
Clinical investigations of immunotherapy for human primary brain tumors

Abstract
Human primary brain cancer is one of the most lethal and clinically challenging malignancies. The failure of conventional therapies to alleviate its poor outcome has prompted efforts to find innovative treatments. Recent breakthroughs in immunotherapy across a variety of solid tumors have set immune-based therapeutics as a pillar for brain cancer treatment. However, the unique features of brain malignancies including intratumoral heterogeneity, immunosuppressive microenvironment, and impervious blood-brain barrier, thwart the success of immunotherapeutic approaches. Yet, seminal findings regarding tumor-driven enrichment of specific immune cells granted the field novel insights to harness the immune cells to fight cancer. This review discusses the anatomical, microenvironmental, and immunobiological features of the human brain and presents an overview of immunotherapies tested for primary brain cancer patients with a special emphasis on registered phase 2, 3, and combinatorial clinical trials. Immune checkpoint inhibitors, immune cell-based therapies, cancer vaccines, oncolytic viral therapy, and combination therapies are investigated in clinical settings for the treatment of human brain tumors. Despite their occasional adverse effects, immune-targeted therapies provide a promising opportunity for primary brain cancer patients to enhance survival and improve prognosis.
Keywords
Introduction
Primary brain tumors are notoriously lethal malignancies. In 2020, primary brain tumors are projected to claim the lives of 18,000 individuals in the United States alone[1]. With more than 220,000 global brain cancer-related deaths in 2016[2], both adult and pediatric populations carry the burden of incident brain and central nervous system (CNS) cancers[3,4]. Brain malignancies can be either intracranial primary tumors arising within the brain or brain metastases most frequently derived from extracranial tumors of the lungs, breast, and skin. Primary brain tumors exhibit salient genomic, molecular, microenvironmental, and histological profiles that result in different types of brain malignancies. In adults, diffuse gliomas are the most common primary tumors of the brain and include astrocytoma, oligodendroglioma, and glioblastoma[5]. Medulloblastomas are the most common malignant embryonal tumors that affect childhood[6]. Other than categorization by cells of origin, brain tumors are assigned a “grade” reflecting growth rate. Low-grade brain tumors present a positive outlook of curable, resectable cancer with an average survival of 7 years[7]. Conversely, high-grade tumors are characterized by poor prognosis in light of their high heterogeneity, diffusiveness, and aggressiveness[8,9]. Low grade gliomas (astrocytomas and oligodendroglioma) are distinguished from high grade gliomas (glioblastoma) by the mutational status of two genes that encode isocitrate dehydrogenase (IDH) isozymes. IDH mutations are enriched in low grade gliomas as compared to glioblastomas and infer a strong positive prognostic marker[5]. The current standard-of-care therapy for gliomas consists of gross total resection followed by a standardized regimen of chemotherapy and radiotherapy. Even following such an aggressive course of treatment, patients with high-grade tumors have a 14.6-month mean survival from the time of diagnosis[10], with less than 7% of patients expected to survive for 5 years[3].
The distinct anatomical, immunobiological, and microenvironmental properties of the brain and its neoplasms offer significant challenges to brain cancer treatment[11]. Brain tumors coerce non-malignant brain components and resident immune cells (e.g., microglia) to promote their growth and help them evade antitumor immune responses[12,13]. Additionally, the impervious blood brain barrier (BBB) severely limits the efficacy and intracranial penetration of the vast majority of conventional chemotherapeutic agents[14]. Crossing the BBB remains a critical consideration in high-grade brain tumors[15], which once resolved can allow the favorable remodeling of the tumor microenvironment (TME)[16]. Thus far, attempts to target conventional pathways deregulated in glioblastoma and other brain tumors have fallen short of expectations[17]. As a result, standard-of-care therapies remain palliative rather than curative with notable side effects, such as acute and long-term neurocognitive decline[18,19]. Therefore, researchers continue to contend with brain tumors to develop novel therapeutics that can enhance the dismal prognosis of brain cancer patients[20].
Historically considered immunologically silent or immunologically privileged, recent studies have debated this concept and revealed that the human brain has afferent and efferent CNS immunity[21,22]. Unchallenged by the BBB, the immune system constitutes a promising opportunity to break the deadlock of brain tumor treatment. The success of immunotherapy in various solid[23,24] and diffuse[25] cancers reflects the potential of harnessing the immune system in replacing current aggressive yet ineffectual treatment protocols. This has spurred investigations to evaluate the efficacy of existing immunotherapeutic agents against primary and metastatic brain tumors[26-28]. The enhancement of systemic and local antitumor immune responses and the subsequent improvement in brain cancer patients’ survival is achievable with immunotherapy of relative safety and tolerability[27,29,30]. While the majority of clinical investigations in primary brain tumors remain in their early stages, available data support the use of immunotherapy alone or in combination with other agents to achieve an intracranial response, disease remission, and improved survival[27,31,32].
In this review, we provide a concise overview of clinical evidence from registered trials detailing immunotherapeutic approaches in the clinical management of primary brain tumors, with an emphasis on available phase 2 and 3 trials. We also discuss a compendium of available immunotherapies and their implication in the immunobiological profile of the brain TME.
