC50 for inhibition of CXCL10 expression). To assess the effects of β-FNA on IL-1β-induced activation of (NF)-κB p65, cells were chronically exposed to β-FNA (3 µmol/L) as described above, then stimulated for 30 min with IL-1β (3 ng/mL). IL-1β-induced NF-κB p65 activation was assessed at 30 min after stimulation (when peak activation is observed).
This was our initial investigation into the effects of β-FNA on chemokine/cytokine expression in C20 microglial cells. Thus, we used our acute exposure model. Consistent with the acute model previously used with astrocytes, we used a higher concentration range of β-FNA (3-30 µmol/L). Briefly, C20 microglial cells were serum deprived for 24 h and then treated with β-FNA (3-30 µmol/L) alone or in combination with IL-1β (20 ng/mL) for 24 h in SFM.
Cell viability
Cell viability was measured using an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously reported[27]. Briefly, cells were exposed to MTT (0.55 mg/mL) for 45 min; next, the medium was removed, and cells were dissolved in 1 mL DMSO. The absorbance was then measured at 492 nm using a BIO-TEK HT spectrophotometer.
Chemokine/cytokine expression
Standard dual-antibody solid-phase immunoassays (ELISA Development Kit, Peprotech) were used for quantitation of cytokines/chemokines in culture medium as previously described[27]. Values were normalized to total protein content, which was determined using the bicinchoninic acid protein assay as previously described[32].
NF-κB activation
Phosphorylated NF-κB p65 in nuclear fractions was measured as an indicator of NF-κB p65 activation. Following experimental treatments, NHA (in 100-mm dishes) were washed two times with PBS; nuclear protein extracts were then collected as previously reported[33], except the lysis buffers contained 1 mmol/L sodium orthovanadate (Sigma, #450243). Total protein (30 µg) was loaded on 7.5% polyacrylamide gels (BioRad TGX FastCast Acrylamide kit, #161-0171), electrophoresed and then transferred to PVDF membranes. Membranes were incubated (with rocking) for 15-18 h at 4 °C. Primary antibodies included anti-phospho-NF-κB p65 (1:1000, Cell Signaling #3033S), anti-NF-κB p65 (1:1000, Cell Signaling #4764S), and anti-β-tubulin (1:1000, Cell Signaling #2146S). Restore Western blot stripping buffer (Thermo Scientific #21059) was used to remove antibodies and allow for re-labelling of membranes. Anti-rabbit IgG, AP-linked (1:1000, Cell Signaling #7054S) was used as the secondary antibody. The blots were scanned in a Typhoon 9410 phosphorimager (GE Healthcare, Uppsala, Sweden) using enhanced chemifluorescence reagent (GE Healthcare, UK). Densitometric analysis was performed using ImageJ software (National Institute of Health, Bethesda, MD, USA).
Statistical analysis
Statistical analyses and figure presentations were performed using PrismTM version 7.04 (GraphPad Inc., San Diego, CA). Dependent measures were analyzed by either one-way or two-way analysis of variance (ANOVA). In those instances where two-way ANOVA was used, stimulus and drug dose were the grouping variables. Data that were > 2 SD from the mean were considered outliers and removed from the analyses. When ANOVA revealed a statistically significant interaction, data were further assessed using a Fisher’s LSD test. The data are all presented as mean ± SEM.
Results
Effects of chronic β-FNA on IL-1β-induced CXCL10 expression in NHA
Exposure of NHA to β-FNA for 72 h resulted in a concentration-dependent inhibition of IL-1β-stimulated CXCL10 expression, with an EC50 = 2.7 µmol/L [Figure 1]. In a subsequent experiment, chronic exposure to a single concentration of β-FNA (3 µmol/L) significantly (P < 0.01) inhibited IL-1β-induced CXCL10 expression as indicated by ANOVA and pairwise comparison by a Fisher’s LSD test. However, neither IL-1β-induced CCL2 nor IL-6 expression was significantly (P ≥ 0.22) affected by β-FNA [Figure 2].
