
What is the role of Brain derived neurotrophic factor in Multiple Sclerosis neuroinflammation?

Abstract
Multiple Sclerosis (MS) is a chronic, inflammatory and degenerative disease of the central nervous system (CNS) with an unknown etiology. The MS pathophysiology is due to altered bidirectional interactions between several immune cell types in the periphery (such as T and B cells, myeloid cells) and resident CNS cells (such as microglia and astrocytes). It is also known that inflammatory responses have both detrimental and neuroprotective effects. The release of brain derived neurotrophic factor (BDNF) by immune cells, in both peripheral blood and into inflammatory lesions in MS, but also by microglia and astrocytes, into the CNS, seems to be a possible mechanism for this neuroprotective effect. So far, the link between BDNF and neuroinflammation has been poorly investigated. A better understanding of this link could help in the development of new therapeutic strategies for MS. In this review, the role of BDNF in MS will be discussed as well as its possible alternative as an innovative therapeutic target.
Keywords
Introduction
Multiple Sclerosis (MS) is a chronic, inflammatory and degenerative disease of the central nervous system (CNS)[1,2] of which the etiology is unknown. The clinical course of MS is characterized by fluctuating neurological symptoms in most patients, with early clinical relapsing-remitting episodes and/or radiological worsening and different degrees of recovery (RRMS)[3]. The relapsing-remitting phase, in most patients, is subsequently followed by a chronic progressive phase; approximately 10%-15% of patients show a progressive form of the disease from onset.
For some authors, MS is an exclusive autoimmune inflammatory disease caused by dysregulated auto-reactive immune cells that traverse the blood-brain barrier (BBB) into the CNS parenchyma, attacking various cell types (the “outside-in” autoimmune hypothesis). For other authors it is a primary degenerative disease (the “inside-out” hypothesis) in which inflammation is secondary to the release of auto-antigens (components of myelin oligodendrocyte glycoprotein, myelin basic protein, and proteolipid protein), promoting autoimmunity[4]. Thus far, it is difficult to discern whether the inflammatory processes of MS are a product or a cause for neurodegeneration with a background autoimmune etiology. In all phases of the disease both immune and degenerative processes appear to coexist and this makes it difficult to definitively resolve the “outside-in” vs. “inside-out” controversy.
Despite this, there is well-documented evidence that, in MS, an uncontrolled inflammatory response in the CNS (neuroinflammation) causes destruction through high levels of pro-inflammatory cytokines, proteases, glutamate, and free radicals. Consequently, immunomodulatory drugs that reduce or suppress the activity of immune cells have been successfully used to reduce clinical relapses in MS and/or neuroradiological “activity”, which are associated with the entry of leukocytes through the BBB[5]. Sustained disability, however, is due to a progressive neurodegenerative process, ending with axonal loss and brain atrophy, primary or secondary to the peripheral and compartmentalized inflammation in the CNS[6]. To date, no approved therapy has provided marked neuroprotective effects nor have commonly anti-inflammatory therapies, used in the treatment of the disease, showed great efficacy in the progressive phase of MS.
Neuroinflammation have not only harmful but also neuroprotective effects[7,8]. In MS and other neurological diseases, the reparative activities of inflammatory response have been demonstrated[9]. Therefore, some authors introduced the concept of “neuroprotective autoimmunity”[10,11]. The release of neurotrophins by immune cells in both peripheral blood and directly into inflammatory lesions in MS[12,13], but also by microglia and astrocytes in the CNS, stimulating neuronal growth and survival, seems to be a possible mechanism for this neuroprotective effect[14]. Among neurotrophins, brain derived neurotrophic factor (BDNF) seems to be a good candidate in promoting the beneficial effects of inflammation in MS.
In this review, the role of BDNF in MS neuroinflammation and as a novel therapeutic target will be discussed.