Brain tumor microenvironment
The brain tumor microenvironment is composed of cancerous cells along with a variety of stromal cells including endothelial cells, fibroblasts, pericytes, and immune cells[33]. The tumor stroma also consists of tissue-specific resident T-cells, including astrocytes, microglia, oligodendritic cells, and neurons[34-36], all of which are physically protected from the systemic circulation by the BBB and embedded in a modified extracellular matrix (ECM) [Figure 1]. In a normal brain, endothelial cells and pericytes interconnect with astrocytes to form the impenetrable BBB. However, neoplastic astrocytes have been shown to communicate with and support cancerous cells through the formation of the connexin43 gap junction, thus mediating the physical transfer of signaling molecules between tumor cells and astrocytes[37]. In a patient-derived glioblastoma xenograft model, active neurons promoted the proliferative activity of glioma cells via secretion of synaptic protein neuroligin-3 (NLGN3) that exerted a mitogenic signal on neoplastic cells, promoting their growth and proliferation[38]. While a wealth of evidence suggests oligodendrocyte precursors as a putative cellular origin for many forms of gliomas[39], a recent study found that these cells provide a supportive niche for glioblastoma cells by inducing their stemness and chemo-radioresistance[40]. The majority of immune cells in the brain TME are tumor-associated macrophages (TAMs) of distinct ontogenicity, including tissue-resident microglia and recruited monocyte-derived macrophages (MDMs). Bidirectional crosstalk prevails between TAMs and cancerous cells in the brain TME. Tumor cells produce factors that recruit TAMs to the local milieu and TAMs in turn supply growth factors and cytokines that induce a protumorigenic microenvironment[12]. The ECM of the brain has a unique composition of proteins that distinguishes it from other non-CNS tissues[41]. Under the effects of cancer cells and their reprogrammed stromal cell neighbors, ECM constituents are heavily modified and promote tumor angiogenesis, progression, and immune evasion[13]. In this manner, brain tumor cells can exploitively alter and reprogram the cellular components of the stroma as well as the ECM into oncogenic drivers that mandate tumor cell proliferation and invasion.
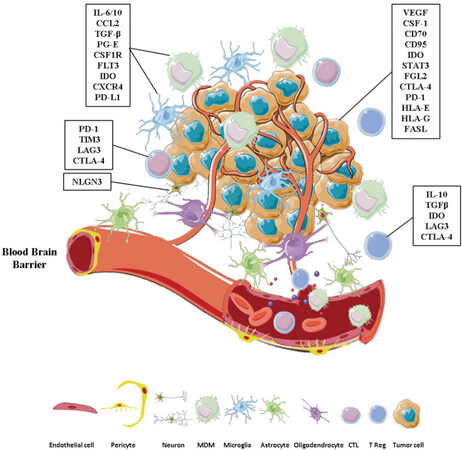
Figure 1. The immune-suppressive brain tumor microenvironment. Tissue-specific resident-cells, including astrocytes, microglia, oligodendrocytes, and neurons form the tumor microenvironment along with infiltrating immune cells. In brain cancers, the blood-brain barrier, formed of a tight junction of pericytes, endothelial cells, astrocyte end feet, and basement membrane, can become leaky. This allows the recruitment and entry of tumor-infiltrating lymphocytes and MDMs. Within the tumor bed, neurons, tumor-associated macrophages, effector T-cells and tumor cells secrete or express various immunoregulatory factors, as illustrated in Figure 1. This leads to the formation of the immunosuppressive microenvironment characteristic of primary brain tumors, thereby promoting tumor proliferation and immune evasion. This figure contains images from Servier Medical Art. MDM: monocyte-derived macrophages; CCL-2: C-C motif chemokine ligand 2; CD: cluster of differentiation; CSF-1: colony stimulating factor 1; CSF-1R: colony stimulating factor 1 receptor; CTL: cytotoxic T lymphocyte; CTLA-4: cytotoxic T-lymphocyte-associated protein 4; CXCR4: CXC-chemokine receptor 4; FASL: FAS ligand; FGL2: fibrinogen-like protein 2; FLT3: FMS-like tyrosine kinase 3; HLA: human leukocyte antigen; IDO: indoleamine 2,3-dioxygenase; IL: interleukin; LAG3: lymphocyte activation gene 3 protein; NLGN3: neuroligin-3; PD-1: programmed cell death 1; PD-L1: programmed death-ligand 1; PG-E: Prostaglandin-E; STAT3: signal transducer and activator of transcription 3; TGF-β: transforming growth factor β; TIM3: T-cell immunoglobulin mucin receptor 3; Treg: regulatory T-cell; VEGF: vascular endothelial growth factor
Immunobiology of the brain