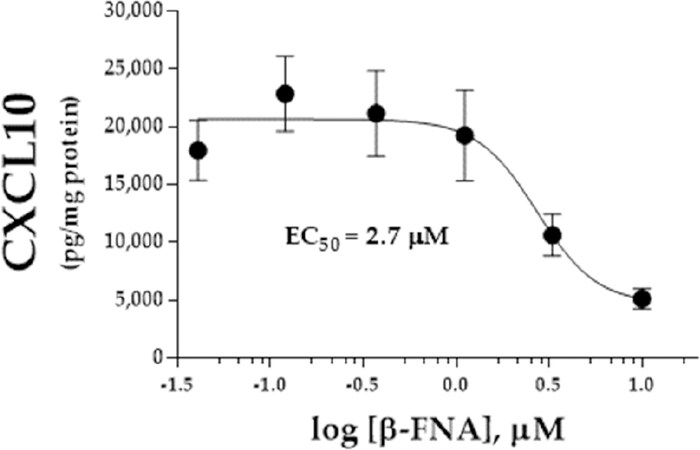
Figure 1. Chronic β-FNA dose-dependently inhibits CXCL10 expression in NHA. Cells were initially cultured in growth medium containing 0.04-10 µmol/L β-FNA for 24 h; the medium was then replaced with serum-free medium containing β-FNA for an additional 48 h. IL-1β (3 ng/mL) was added to cultures for the final 24 h. CXCL10 in the medium was measured by ELISA. Data are presented as mean ± SEM (n = 8-9). CXCL10: interferon-γ inducible protein-10; NHA: normal human astrocytes; ELISA: enzyme-linked immunoabsorbant assay; SEM: standard error of the mean; β-FNA: beta-funaltrexamine; IL-1β: interleukin-1β
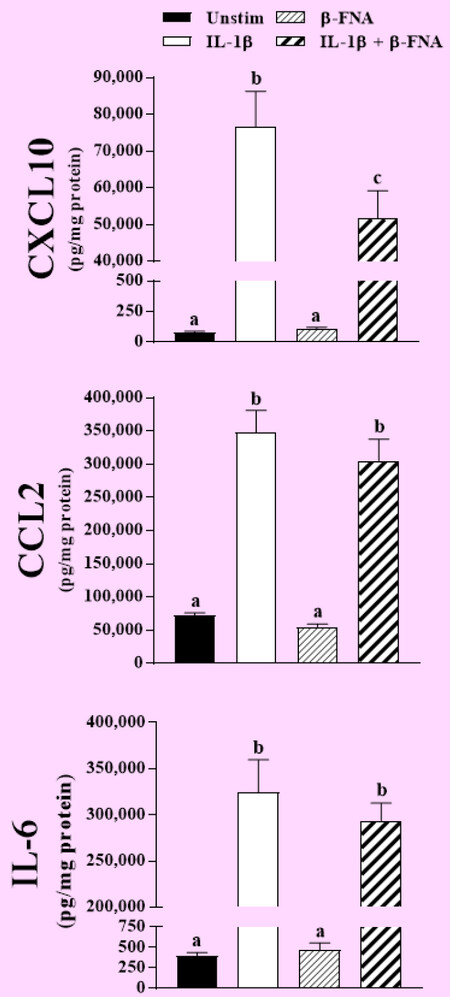
Figure 2. Chronic β-FNA differentially affects chemokine/cytokine expression in NHA. Cells were cultured in growth medium containing 3 µmol/L β-FNA for 24 h; the medium was then replaced with serum-free medium containing β-FNA for an additional 48 h. IL-1β (3 ng/mL) was added to cultures for the final 24 h. Chemokine/cytokine levels in the medium were measured by ELISA. Data are presented as mean ± SEM (n = 11-16). Differing letters above the bars indicate the means are significantly (P < 0.01) different as determined by ANOVA and subsequent Fisher’s LSD. NHA: normal human astrocytes; ELISA: enzyme-linked immunoabsorbant assay; ANOVA: analysis of variance; LSD: least significant difference; SEM: standard error of the mean; β-FNA: beta-funaltrexamine; IL-1β: interleukin-1β
Effects of chronic β-FNA on NHA viability
Cell viability was not significantly (P = 0.34) affected by chronic exposure to β-FNA alone or in combination with IL-1β, as revealed by ANOVA [Figure 3].