Neuroinflammation: the detrimental effect
MS pathophysiology is characterized by altered bidirectional interactions between several immune cell types in the periphery and resident cells of the CNS, such as microglia and astrocytes[15]. The MS relapses, typical in the early phases of disease, are characterized by the infiltration of pro inflammatory CNS-specific effector T cells (CD4+ and CD8+ T cells), B cells and myeloid cells into the CNS parenchyma, that are activated and/or regulated in an aberrantly way[16]. The altered function of regulatory T (Treg) cells and resistance of CNS-specific effector T cells to Treg cell-mediated regulation could be a possible cause of the neuroinflammation[17-21]. Furthermore, CNS-resident cells, that secrete many inflammatory mediators, recruit inflammatory cells into the CNS. Microglia and astrocytes in particular, can also produce cytokines, chemokines and reactive oxygen species in the presence of homeostatic disturbance, promoting and sustaining axonal damage and neurodegeneration in MS[16]. Therefore, both peripheral and CNS-compartmentalized inflammatory mechanisms contribute to MS pathogenesis[22].
In the advanced stages of the disease, the infiltration of immune cells into the CNS is reduced, whereas ongoing CNS-compartmentalized inflammation seems to dominate progressive phases of MS. During this progressive phase, the role of B cells in driving inflammation seems to be prominent, particularly within meningeal inflammation[23]. In this phase, the B cell functions are antibody production, cytokine secretion, antigen presentation and ectopic formation of follicle-like structures[23-25]. The latter seem to maintain a high level of humoral response and other autoimmune mechanisms, in the CNS, independently from peripheral inflammation. This is very relevant during progressive MS phase, with the BBB being relatively intact and the contribution to disease from entry of peripheral immune cells into the brain fairly exiguous[26].
Activated microglia also plays a central role in neuroinflammation because it can sustain ongoing inflammation[27]. Microglia activation, in MS, is diffusely present in the lesions, in normal-appearing white and in grey matter[28]. Activated microglia, secreting pro-inflammatory cytokines, such as IL-1, IL-6, TNF-alfa, and IFN, promoting phagocytic activity, and presenting antigens via MHC Class II to CD4+ T cells[27], causes damage to oligodendrocytes; moreover, microglia inducing mitochondrial dysfunction, through reactive oxygen and nitrogen species, contributes to neuronal damage[29].
In addition, activation of astrocytes into demyelinating lesions contribute to oligodendrocyte injury and axonal degeneration[30]. So far, theories of either innate immune cells in the CNS are dysregulated and drive primary degeneration, or react against an unidentified primary injury causing tissue damage remain unknown.
Neuroinflammation in MS: the neuroprotective effect
Evidence for neuroprotective functions of immune cells
Some evidences have unexpectedly shown that some cells of the immune system might have a protective function during inflammation. This neuroprotective effect may be partially mediated with the production of anti-inflammatory cytokines (TGF-beta, IL-10 etc.) and secretion of pro-inflammatory cytokines (such as IL-6, IFN-gamma, TNF-alfa) in a dose- and time-dependent manner[31-33]. Immune cells also induce neuroprotection by production and local secretion of neurotrophic factors[11,12,34]. neural growth factor (NGF) was the first neurotrophin shown to be produced by T and B lymphocytes, macrophages, and mastcells[35]. The expression of BDNF by immune cells was also subsequently described[12]. In particular, CD4+ and CD8+ T lymphocytes, B lymphocytes, and monocytes in the human peripheral immune system can produce BDNF[12]. Moreover, neurotrophin receptors expressed by immune cells can also be targeted by autocrine or paracrine neurotrophin actions. Therefore, neurotrophins seem to mediate bidirectional cross-talk between the immune and nervous systems[11].