Translation of available immunotherapeutic approaches to brain tumors necessitates a deep understanding of the immunobiology of the brain microenvironment. The unique features of brain tumors generate an exceptionally immunosuppressive microenvironment characterized by immunoinhibitory cytokines [Figure 1]. These factors, including interleukin (IL)-6, IL-10, CCL2, TGF-β, and prostaglandin-E, are secreted by both brain cancer cells and immune cells. The complex interactions among these cells contribute to an anti-inflammatory environment[42] that supports tumor proliferation and drug-resistance[43,44]. CCL2 chemokine recruits regulatory T-cells (Tregs) and myeloid-derived suppressor cells (MDSC) to the peritumor environment[45,46]. Tregs, in turn, release the anti-inflammatory mediators IL-10 and TGF-β[47]. These two mediators potentiate immune evasion, enhance tumor growth, and contribute to poor prognosis in glioblastoma patients[48,49]. Other factors secreted by brain tumor cells include vascular endothelial growth factor (VEGF) and colony-stimulating factor-1. Both factors are implicated in hypoxia-mediated suppression of immune cells achieved through the deregulation of immune cell maturation, the skew of TAMs to a tumor-supportive M2 phenotype, and the recruitment of both Tregs and MDSC[50-52]. Recently, two studies have interrogated the simplified view of the immune cells’ roles in the context of brain tumors and the M1/M2 dichotomy[53,54]. They found that the immune landscape of brain tumors is much more complicated and multifaceted than previously envisioned. The TME of each brain malignancy dictates the type of immune cells that will predominate. While glioma TMEs are enriched with microglia-derived TAMs, brain metastases originating from extracranial tumors are dominated by neutrophils[54] and tumor infiltrating leukocytes, specifically Tregs[53]. In brain metastases, TAMs are mainly tissue-infiltrating MDMs and are phenotypically distinct from MDMs identified in gliomas[53]. Intriguingly, the transcriptomic signature of TAMs did not reflect the M1/M2 polarization status in all brain tumors tested, but rather a multifaceted phenotype of TAMs sculpted by the underlying tumor type[53,54]. Collectively, these studies uncover the disease-specific alterations of immune cells, explaining the discrepancy in clinical outcomes of immunotherapy in patients with brain tumors.
Brain tumor cells not only secrete immunosuppressive factors, but they also express them on their surface. CD95 and CD70 are two cell surface markers associated with primary brain tumor stemness, aggressiveness, and progression through their inherent apoptotic and immunomodulatory effects on TAMs and infiltrating T-cells[55,56]. The rapid induction of indoleamine 2,3-dioxygenase (IDO) expression on brain tumor cells recruits Tregs and enhances tumor growth[57,58]. The hypoxic environment of brain tumors exhibits persistent activation of the signal transducer and activator of the transcription 3 pathway, an immunosuppressive pathway associated with poor survival[59,60]. Fibrinogen-like protein 2 expression in gliomas leads to the inhibition of immune cell differentiation while increasing the expression of M2 macrophages and programmed cell death protein-1 (PD-1)[61,62]. Increased expression of both PD-1 and cytotoxic T lymphocyte protein-4 (CTLA-4) on brain tumor cells affects tissue-resident T-cells (TRM) and TAMs by limiting their phagocytic capacity[63] and causing effector cell exhaustion[64]. The immunosuppressive nature of the brain TME is one of the challenges that needs to be addressed and targeted in immunotherapy.
Immunotherapeutic strategies for brain cancer
Checkpoint inhibition
The concept of immune surveillance was proposed in the 1900s and defined as the host’s immunologic defense mechanisms to get rid of unhealthy cells. Immune checkpoints are molecules located on the surfaces of activated T-cells whose primary purpose is to inhibit uncontrolled inflammatory responses. The interaction of PD-1 and CTLA-4, the classical immune checkpoints, with their ligands signals T-cell inactivation and apoptosis, respectively. Therefore, checkpoint blockade can remove immune tolerance and restore activated T-cells [Figure 2A][65].
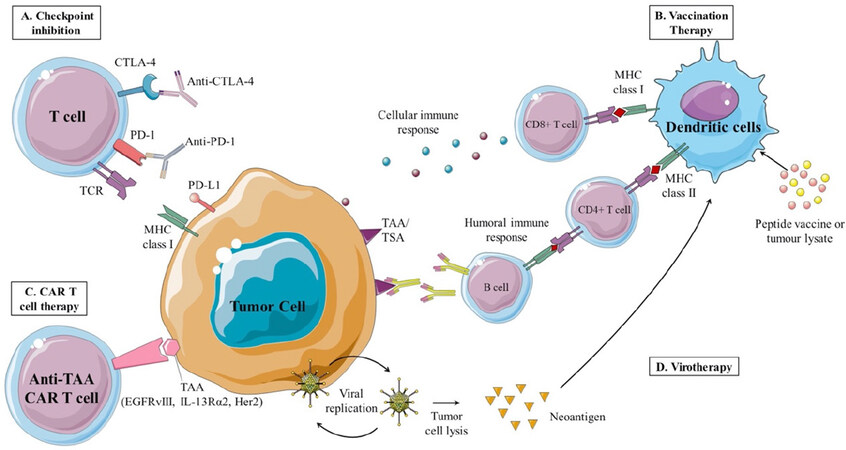