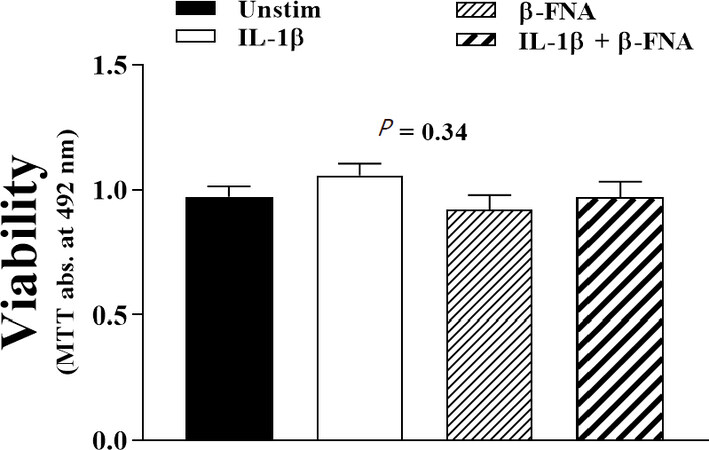
Figure 3. Effects of chronic β-FNA on viability of NHA. Cells were cultured in growth medium containing 3 µmol/L β-FNA for 24 h; the medium was then replaced with serum-free medium containing β-FNA for an additional 48 h. IL-1β (3 ng/mL) was added to cultures for the final 24 h. Cell viability was assessed using the MTT assay. Data are presented as mean ± SEM (n = 8). ANOVA did not reveal any significant differences. MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SEM: standard error of the mean; β-FNA: beta-funaltrexamine; NHA: normal human astrocytes; IL-1β: interleukin-1β
Effects of chronic β-FNA on NF-κB activation in NHA
One-way ANOVA and pairwise comparison by a Fisher’s LSD test revealed that chronic exposure to β-FNA significantly (P < 0.05) inhibited IL-1β-induced phosphorylation of nuclear NF-κB p65 in NHA [Figure 4]. The expression of total nuclear NF-κB p65 in NHA was not significantly (P = 0.2) affected by IL-1β or β-FNA alone, or the combination of chronic β-FNA plus stimulation with IL-1β [Figure 4].
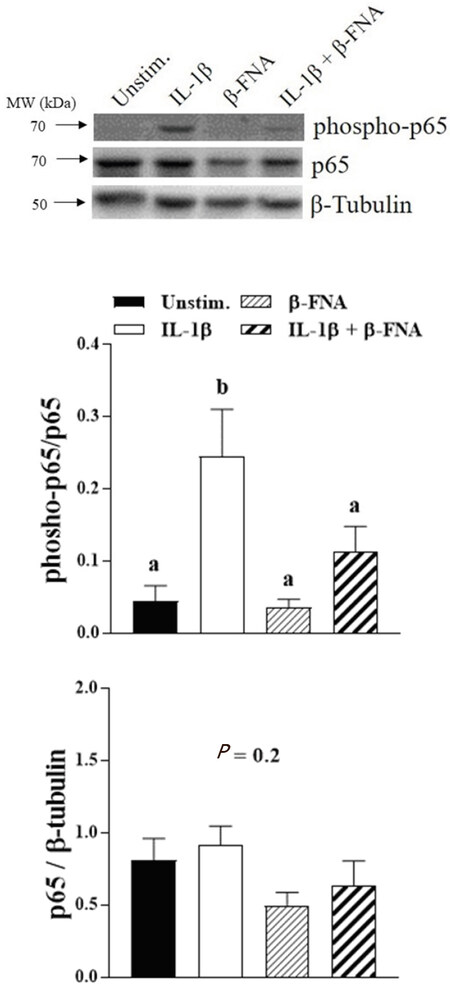
Figure 4. Chronic β-FNA inhibits NF-κB activation in NHA. Cells were cultured in growth medium containing 3 µmo/L β-FNA for 24 h; the medium was then replaced with serum-free medium containing β-FNA for an additional 48 h. IL-1β (3 ng/mL) was added to cultures for the final 30 min. Western blot was used to measure levels of phospho-NF-κB p65, NF-κB p65, and β-tubulin in nuclear extracts (a representative blot is presented at the top of the figure). Data are presented as mean ± SEM (n = 5). Differing letters above the bars indicate the means are significantly (P < 0.05) different as determined by ANOVA and subsequent Fisher’s LSD. LDS: least significant difference; ANOVA: analysis of variance; SEM: standard error of the mean; IL-1β: interleukin-1β; β-FNA: beta-funaltrexamine; NF-κB: nuclear factor-kappaB; NHA: normal human astrocytes
Effects of acute β-FNA on IL-1β-induced chemokine/cytokine expression in C20 microglial cells
Two-way ANOVA revealed that IL-1β significantly increased CXCL10 (F1,79 = 51.4; P < 0.0001), CCL2 (F1,66 = 0.71; P < 0.0001), and IL-6 (F1,51 = 55.6; P < 0.0001) levels in C20 microglial cells [Figure 5]. Acute (24 h) exposure to β-FNA did not significantly affect expression of CXCL10 (F3,79 = 0.19; P = 0.9), CCL2 (F3,66 = 0.71; P = 0.54), or IL-6 (F3,51 = 0.065; P = 0.98) in C20 microglial cells. Furthermore, there was no significant interaction between stimulation and β-FNA dose for CXCL10, CCL2, or IL-6 (F3,79 = 0.24; P = 0.86; F3,66 = 0.60; P = 0.62; F3,51 = 0.07; P = 0.98, respectively.