Evidence for neuroprotective function of microglia and astrocytes
Resident CNS cells also exercise a defensive action against immune-mediated attacks, aside from being involved in neuroinflammation. Microglia has an important role in neuroprotection and this action seems to be time-dependent. Acutely activated microglia produces inflammatory mediators that recruit other activated immune cells, amplifying the inflammatory damage, but chronically activated microglia may have a neuroprotective effect supporting the growth and survival of neural progenitor stem cells[31,36]. On oligodendrocyte precursor cells microglia seems to have always direct protective action, being the detrimental action mediated by astrocytes[37].
The neuroprotective function of microglia is mediated by different mechanisms such as debris clearance, production of growth factors (overall BDNF), production of the immunosuppressive cytokine IL-10 and neuronal circuit-shaping[27,38].
TGF-beta secretion and CTLA-4 expression produced by neurons induce CD4+CD25-effector T cells to take regulatory phenotype that exerts bystander suppression in experimental autoimmune encephalomyelitis (EAE)[39]. Self-associated molecular patterns expressed by resident neurons and astrocytes drive innate cell immune responses toward a less inflammatory response[40]. Astrocytes also cause apoptosis of activated immune cells and drive microglial activity towards a less inflammatory pattern[41,42].
The role of BDNF in MS neuroinflammation
BDNF
BDNF is a member of the neurotrophins gene family that includes also NGF and neurotrophins 3 and 4 (NT3 and NT4)[43] and is the neurotrophin most expressed in the brain by numerous cell types[44,45]. It plays a critical role on neuronal and oligodendroglial growth and survival, in healthy brains and in several neurologic diseases[46]. Interestingly, BDNF also modulates inflammatory homeostasis in the injured CNS[47,48].
The BDNF gene consist of a common 3ʹ-exon that encodes the pro-BDNF region of the protein, and several species-dependent 5ʹ-noncoding, promoter-regulated regions, terminating in a coding 5ʹ-exon that contain the gene expression[49,50]. BDNF is translated as a proneurotrophin (pro-BDNF) that can be cut in the mature form. Both mature BDNF and pro-BDNF bind to the low affinity p75 neurotrophin receptor, activating the apoptosis cascade[51,52]. Mature BDNF binds to its high-affinity receptor tyrosine kinase B (TrkB), activating several signalling cascades[53,54][Figure 1]. Among these, an increase in Ca2+ intake, phosphorylation of transcription factors, and de novo expression of the BDNF gene can be induced[53]. The nuclear factor-kappa B (NF-κB), a transcription factor with the ability to increase the expression of several pro- and antiapoptotic genes, including BDNF, is one of the main factors of inflammatory activation[55]. The binding of BDNF to the TrkB receptor can also induce the expression of NF-κB but, the pathways for this modulation are not yet completely understood[54]. Thus, the role of BDNF in neuroinflammation, is strongly linked to its ability to induce, and being induced by, NF-κB.
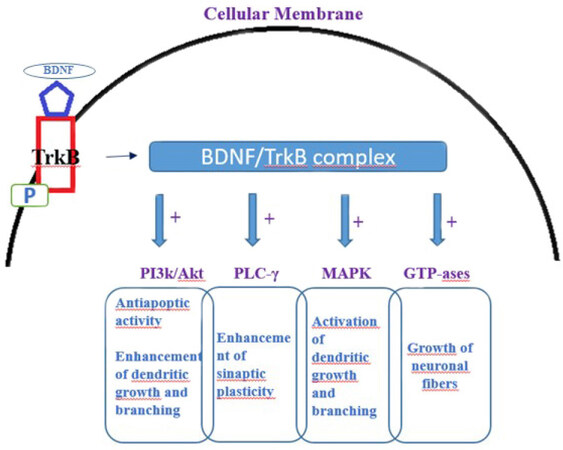
Figure 1. Intracellular signaling cascades induced by interaction of mature (m-)BDNF with TrkB receptor. Binding of BDNF to TrkB receptor induces its phosphorylation and translocation to cellular membrane. The BDNF/TrkB receptor complex triggers signaling pathways mediated by activation of PI3K, MAPK, PLC-γ, and GTP-ases. All these pathways induced by BDNF cause the enhancement/activation of dendritic growth and branching and growth of neuronal fibers. TrkB: tyrosine kinase B; BDNF: brain derived neurotrophic factor; PI3K: phosphoinositide 3-kinase; Akt: Protein kinase B; PLC: phospholipase C; MAPK: mitogen-activated protein kinase; TrkB: tropomyosin receptor kinase B
Lai et al.[56] recently demonstrated that BDNF modulates inflammatory homeostasis, reducing inflammatory activity on microglia also through the erythropoietin and sonic hedgehog signalling pathways. BDNF may also influence the microglia inflammatory response differently in male and females probably by driving activated microglia responses toward a less inflammatory pattern in females[57].