Figure 2. Immunotherapeutic strategies for treatment of brain cancers. A: checkpoint blockade with monoclonal antibodies against PD-1 and CTLA-4 can block the interaction of these receptors with their ligands expressed on tumor cells leading to sustained T-cell activation and anti-tumor responses; B: vaccination therapy depends on dendritic cell antigen presentation of peptides or antigens derived from tumor lysates. In peptide vaccine therapy, peptides can be antigens encoded by mutant genes and thus be expressed only on tumor cells. These neoantigens are called TSA. Other peptides can be antigens derived from the modulated expression of normal peptides and are termed TAA. In cell-based vaccination, dendritic cells are expanded and activated ex vivo with patient-tumor lysates prior to administration. Both vaccine modalities aim to stimulate anti-tumor humoral and cellular immune responses; C: in CAR T-cell therapy, autologous T-cells are isolated and transfected with CAR construct, and transferred back into patients. These T-cells can recognize tumor-associated antigens obviating the need for MHC-dependent antigen presentation; D: oncolytic virus therapy allows the preferential replication of the virus in tumor cells causing tumor cell lysis and release of antigens that can induce an immune response. This figure contains images from Servier Medical Art. CAR: chimeric antigen receptor; CD: cluster of differentiation; CTLA-4: cytotoxic T-lymphocyte-associated protein-4; EGFRvIII: epidermal growth factor receptor variant III; HER2: human epidermal growth factor receptor 2; IL-13Rα2: interleukin-13 receptor alpha 2; MHC: major histocompatibility complex; PD-1: programmed cell death-1; PD-L1: programmed death-ligand 1; TAA: tumor-associated antigens; TCR: T-cell receptor; TSA: tumor-specific antigen
PD-1 and CTLA-4 are inhibitory immune receptors, or checkpoint inhibitors, predominately expressed on T-cells and functionally dysregulated in the TME[66,67]. PD-1 is an immune checkpoint inhibitor expressed on T-cells, myeloid cells, natural killer (NK) cells, and B lymphocytes[68]. The interaction of PD-1 with its ligands PD-L1 or PD-L2 present on antigen-presenting cells (APC) and tumor cells induces immune tolerance and suppression of T-cell activation[68-70]. Tumor cells utilize this mechanism to circumvent immune surveillance through the attenuation of T-cell responses[69]. CTLA-4, also known as CD152, is a member of the immunoglobulin superfamily and is the first coinhibitory molecule identified[71]. It is expressed on regulatory and activated T-cells but not on naïve T-cells[72,73]. Once CTLA4 interacts with its ligands CD80/CD86 on the surface of APCs, it inhibits costimulatory cell signaling pathways and decreases T-cell activation and responsiveness[74].
A recent study shows that PD-1 expression in glioma patients’ tumor-infiltrating lymphocytes is a marker of T-cell exhaustion[64]. High expression of PD-1 and its ligands has been associated with lower prognosis in gliomas[75,76] and various pediatric brain tumors (e.g., ependymoma and high-grade glioma)[77]. Similarly, gliomas with higher CTLA-4 expression exhibited more immune cell infiltration and worse overall survival (OS)[78]. Therefore, targeting immune checkpoint molecules offers an effective T-cell based immunotherapeutic approach for brain cancer treatment. The therapeutic efficacy of blocking the interaction of PD-1 and its ligand PD-L1, alone or in combination with CTLA-4 inhibitors, has been demonstrated in melanoma[23,79], non-small cell lung cancer[24,80], and renal cell carcinoma[81]. The success of checkpoint inhibitors in different cancers, particularly in untreated brain metastases[82], motivated efforts to investigate their potential in the improvement of brain cancer outcomes.
The mounting interest in checkpoint inhibitors for brain cancer treatment has encouraged a series of clinical trials. Table 1 lists select completed clinical trials of PD-1 inhibitors in brain tumors. No completed trials were documented using CTLA-4 inhibitors as a monotherapy in brain cancers. The PD-1 inhibitor pembrolizumab is well-tolerated in refractory high-grade glioma patients, with a low response rate and increased progression-free survival (PFS) even in combination with bevacizumab[83]. Bevacizumab is a VEGF-A inhibitor and is the only monoclonal antibody approved for use in recurrent human glioblastoma. However, a retrospective analysis reported that nivolumab (anti-PD-1) monotherapy did not afford a significant improvement in survival when administered as salvage therapy in recurrent glioblastoma progressing on bevacizumab[84].