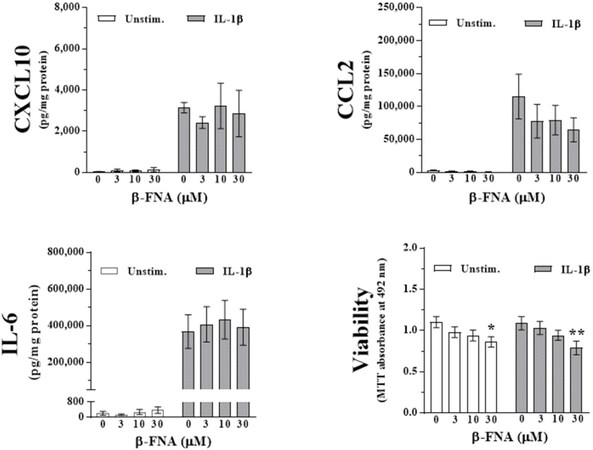
Figure 5. β-FNA differentially affects chemokine/cytokine expression in C20 human microglial cells. Cells were serum deprived for 24 h and then exposed to β-FNA (3-30 µmol/L) alone or in combination with IL-1β (20 ng/mL) for 24 h. Chemokine/cytokine levels in the medium were measured by ELISA; and viability was determined using the MTT assay. Data are presented as mean ± SEM (n = 8-12) and were analyzed by two-way ANOVA and subsequent Fisher’s LSD. *P < 0.05 vs. unstimulated, 0 µmol/L β-FNA; **P < 0.005 vs. IL-1β, 0 µmol/L β-FNA. ELISA: enzyme-linked immunoabsorbant assay; LSD: least significant difference; IL-1β: interleukin-1β; β-FNA: beta-funaltrexamine; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SEM: standard error of the mean
Effects of acute β-FNA on C20 microglial cell viability
Two-way ANOVA indicated that the viability of C20 microglial cells was not significantly affected by IL-1β (F1,86 = 0.025; P = 0.87), yet there was a significant main effect of β-FNA (F3,86 = 4.99; P < 0.005) [Figure 5]. Additionally, there was no significant interaction between stimulation and β-FNA (F3,86 = 0.26; P = 0.85).
Discussion
Astrocytes and microglia are instrumental in neuroinflammation and both cytokines and chemokines are among the inflammatory molecules released by these cells during neuroinflammation[34-36]. Hence, there is substantial interest in targeting neuroinflammation as a therapeutic strategy for selected brain disorders. The therapeutic potential of β-FNA is of particular interest to our group, and the results of this study are in line with our previous findings and advance our understanding of the anti-inflammatory effects of β-FNA. In our earlier studies, we found that β-FNA inhibition of IL-1β-stimulated CXCL10 expression was at least in part transcriptional, given that both protein and mRNA levels were significantly reduced by β-FNA[37]. We now show that CXCL10 inhibition in human astrocytes is even more sensitive to chronic (3 day) β-FNA as indicated by an approximately 3-fold lower EC50 compared to that observed in acutely treated cells. Importantly, and consistent with our previous findings[27], the inhibitory effects of β-FNA are not due to cytotoxicity, as we have shown that even extended exposure to β-FNA does not reduce the viability of human astrocytes. These preclinical findings that astrocyte viability is not compromised are quite important as future therapeutic potential will likely involve extended exposure to this drug.