The understanding of BDNF function in humans has greatly benefited from the identification of an SNP in the BDNF gene that causes a valine (Val) to methionine (Met) substitution at codon 66 (Val66Met, c.196G>A, dbSNP: rs6265). In this Met variant form of BDNF carriers, that is, BDNF Val/Met heterozygotes and Met/Met homozygotes, the pro-domain structure of the gene is altered[58]. The polymorphism can potentially alter BDNF protein-protein interactions, binding affinities, localisation, or conformational stability of the protein. Whether the polymorphism has any significant impact on the proteome profile or posttranslational modifications of various proteins in the neuronal tissues or body fluids is currently unknown[58]. Several studies have emerged implicating the association or otherwise of this polymorphism with MS. So far, no conclusive data have been published[59]. New advances in the epigenetic field, highlight the role of BDNF antisense RNA (BDNF-AS), a naturally conserved long noncoding RNA, and of DNA methylation, in the regulation of BDNF expression in MS and in several neurological diseases[60-63]. So far, only few studies have been published on this argument[64].
BDNF in MS
BDNF is the neurotrophin which is expressed more in inflammatory brain lesions of MS patients[12,13]. A significant amount of BDNF was found in infiltrating immune cells, overall in T cells and macrophages, and in neurons and astrocytes[11]. BDNF is expressed by immune cells in actively demyelinating areas of MS lesions but not in lesions without ongoing myelin breakdown. Moreover, the neurotrophin is expressed more in the actively demyelinating edge of the plaque in the early phase of its development. It is released near to axons, not directly attacked by activated immune system cells but is at high risk of bystander damage[13]. Outside MS lesions, neurons are the major source of BDNF[13]. The literature data agree in showing that neurons are the major targets for neurotrophic interactions in the CNS. In particular, the full-length isoforms of TrkB (receptor for BDNF and NT4/5) and TrkC (receptor for NT3) are usually expressed on neuronal cells. Neurons close to MS plaques showed a prominent expression of full-length TrkB (gp145TrkB)[11]. Moreover, TrkB is upregulated in a part of damaged neurons. It is known that BDNF can be anterogradely transported and released by neurons. This process is up-regulated after axonal injury and transection[65]. The common occurrence of axonal damage in MS suggests that neuronal BDNF might contribute to endogenous neurotrophic support in MS plaques[66,67].
In older and chronic MS plaques, endogenous neurotrophins are low[13]. This may be one cause for the ongoing axonal degeneration in the chronic progressive stage of MS[68-70].
In the relapsing phase, levels of BDNF are generally reported to be increased in peripheral blood mononuclear cells (PBMC) and serum[71,72], but Azoulay et al.[73] found less BDNF in the serum of RRMS patients with no difference in remission and relapse phases. In MS patients, serum and CSF levels and PBMC secretion of BDNF are reduced compared to healthy controls[74,75]. In line with neuropathological findings[68-70], BDNF production by immune cells in RRMS patients is higher compared to progressive MS, suggesting again that progression of MS may be due to a failure of neuroprotection and neurorepair functions under chronic injury[72,75].