Select completed clinical trials of immunotherapeutic approaches in brain cancer patients
| Therapeutic name | Intervention | Patient population | Study phase | Tumor type | Median OS
(months) | Median PFS
(months) | Study identifier | |
|---|---|---|---|---|---|---|---|---|
| Checkpoint inhibitors | Nivolumab | Anti-PD-1 | Child, Adult, Older Adult | 2 | Resectable Glioblastoma | 7.3 | 4.1 | NCT02550249 |
| Pembrolizumab | Anti-PD-1 | Adult | - | Recurrent Glioblastoma | Adjuvant: 7.5
Neoadjuvant: 13.7 | Adjuvant: 2.4
Neoadjuvant: 3.3 | N/A | |
| Cancer vaccines | mRNA-transfected DC-based vaccine | Anti-cancer stem cells vaccine | Adult, Older Adult | 1/2 | Glioblastoma | 25 | 22.8 | NCT00846456 |
| DC-based Immunotherapy | Child, Adult, Older Adult | 2 | CNS tumors | N/A | N/A | NCT00576537 | ||
| HSPPC-96 | Autologous heat-shock protein peptide complex-96 (HSPPC-96) vaccine | Adult, Older Adult | 2 | Recurrent Glioblastoma | 42.6 | 19.1 | NCT00293423 | |
| - | Autologous tumor lysate-loaded DCs | Adult, Older Adult | 2 | Glioblastoma | 28 | 9.5 | NCT00323115 | |
| ICT-107 | DCs pulsed with synthetic peptides | Adult, Older Adult | 2 | Newly diagnosed Glioblastoma | 18.3 | 11.2 | NCT01280552 | |
| Cell-based immunotherapy | - | Anti-EGFRvIII CAR T-cells | Adult, Older Adult | 1/2 | Malignant EGFRvIII-positive Gliomas | N/A | N/A | NCT01454596 |
| - | Aldesleukin-stimulated lymphokine-activated killer cells + IL-2 | Child, Adult, Older Adult | 2 | Glioblastoma | 14.5 | N/A | NCT00331526 | |
| - | Anti-EphA2-CAR T-cell immunotherapy | Adult, Older Adult | 1/2 | EphA2-Positive Malignant Glioma | N/A | N/A | NCT02575261 | |
| - | Anti-GD2-CAR-T-cell immunotherapy | Adult, Older Adult | 1/2 | GD2 Positive Glioma | N/A | N/A | NCT03252171 | |
| Oncolytic viruses | G207 | Oncolytic herpes simplex virus | Adult, Older Adult | 1/2 | Recurrent malignant glioma | 7.5 | N/A | NCT00028158 |
| Delta24-RGD | Oncolytic adenovirus | Adult, Older Adult | 1/2 | Recurrent Glioblastoma | N/A | N/A | NCT01582516 | |
| ParvOryx | H-1 parvovirus | Adult, Older Adult | 1/2 | Progressive Primary or Recurrent Glioblastoma | 15.25 | 3.64 | NCT01301430 |
While clinical benefit is not always evident following monotherapeutic PD-1 blockade with nivolumab in resectable glioblastoma (NCT02550249), favorable changes are detected in the TME in terms of immune cell infiltration[85]. The KEYNOTE-028 clinical trial reported relative tolerability of pembrolizumab monotherapy in a cohort of recurrent glioblastoma patients, with a median OS exceeding that afforded by conventional treatments[86]. The significant extension of overall and PFS in recurrent glioblastoma patients treated with pembrolizumab as adjuvant or neoadjuvant therapy was associated with improvements in antitumor immunity, particularly with enhanced clonal expansion of T-cells[27].
Few immunotherapeutic agents have reached advanced clinical stages in brain cancer patients. To date, no phase 3 clinical trials have been completed. Only one active trial is investigating the efficacy of PD-1 monotherapeutic blockade in comparison with VEGF-A inhibition in patients with recurrent glioblastoma (NCT02017717). While OS was comparable in both approaches, preliminary data showed that treatment with a PD-1 inhibitor ensured more durable responses than those with VEGF-A blockade[29]. The majority of clinical investigations examining the efficacy of checkpoint inhibitors are either ongoing or recruiting patients. Two phase 2 clinical trials studying the efficacy of nivolumab in selected rare CNS cancers and high-grade mutated gliomas (NCT03173950; NCT03925246) are currently recruiting patients. Other immune inhibitors, such as ipilimumab, pembrolizumab, and bevacizumab are also set to be evaluated in adult glioma patients at different disease stages (NCT02852655; NCT03425292). Pre-clinical studies support the therapeutic relevance of targeting IDO, a clinically relevant marker of poor survival which enhances Tregs-mediated immunosuppression in brain tumors[57,58]. In pediatric populations, the efficacy of indoximod, an anti-IDO agent, is being investigated in combination with chemotherapy and/or radiation for children with progressive brain tumors or newly diagnosed diffuse intrinsic pontine glioma in a phase 2 trial (NCT04049669). Other clinical efforts remain in earlier phase 1 stages and mainly examine the efficacy of PD-1 blockade in younger brain cancer cohorts (NCT02359565; NCT02359565).
Vaccines
Therapeutic immunization against brain tumors aims to stimulate patient antitumor immunity through the recognition of tumor-associated antigens by T-cells [Figure 2B]. This is expected to elicit an immune response, which eventually leads to the eradication of cancer cells. The development of cancer vaccines differs from the conventional prophylactic preventative vaccines against infections at multiple levels. The immunocompromised state of the patients, the poor immunogenicity of self-antigens, and the capability of tumor cells to evade and suppress the immune system are challenges that cancer vaccination needs to overcome. The immunization of patients is usually achieved with either tumor-specific peptide(s) or autologous/allogeneic ex vivo-modified cells primed with patient’s own tumor antigens. So, cancer vaccines include peptide vaccines and cell-based vaccines. In peptide vaccination, tumor specific antigens (e.g., EGFRvIII, heat shock proteins) are injected into the patient to elicit an immune response. Cell-based vaccines employ the injection of ex vivo-modified cells (tumor cells, dendritic cells) to induce immunogenic tumor cell destruction.