Interestingly, β-FNA did not inhibit IL-1β-induced expression of either CCL2 or IL-6 in NHA, suggesting a level of selectivity for CXCL10. However, it may be that higher concentrations would inhibit the expression of CCL2 and IL-6. Therefore, further investigation, including a dose-response, is necessary.
Mechanistically, there is still more to learn about the anti-inflammatory effects of β-FNA. Importantly, as stated above and discussed previously in detail, we have determined that the anti-inflammatory effects in astrocytes do not seem to be due to actions at MOR (or other opioid receptors)[26-28]. Overall, our previous findings on the acute effects of β-FNA suggest that that the anti-inflammatory actions are likely mediated, at least in part, by disrupting the NF-κB signaling pathway[26-28,37]. In this study, we found that NF-κB signaling is also inhibited by chronic exposure to a lower concentration of β-FNA. Interestingly, the overall level of NF-κB p65 in the nucleus was not impacted by β-FNA; rather, phosphorylation of NF-κB p65 was reduced in the presence of β-FNA. Together, these findings suggest that β-FNA acts at a common factor in the signaling pathways activated by these diverse stimuli, in turn implicating the NF-κB signaling pathway. We hypothesize that β-FNA exerts these anti-inflammatory effects via alkylation of one or more lysines of the signaling proteins in the NF-κB pathway, and we are currently testing this hypothesis using in vitro approaches. Further studies are warranted to determine the mechanism by which β-FNA inhibits the phosphorylation of NF-κB p65.
We have also determined that β-FNA exerts anti-inflammatory actions in vivo[29,37]. For instance, LPS-induced neuroinflammation and sickness behavior in mice were attenuated by peripherally administered β-FNA[29]. More specifically, β-FNA inhibited LPS-induced expression of both CXCL10 and CCL2 in the brain, but had no effect on IL-6 levels. Furthermore, β-FNA did not impact plasma levels of CXCL10, CCL2, or IL-6. These in vivo findings are largely in line with our in vitro findings in human astrocytes, except that CCL2 expression in NHA was inhibited by β-FNA. Certainly, the differential sensitivity of CCL2 to β-FNA could be related to species differences or model systems (in vitrovs. in vivo). However, it may also reflect cell type-specific differences in sensitivity to β-FNA. For example, microglial cells (at least IL-1β-stimulated human microglial cells) are not sensitive to the anti-inflammatory actions of β-FNA. Because relatively high concentrations of β-FNA had no effect on chemokine/cytokine expression in C20 microglial cells and because of the observed cytotoxicity, we did not pursue the chronic effects of β-FNA on these cells at this point. However, in future experiments, we expect to assess chronic exposure to lower concentrations of β-FNA. Together, it is conceivable that the protective effects of β-FNA in vivo are largely due to modulatory effects on astrocytes; however, further investigation is needed to clearly establish the cell types affected and mechanisms involved.
In summary, we advanced our understanding of the anti-inflammatory effects of β-FNA by demonstrating that chronic exposure inhibits NF-κB p65 activation and CXCL10 expression in astrocytes more effectively than does acute treatment. We also found that expression of neither CCL2 nor IL-6 in astrocytes is affected by chronic β-FNA. Lastly, we provided evidence of cell type-specific effects of β-FNA, as indicated by the relative resistance of C20 human microglial cells to the anti-inflammatory effects of β-FNA. Further study is warranted and expected to advance the therapeutic potential of β-FNA, or related compounds, in the treatment of brain disorders that involve neuroinflammation.
Declarations
AcknowledgmentsWe greatly appreciate the statistical expertise and insights provided by J. Thomas Curtis, PhD.