Numerous studies tried to correlate the role of rs6265 BDNF polymorphism with the prognosis of MS patients, with conflicting data so that other mechanisms could be involved in the modulation of BDNF gene[59]. Latest advances in the field of epigenetics highlight the role of epigenetic mechanisms, such as methylation, in controlling key biological processes. The presence of rs6265 SNP is not a prognostic factor for reaching a more severe Expanded Disability Status Scale (EDSS)[64]. When the percentage of methylation of the BDNF gene is considered, a lower percentage is associated with higher odds in achieving significant disability regardless of its polymorphism. Being a higher methylation a “silencer” of the gene, a lower inhibition of the gene correlates with a high probability in achieving an EDSS score of 6.0. Patients with more severe inflammation could appeal to a de-methylation to have a higher secretion of BDNF, preserving better CNS functions. The same patients tend to reach a more severe disability score by depleting the functional reserves of the brain at a faster rate[64]. If BDNF methylation is considered as an epiphenomenon of the disease activity (or better of the neuroinflammation status), it might help to differentiate patients with a higher degree of inflammation from patients with a lower ones. If these data will be confirmed by other studies, BDNF rs6265 polymorphism methylation could become a valid prognostic factor in MS to precociously recognise patients with a more severe disease from those with a milder one[64].
BDNF as promising therapy in MS
MS, but also many other CNS diseases, are tricky to treat due to the difficulty of drugs to cross the BBB. To do this, a drug must have the appropriate physicochemical properties. Alternatively, some drugs may be directly injected into the CNS but these invasive procedures are not risk-free.
Most available MS treatment have an exclusive anti-inflammatory effect helpful in reducing clinical and neuroradiological relapses but ineffective in preventing axonal loss and neurodegeneration. On the other hand, neuroprotective and/or remyelinating molecules failed to achieve the primary endpoint in clinical trials[76,77]. Conversely, brain delivery of BDNF has a potential role in reversing neurodegenerative diseases[78,79] but, so far, not through systemic administration. Therefore, there is an urgent need for development of a non-invasive trans-BBB delivery method. All the therapeutic strategies designed for delivery of neurotrophins are well summarised in the review by Huang and Dreyfus[80].
Recently, the possibility to deliver BDNF in a non-invasive way into the CNS through a BBB modulator, the ADTC5, has been found[81]. BDNF + ADTC5 delivered to the brains of mice with RR-EAE via systemic administration, significantly improve the clinical body scores of EAE mice and induce remyelination, compared to controls. Further studies are needed to confirm these data and to definitively find the best way to delivery BDNF in CNS via systemic administration.
Conclusion
Few studies investigated the link between BDNF and neuroinflammation even if, in many brain disorders, neuroinflammation and altered BDNF expression are commonly found. Better understanding of the interaction between BDNF and neuroinflammation could help in improving the knowledge of diseases pathogenesis and in developing of new therapeutic strategies for CNS disorders.
In MS, a large body of neuropathological, experimental and clinical evidences shows that BDNF may play an important role in neuroinflammation modulation, neuroprotection and neurorepair. These data make BDNF a good candidate for new therapeutic strategies in MS. But, when growth factors are considered as possible treatments in brain disease, some issues have to be taken into account: first, how to increase growth factors levels within specific regions of the CNS; second, how to optimize entry of growth factors from the periphery; third, to define the rate at which BDNF is taken up by the brain; fourth, the need to better understand the pharmacological characteristics of BDNF-based substances. Further studies are needed to define these aspects.
However, the challenge for neuroprotection in MS is greater than in other brain disease, because MS requires the association of both neuroprotective and immunomodulation therapies. Any exclusive inflammatory suppression is likely required to abolish both destructive and protective components and any neuroprotective treatment cannot work without a powerful anti-inflammatory therapy.
Declarations
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Naegele M, Martin R. The good and the bad of neuroinflammation in multiple sclerosis. Handb Clin Neurol 2014;122:59-87.
2. Gold R, Montalban X. Multiple sclerosis: more pieces of the immunological puzzle. Lancet Neurol 2012;11:9-10.
3. Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292-302.
5. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet 2018;391:1622-36.
6. Machado-Santos J, Saji E, Tröscher AR, Paunovic M, Liblau R, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 2018;141:2066-82.
7. Hammarberg H, Lidman O, Lundberg C, Eltayeb SY, Gielen AW, et al. Neuroprotection by encephalomyelitis: rescue of mechanically injured neurons and neurotrophin production by CNS-infiltrating T and natural killer cells. J Neurosci 2000;20:5283-91.
8. Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, et al. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 1999;5:49-55.
9. Graber JJ, Dhib-Jalbut S. Protective autoimmunity in the nervous system. Pharmacol Ther 2009;121:147-59.
10. Hohlfeld R, Kerschensteiner M, Stadelmann C, Lassmann H, Wekerle H. The neuroprotective effect of inflammation: implications for the therapy of multiple sclerosis. Neurol Sci 2006;27 Suppl 1:S1-7.
11. Kerschensteiner M, Stadelmann C, Dechant G, Wekerle H, Hohlfeld R. Neurotrophic cross-talk between the nervous and immune systems: implications for neurological diseases. Ann Neurol 2003;53:292-304.
12. Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med 1999;189:865-70.
13. Stadelmann C, Kerschensteiner M, Misgeld T, Brück W, Hohlfeld R, et al. BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain 2002;125:75-85.
14. De Santi L, Polimeni G, Cuzzocrea S, Esposito E, Sessa E, et al. Neuroinflammation and neuroprotection: an update on (future) neurotrophin-related strategies in multiple sclerosis treatment. Curr Med Chem 2011;18:1775-84.
15. Li R, Patterson K, Bar-Or A. Reassessing the contributions of B cells in multiple sclerosis. Nat Rev Immunol 2018;19:696-707.
16. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol 2015;15:545-58.
17. Kaskow BJ, Baecher-Allan C. Effector T cells in multiple sclerosis. Cold Spring Harb Perspect Med 2018;8:a029025.
18. Kitz A, Singer E, Hafler D. Regulatory T cells: from discovery to autoimmunity. Cold Spring Harb Perspect Med 2018;8:a029041.
19. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+ CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 2004;199:971-9.
20. Venken K, Hellings N, Thewissen M, Somers V, Hensen K, et al. Compromised CD4+ CD25(high) regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology 2008;123:79-89.
21. Frisullo G, Nociti V, Iorio R, Patanella AK, Caggiula M, et al. Regulatory T cells fail to suppress CD4T+-bet+ T cells in relapsing multiple sclerosis patients. Immunology 2009;127:418-28.
22. Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, et al. Multiple sclerosis. Nat Rev Dis Primers 2018;4:43.
23. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007;130:1089-104.
24. Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol 2006;6:205-17.
25. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 2004;14:164-74.
26. Correale J, Gaitán MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain 2017;140:527-46.
27. Correale J. The role of microglial activation in disease progression. Mult Scler 2014;20:1288-95.
28. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005;128:2705-12.
29. Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 2011;17:495-9.
30. Correale J, Farez MF. The Role of Astrocytes in Multiple Sclerosis Progression. Front Neurol 2015;6:180.
31. Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, et al. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci 2006;31:149-60.
32. Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, et al. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 2001;4:1116-22.
33. Arnett HA, Wang Y, Matsushima GK, Suzuki K, Ting JP. Functional genomic analysis of remyelination reveals importance of inflammation in oligodendrocyte regeneration. J Neurosci 2003;23:9824-32.
34. Torcia M1, Bracci-Laudiero L, Lucibello M, Nencioni L, Labardi D, et al. Nerve growth factor is an autocrine survival factor for memory B lymphocytes. Cell 1996;85:345-56.
35. Ehrhard PB, Erb P, Graumann U, Otten U. Expression of nerve growth factor and nerve growth factor receptor tyrosine kinase Trk in activated CD4-positive T-cell clones. Proc Natl Acad Sci U S A 1993;90:10984-8.