Early studies have reported the potential immunological and clinical benefit as well as the relative tolerability of integrating vaccine therapy into malignant glioma treatment regimens [Table 1][87,88]. The identification of the tumor-associated antigen, epidermal growth factor receptor variant III (EGFRvIII), led to the development of the EGFRvIII-specific peptide (PEPvIII) vaccine. PEPvIII induced a significant increase in humoral response against EGFRvIII in glioblastoma patients[89]. An EGFRvIII-targeted vaccine also seems promising in patients with glioblastoma subsets expressing this receptor as it has increased OS when compared to unvaccinated matched controls[90]. Despite the possible benefits of EGFRvIII-targeting vaccines, the heterogeneity of brain tumors and the immunological escape at recurrence may restrict the efficacy of this therapeutic approach claiming further clinical investigations[90].
Heat shock proteins (HSPs) are a family of stress-related chaperone molecules that possess the capacity to bind to tumor-associated antigens and induce targeted antitumor immune responses[91]. The HSP vaccine is made up of a complex of HSP and autologous tumor antigen. The heat shock protein peptide complex-96 (HSPPC-96) is the HSP vaccine mainly used in brain cancer treatment. Vaccination using this peptide in newly diagnosed glioblastoma was proven safe and effective in early clinical findings with more than two-fold increase in tumor-specific immune response and a significant increase in OS[92]. Vaccinated patients survived more than 40 months, compared to an average of 14.6 months among their unvaccinated low tumor-specific immune response counterparts[92]. HSPPC-96 vaccination after gross total resection of recurrent glioblastoma also showed tolerability in a phase 2 clinical trial. The majority of patients (90.2%) survived 6 months after treatment, and 29.3% were alive after 1 year[93].
Cell-based vaccines made from autologous or allogeneic ex vivo-modified cells have also attracted their share of clinical interest. The cells can be dendritic cells, tumor cells, or other immune cells. The most common approach comprises blood-isolated dendritic cells, which are pulsed with tumor lysates, activated, and reinjected into patients as vaccines. Phase 1 studies have demonstrated the feasibility, safety, and bioactivity of tumor lysate-pulsed dendritic cells for the treatment of malignant gliomas[94,95]. Subsequently, multiple phase 2 studies have been completed but not published in brain cancer patients (e.g., NCT00576537). A dendritic cell-based vaccine was also used in a phase 1/2 trial to target cancer stem cells and was found to cause a 2.9-fold increase in PFS in glioblastoma patients[96]. Vaccination with autologous tumor lysate-loaded DCs favored the survival of glioblastoma patients treated with radiotherapy and chemotherapy and induced tumor-specific responses[97]. However, the variability in vaccine-induced immunogenicity shows that much remains to be learned about the optimal preparation, contents, and dose of dendritic cell-based vaccines in brain cancer patients[98]. Another cell-based vaccine, ICT-107, was generated using autologous DCs pulsed with synthetic peptides targeting antigens (e.g., MAGE-1, HER-2, AIM-2, TRP-2, gp100, and IL13Rα2) associated with either glioblastoma tumor cells or stem cells. ICT-107 was used for the therapeutic vaccination of adult patients with newy diagnosed glioblastoma in a phase 2b clinical trial. Patients treated with ICT-107 showed a significant improvement in PFS and maintenance of quality of life, particularly those exhibiting HLA-A2 primary tumor antigen expression[99].
Cell-based immunotherapy
Adoptive T-cell therapy (ACT) is emerging as a novel and promising immunotherapeutic strategy against brain tumors. The most successful ACT approach involves the transfection of a chimeric antigen receptor (CAR) construct directed at various tumor antigens into autologous T-lymphocytes [Figure 2C][100]. Independent of MHC co-stimulation, the engineered T-cells can home in to the tumor cells once they are infused in the patient. This alleviates the immunosuppressive TME characteristic of brain tumors and induces the destruction of malignant T-cells. The leading CAR candidates in brain cancer include interleukin-13 receptor alpha 2 (IL-13Rα2), EGFRvIII, and human epidermal growth factor receptor 2 (HER2)[101].
IL13Rα2 is the first CAR target clinically developed and optimized for adoptive cell therapy in brain cancers. Expressed on glioma stem-like cells (GSC) and differentiated cancer cells, IL13Rα2 proves to be an effective immunotherapeutic target and renders both cell populations susceptible to cytotoxic activity of the first generation IL13Rα2-CAR T-cells, termed IL13-zetakine. Engineered IL13-zetakine T-cells were shown to have abolished GSC and differentiated tumor cells both in vitro and in vivo[102]. A preclinical study in a murine model demonstrated the ability of IL13Rα2-CAR T-cells to efficiently kill glioma cells and to shift the glioblastoma TME to a pro-inflammatory landscape[103]. In a clinical setting, a pilot study of three patients with refractory glioblastoma was conducted and showed an increase in tumor necrosis following the intracranial administration of IL13Rα2-specific CAR-T-cells[104]. To improve the antitumor potential of this adoptive cell therapy, a patient suffering from recurrent multifocal glioblastoma received an IL13Rα2-CAR T-cell treatment and experienced dramatic regression of his tumors[105]. Donor-derived allogenic CAR CD8+ T-cells can also be induced to express IL13 zetakine/HyTk CAR and used to treat refractory high-grade glioma in combination with IL-2 (NCT01082926).