Authors’ contributionsConcept, experimental design, literature review, statistical analysis, manuscript preparation: Davis RL
Performed experiments, data acquisition and analysis, and manuscript editing: McCracken K, Buck DJ
Availability of data and materialsData can be made available upon valid request.
Financial support and sponsorshipThis work was supported in part by Oklahoma Health Research Program (HR 14-007, HR 18-033); Oklahoma State University Center for Health Sciences, Intramural funds (RLD).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethics approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell 2010;140:918-34.
2. Kolb SA, Sporer B, Lahrtz F, Koedel U, Pfister HW, et al. Identification of a T cell chemotactic factor in the cerebrospinal fluid of HIV-1-infected individuals as interferon-gamma inducible protein 10. J Neuroimmunol 1999;93:172-81.
3. Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010;7:22-30.
4. Khansari PS, Sperlagh B. Inflammation in neurological and psychiatric diseases. Inflammopharmacology 2012;20:103-7.
5. Whitton PS. Neuroinflammation and the prospects for anti-inflammatory treatment of Parkinson’s disease. Curr Opin Invest Drugs 2010;11:788-94.
6. Dodd S, Maes M, Anderson G, Dean OM, Moylan S, et al. Putative neuroprotective agents in neuropsychiatric disorders. Prog Neuropsychopharm Biol Psych 2013;42:135-45.
7. Chen SL, Lee SY, Chang YH, Chen SH, Chu CH, et al. Inflammation in patients with schizophrenia: the therapeutic benefits of risperidone plus add-on dextromethorphan. J Neuroimmune Pharmacol 2012;7:656-64.
8. Ohgi Y, Futamura T, Kikuchi T, Hashimoto K. Effects of antidepressants on alternations in serum cytokines and depressive-like behavior in mice after lipopolysaccharide administration. Pharmacol Biochem Behav 2013;103:853-9.
9. Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, et al. A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain Behav Immun 2012;26:469-79.
10. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest 2012;122:1164-71.
11. Blank T, Prinz M. Microglia as modulators of cognition and neuropsychiatric disorders. Glia 2013;61:62-70.
12. Shie FS, Chen YH, Chen CH, Ho IK. Neuroimmune pharmacology of neurodegenerative and mental diseases. J Neuroimmune Pharmacol 2011;6:28-40.
13. Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, et al. Glial cells in (patho)physiology. J Neurochem 2012;121:4-27.
14. Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol 2007;28:138-45.
15. Moynagh PN. The interleukin-1 signalling pathway in astrocytes: a key contributor to inflammation in the brain. J Anat 2005;207:265-9.
16. John GR, Lee SC, Song X, Rivieccio M, Brosnan CF. IL-1-regulated responses in astrocytes: relevance to injury and recovery. Glia 2005;49:161-76.
17. Sui Y, Stehno-Bittel L, Li S, Loganathan R, Dhillon NK, et al. CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur J Neurosci 2006;23:957-64.
18. Bhattacharya A, Derecki NC, Lovenberg TW, Drevets WC. Role of neuro-immunological factors in the pathophysiology of mood disorders. Psychopharmacology 2016;233:1623-36.
19. Elsayed M, Magistretti PJ. A new outlook on mental illnesses: glial involvement beyond the glue. Front Cell Neurosci 2015;9:468.
20. Pfau ML, Menard C, Russo SJ. Inflammatory mediators in mood disorders: therapeutic opportunities. Annu Rev Pharmacol Toxicol 2018;58:411-28.
21. Sild M, Ruthazer ES, Booij L. Major depressive disorder and anxiety disorders from the glial perspective: Etiological mechanisms, intervention and monitoring. Neurosci Biobehav Rev 2017;83:474-88.
22. Portoghese PS, Larson DL, Sayre LM, Fries DS, Takemori AE. A novel opioid receptor site directed alkylating agent with irreversible narcotic antagonistic and reversible agonistic activities. J Med Chem 1980;23:233-4.
23. Chen C, Yin J, Riel JK, DesJarlais RL, Raveglia LF, et al. Determination of the amino acid residue involved in [3H] beta-funaltrexamine covalent binding in the cloned rat mu-opioid receptor. J Biol Chem 1996;271:21422-9.