36. Aarum J, Sandberg K, Haeberlein SL, Persson MA. Migration and differentiation of neural precursor cells can be directed by microglia. Proc Natl Acad Sci U S A 2003;100:15983-8.
37. Lombardi M, Parolisi R, Scaroni F, Bonfanti E, Gualerzi A, et al. Detrimental and protective action of microglial extracellular vesicles on myelin lesions: astrocyte involvement in remyelination failure. Acta Neuropathol 2019;138:987-1012.
38. Kotter MR, Setzu A, Sim FJ, Van Rooijen N, Franklin RJ. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia 2001;35:204-12.
39. Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med 2006;12:518-25.
40. Griffiths M1, Neal JW, Gasque P. Innate immunity and protective neuroinflammation: new emphasis on the role of neuroimmune regulatory proteins. Int Rev Neurobiol 2007;82:29-55.
41. Pender MP1, Rist MJ. Apoptosis of inflammatory cells in immune control of the nervous system: role of glia. Glia 2001;36:137-44.
42. Aloisi F, Penna G, Cerase J, Menéndez Iglesias B, Adorini L. IL-12 production by central nervous system microglia is inhibited by astrocytes. J Immunol 1997;159:1604-12.
46. Hu Y, Russek SJ. BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J Neurochem 2008;105:1-17.
47. Asami T, Ito T, Fukumitsu H, Nomoto H, Furukawa Y, et al. Autocrine activation of cultured macrophages by brain-derived neurotrophic factor. Biochem Biophys Res Commun 2006;344:941-7.
48. Mizoguchi Y, Monji A, Kato T, Seki Y, Gotoh L, et al. Brain-derived neurotrophic factor induces sustained elevation of intracellular Ca2+ in rodent microglia. J Immunol 2009;183:7778-86.
49. Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res 2007;85:525-35.
50. Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T. Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007;90:397-406.
54. Lima Giacobbo B, Doorduin J, Klein HC, Dierckx RAJO, Bromberg E, et al. Brain-derived neurotrophic factor in brain disorders: focus on neuroinflammation. Mol Neurobiol 2019;56:3295-312.
55. Marini AM, Jiang X, Wu X, Tian F, Zhu D, et al. Role of brain-derived neurotrophic factor and NF-kappaB in neuronal plasticity and survival: From genes to phenotype. Restor Neurol Neurosci 2004;22:121-30.
56. Lai SW, Chen JH, Lin HY, Liu YS, Tsai CF, et al. Regulatory effects of neuroinflammatory responses through brain-derived neurotrophic factor signaling in microglial cells. Mol Neurobiol 2018;55:7487-99.
57. Rossetti AC, Paladini MS, Trepci A, Mallien A, Riva MA, et al. Differential neuroinflammatory response in male and female mice: a role for BDNF. Front Mol Neurosci 2019;12:166.
58. Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003;112:257-69.
59. Shen T, You Y, Joseph C, Mirzaei M, Klistorner A, et al. BDNF polymorphism: a review of its diagnostic and clinical relevance in neurodegenerative disorders. Aging Dis 2018;9:523-36.
60. D’Addario C, Dell’Osso B, Galimberti D, Palazzo MC, Benatti B, et al. Epigenetic modulation of BDNF gene in patients with major depressive disorder. Biol Psychiatry 2013;73:e6-7.
61. Rao JS, Keleshian VL, Klein S, Rapoport SI. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl Psychiatry 2012;2:e132.
62. Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 2008;82:696-711.
63. Ursini G, Cavalleri T, Fazio L, Angrisano T, Iacovelli L, et al. BDNF rs6265 methylation and genotype interact on risk for schizophrenia. Epigenetics 2016;11:11-23.