As a tumor-specific antigen, EGFRvIII seems to be a promising CAR target for brain tumors, and has attracted clinical attention. The first phase 1 trial (NCT02209376) of CAR T-cells directed against EGFRvIII showed trafficking of engineered T-cells to the tumor site, T-cell expansion, and downregulation of EGFRvIII expression in tumors suggesting antigen-directed cytolytic activity[106]. However, another recent phase 1 pilot study (NCT01454596) using anti-EGFRvIII CAR-T-cell therapy did not show clinical significance in patients with recurrent glioblastoma. The transduced T-cells were administered in a dose-escalation pattern with intravenous IL-2 support. They caused severe pulmonary toxicity in two patients and the death of another patient upon administration of T-cells at the highest dose[107]. These studies highlight the barriers associated with EGFRvIII-CAR T-cell therapy in the treatment of brain tumors. Since no significant antitumor responses were shown, these clinical trials mandate the need to better understand the challenges associated with adoptive cell therapy; in particular, the heterogeneity of EGFRvIII expression and the immunosuppressive environment of brain tumors.
HER2-CAR T-cell immunotherapy remains in the early stages of brain cancer treatment. Safety became an issue while targeting HER2 after the first patient receiving HER2-CART-cell immunotherapy died. Recently, a phase 1 trial (NCT01109095) using bispecific CAR-T-cells aimed to improve the persistence and safety of HER2-CAR T-cells. The engineered T-cells expressed CARs specific for cytomegalovirus and HER-2. This trial demonstrated a clinical benefit in 38% of patients with progressive glioblastoma, and the therapy was well tolerated[108].
Although targeting the aforementioned tumor-associated antigens (i.e., IL13Rα2, EGFRvIII, and HER2) has shown clinical benefit in patients with brain tumors, tumor heterogeneity and subsequent resistance against CAR T-cells underscore the need to discover a new repertoire of targeted antigens. In this context, preclinical investigations for CAR therapy in brain cancers have been initiated and a set of new antigens [e.g., B7-H3, CD133, CSPG4, ephrin A2 (EphA2), and GD2] are being intensively explored[109]. Some of these antigens have already found their way to clinical trials but the results are not published yet, such as EphA2 (NCT02575261), GD2 (NCT03252171), and B7-H3 (NCT04185038)[109].
Although CAR-T-cell immunotherapies have received FDA approval in certain settings, they remain associated with significantly high costs, off-target effects on healthy tissues, as well as other toxicities such as cytokine release syndrome[110]. In addition, failures of many immunotherapeutic approaches for the treatment of glioblastoma pleaded the need for alternative strategies. One promising approach includes NK cells, the lymphocytes of innate immunity, which are capable of eliminating malignant T-cells and coordinating the crosstalk between DCs and T-cells without the need for antigen presentation. The clinical effectiveness of NK-based immunotherapy lies in the ability of NK cells to leverage antitumor immunity and DC-mediated T-cell activation through identification and elimination of cells that lack self-MHC class I molecules[111]. This favors the utility of allogeneic NK cells from healthy donors as they will not recognize tumor cells as self and will not induce graft-vs-host-disease. Engineering NK cells instead of T-cells is therefore expected to have higher clinical efficacy and safety (reviewed extensively in Burger et al.[112]). So, ongoing efforts are directed to enhance NK-cell therapeutics ranging from blockade of their inhibitory receptor pathways to specifically targeting tumor cells using bispecific antibodies or CARs. Clinical studies remain in their earlier stages in glioblastoma[113], with ErbB2 (HER2)-specific CAR-NK cells currently under investigation in the CAR2BRAIN phase 1 study (NCT03383978).
CAR-T and NK cell therapy is not the only weapon in the arsenal of cellular immunotherapy [Table 1]. Other methods include the generation of intracavitary alloreactive cytotoxic T lymphocytes sensitized to MHC and their administration with interleukin-2 (IL-2). This strategy proved to be well-tolerated in recurrent malignant glioma patients[114]. Intralesional exposure of resectable glioblastoma tumors to autologous lymphokine-activated killer cells is another safe cell-based immunotherapy with promising results in terms of patient survival[115]. Systemic adoptive cell therapy has also been reported in clinical trials and involves the administration of ex-vivo activated T or NK cells to treat tumors. A phase 2 trial examined the inoculation of irradiated tumor cells in malignant glioma patients, followed by isolation and transfer of ex-vivo expanded T-cells. The clinical results were not conclusive and warranted further investigation[116]. Recent efforts have examined the safety, tolerability, and preliminary efficacy of autologous NK cell-based immunotherapy both in adult (NCT01588769) and pediatric (NCT01875601) brain cancer patients, but results are not yet published. Cellular and adoptive immunotherapies are also being investigated in a phase 2 trial in solid tumor patients receiving NK cells from haploidentical donors (NCT02100891). Another phase 1 study is also set to determine the feasibility and safety of administering gene-modified drug-resistant γδ T-cells. These cells will be generated from newly diagnosed glioblastoma patients’ own lymphocytes and will be administered with temozolomide (TMZ) chemotherapy (NCT04165941).