24. Ward SJ, Portoghese PS, Takemori AE. Pharmacological characterization in vivo of the novel opiate, beta-funaltrexamine. J Pharmacol Exp Ther 1982;220:494-8.
25. Liu-Chen LY, Li SX, Tallarida RJ. Studies on kinetics of [3H] beta-funaltrexamine binding to mu opioid receptor. Mol Pharmacol 1990;37:243-50.
26. Davis RL, Buck DJ, Saffarian N, Mohan S, DeSilva U, et al. Beta-funaltrexamine inhibits inducible nitric-oxide synthase expression in human astroglial cells. J Neuroimmune Pharmacol 2008;3:150-3.
27. Davis RL, Buck DJ, Saffarian N, Stevens CW. The opioid antagonist, beta-funaltrexamine, inhibits chemokine expression in human astroglial cells. J Neuroimmunol 2007;186:141-9.
28. Davis RL, Das S, Buck DJ, Stevens CW. Beta-funaltrexamine inhibits chemokine (CXCL10) expression in normal human astrocytes. Neurochem Int 2013;62:478-85.
29. Davis RL, Stevens CW, Thomas Curtis J. The opioid antagonist, beta-funaltrexamine, inhibits lipopolysaccharide-induced neuroinflammation and reduces sickness behavior in mice. Physiol Behav 2017;173:52-60.
30. Garcia-Mesa Y, Jay TR, Checkley MA, Luttge B, Dobrowolski C, et al. Immortalization of primary microglia: a new platform to study HIV regulation in the central nervous system. J Neurovirol 2017;23:47-66.
31. Davis RL, Buck DJ, McCracken K, Cox GW, Das S. Interleukin-1β-induced inflammatory signaling in C20 human microglial cells. Neuroimmunol Neuroinflammation 2018;5:50.
32. Davis RL, Dertien J, Syapin PJ. Ethanol-induced modulation of inducible nitric-oxide synthase activity in human A172 astrocytoma cells. Alcohol Clin Exp Res 2002;26:1404-11.
33. Sanchez AC, Davis RL, Syapin PJ. Identification of cis-regulatory regions necessary for robust Nos2 promoter activity in glial cells: indirect role for NF-kappaB. J Neurochem 2003;86:1379-90.
34. Lozano R, Stevens L, Thompson BC, Gilmore KJ, Gorkin R, et al. 3D printing of layered brain-like structures using peptide modified gellan gum substrates. Biomaterials 2015;67:264-73.
35. Mangus LM, Dorsey JL, Laast VA, Hauer P, Queen SE, et al. Neuroinflammation and virus replication in the spinal cord of simian immunodeficiency virus-infected macaques. J Neuropathol Exp Neurol 2015;74:38-47.
36. Noto C, Maes M, Ota VK, Teixeira AL, Bressan RA, et al. High predictive value of immune-inflammatory biomarkers for schizophrenia diagnosis and association with treatment resistance. World J Biol Psychia 2015;16:422-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Davis RL, McCracken K, Buck DJ. β-funaltrexamine differentially modulates chemokine and cytokine expression in normal human astrocytes and C20 human microglial cells. Neurosciences 2020;7:300-10. http://dx.doi.org/10.20517/2347-8659.2020.19
AMA Style
Davis RL, McCracken K, Buck DJ. β-funaltrexamine differentially modulates chemokine and cytokine expression in normal human astrocytes and C20 human microglial cells. Neuroimmunology and Neuroinflammation. 2020; 7(4): 300-10. http://dx.doi.org/10.20517/2347-8659.2020.19
Chicago/Turabian Style
Davis, Randall L., Kelly McCracken, Daniel J. Buck. 2020. "β-funaltrexamine differentially modulates chemokine and cytokine expression in normal human astrocytes and C20 human microglial cells" Neuroimmunology and Neuroinflammation. 7, no.4: 300-10. http://dx.doi.org/10.20517/2347-8659.2020.19
ACS Style
Davis, RL.; McCracken K.; Buck DJ. β-funaltrexamine differentially modulates chemokine and cytokine expression in normal human astrocytes and C20 human microglial cells. Neurosciences. 2020, 7, 300-10. http://dx.doi.org/10.20517/2347-8659.2020.19
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 8
likes
Like This Article 8
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.