64. Nociti V, Santoro M, Quaranta D, Losavio FA, De Fino C, et al. BDNF rs6265 polymorphism methylation in Multiple Sclerosis: a possible marker of disease progression. PLoS One 2018;13:e0206140.
65. von Bartheld CS, Byers MR, Williams R, Bothwell M. Anterograde transport of neurotrophins and axodendritic transfer in the developing visual system. Nature 1996;379:830-3.
66. Tonra JR, Curtis R, Wong V, Cliffer KD, Park JS, et al. Axotomy upregulates the anterograde transport and expression of brain-derived neurotrophic factor by sensory neurons. J Neurosci 1998;18:4374-83.
67. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998;338:278-85.
69. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med 2000;343:938-52.
70. Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol 2001;14:271-8.
71. Caggiula M, Batocchi AP, Frisullo G, Angelucci F, Patanella AK, et al. Neurotrophic factors and clinical recovery in relapsing-remitting multiple sclerosis. Scand J Immunol 2005;62:176-82.
72. Sarchielli P, Greco L, Stipa A, Floridi A, Gallai V. Brain-derived neurotrophic factor in patients with multiple sclerosis. J Neuroimmunol 2002;132:180-8.
73. Azoulay D, Urshansky N, Karni A. Low and dysregulated BDNF secretion from immune cells of MS patients is related to reduced neuroprotection. J Neuroimmunol 2008;195:186-93.
74. Azoulay D, Vachapova V, Shihman B, Miler A, Karni A. Lower brain-derived neurotrophic factor in serum of relapsing remitting MS: reversal by glatiramer acetate. J Neuroimmunol 2005;167:215-8.
75. Hamamcioglu K, Reder AT. Interferon-beta regulates cytokines and BDNF: greater effect in relapsing than in progressive multiple sclerosis. Mult Scler 2007;13:459-70.
76. Ineichen BV, Plattner PS, Good N, Martin R, Linnebank M, et al. Nogo-A antibodies for progressive multiple sclerosis. CNS Drugs 2017;31:187-98.
77. Ruggieri S, Tortorella C, Gasperini C. Anti lingo 1 (opicinumab) a new monoclonal antibody tested in relapsing remitting multiple sclerosis. Expert Rev Neurother 2017;17:1081-9.
78. Crozier RA, Bi C, Han YR, Plummer MR. BDNF modulation of NMDA receptors is activity dependent. J Neurophysiol 2008;100:3264-74.
79. Obermeyer JM, Tuladhar A, Payne SL, Ho E, Morshead CM, et al. Local delivery of brain-derived neurotrophic factor enables behavioral recovery and tissue repair in stroke-injured rats. Tissue Eng Part A 2019;25:1175-87.
80. Huang Y, Dreyfus CF. The role of growth factors as a therapeutic approach to demyelinating disease. Exp Neurol 2016;283:531-40.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Nociti V. What is the role of Brain derived neurotrophic factor in Multiple Sclerosis neuroinflammation?. Neurosciences 2020;7:291-9. http://dx.doi.org/10.20517/2347-8659.2020.25
AMA Style
Nociti V. What is the role of Brain derived neurotrophic factor in Multiple Sclerosis neuroinflammation?. Neuroimmunology and Neuroinflammation. 2020; 7(3): 291-9. http://dx.doi.org/10.20517/2347-8659.2020.25
Chicago/Turabian Style
Nociti, Viviana. 2020. "What is the role of Brain derived neurotrophic factor in Multiple Sclerosis neuroinflammation?" Neuroimmunology and Neuroinflammation. 7, no.3: 291-9. http://dx.doi.org/10.20517/2347-8659.2020.25
ACS Style
Nociti, V. What is the role of Brain derived neurotrophic factor in Multiple Sclerosis neuroinflammation?. Neurosciences. 2020, 7, 291-9. http://dx.doi.org/10.20517/2347-8659.2020.25
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 28 clicks
Cite This Article 28 clicks

 Like This Article 8
likes
Like This Article 8
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.