Oncolytic viral therapy
Powered by advancements in DNA recombinant technology, oncolytic virotherapy has emerged as a promising cancer therapeutic candidate. During infection, viruses use host replication machinery to amplify their genetic code and expand in numbers. This results in the inevitable lysis of the host T-cell and the spreading of the viral progeny [Figure 2D]. In virotherapy, viruses are engineered to selectively infect cancer cells and to minimize immunosuppression of viral agents[117]. Oncolytic viruses are seen as a potential therapeutic approach in the notoriously challenging field of brain tumor treatment. Completed clinical trials have mainly utilized one of the two genetically modified viruses, adenovirus and herpes simplex virus (HSV), to target brain cancer cells [Table 1]. Other virotherapeutic agents with anti-glioma effects are currently under investigation in phase 1/2 clinical trials including reovirus, poliovirus, parvovirus, measles, and Newcastle disease virus[118].
DNX-2401 (Delta-24-RGD; tasadenoturev) is an oncolytic adenovirus designed to be replication-competent and glioma-selective. The oncolytic efficacy of DNX-2401 has been proven in both adult and pediatric glioma models along with its capacity to induce antitumor immune responses[119]. In a phase 1 clinical study, intratumoral DNX-2401 demonstrated satisfactory clinical activity, tolerability, and safety in recurrent high-grade glioma patients[120]. These promising results fueled phase 2 trials of this oncolytic virus in either glioblastoma alone (NCT01582516) or in combination with a PD-1 inhibitor (CAPTIVE/KEYNOTE-192; NCT02798406), and a phase 1 study in newly diagnosed pediatric glioma (NCT03178032). In another phase 1 trial, neural stem cells loaded with a conditionally replicative oncolytic adenoviral vector (CRAd), NSC-CRAd-Spk7, were injected in patients with malignant gliomas. Following viral infection with NSC-CRAd-Spk7, the tumor-specific survivin promoter induces the lysis of malignant T-cells. While the clinical investigations of this virotherapy are completed, the therapeutic value of this approach is not yet reported (NCT03072134).
Oncolytic HSV is the first recombinant virus developed and armed with various therapeutic genes to be used in the treatment of different cancers[121]. G207 is a conditionally replicating derivative of HSV widely used in brain cancer therapy. Inoculation of glial tumors with G207 induced radiographically and neuropathologically evident activity against tumor cells[122]. More importantly, G207 viral infection seems to be durable without eliciting any HSV encephalitis, toxicity, or serious adverse events[122]. The promising therapeutic activity of G207 has warranted investigations in pediatric cohorts prevalent with refractory cerebellar brain tumors (NCT03911388) as well as recurrent supratentorial brain tumors with or without a single radiation dose (NCT02457845). The results of these two studies are not published yet.
Other clinical efforts remain in their early stages and seem to predominately assess the safety and efficacy of other genetically engineered HSVs (NCT02062827; NCT03152318; NCT03657576) and a Polio/Rhinovirus recombinant (NCT01491893) in adult malignant recurrent brain tumors. Evidence from a published phase 1/2a trial reflect the safety, clinical efficiency, and immunostimulatory capacity of ParvOryx01, an H-1 parvovirus, when administered in recurrent glioblastoma patients[123]. Pediatric gliomas and other primary brain tumors have not been excluded from the clinical investigations of oncolytic viruses. Viral strains under investigation include a Polio/Rhinovirus Recombinant (NCT03043391) and a Replication Competent Reovirus (Reolysin®) (NCT02444546), which has reached phase 3 stage in the clinical course of treating head and neck cancers (NCT01166542).
Combination therapy
Single immunotherapeutic strategies have failed to result in effective clinical responses in patients with brain tumors. This is attributed to the multiple immune evasion mechanisms of brain cancers and their tumor heterogeneity. To surmount this issue, combinatorial immune-based approaches that can target multiple branches of the immune system have been suggested. Immune checkpoint inhibitors provide brain cancer patients with ever fleeting improvements in OS when combined with other agents or therapies (e.g., radiotherapy or chemotherapy) [Table 2]. Administered with standard radiotherapy, the anti-PD-L1 agent Durvalumab prolonged survival up to 34.9 months in patients with newly diagnosed unmethylated MGMT (Tumor O-6-methylguanine DNA Methyltransferase) glioblastoma[124]. Another PD-L1 inhibitor called avelumab did not exhibit satisfactory clinical activity in a phase 2 trial investigating its combination with a tyrosine kinase inhibitor in recurrent glioblastoma patients[125]. PD-1 blockade using pembrolizumab showed satisfactory tolerability in recurrent glioblastoma patients regardless of their treatment arm and demonstrated superior clinical efficiency when administered in combination with bevacizumab than when administered alone[126]. The preliminary results of a phase 1 clinical trial examining the efficacy of nivolumab (anti-PD-1) in combination with a dendritic cell vaccine prior to or after surgical resection of recurrent brain tumors, have recently been reported. The combination resulted in prolonged OS and PFS when administered before surgery, albeit with more reported adverse effects (NCT02529072). Another completed clinical trial (NCT02052648) evaluated the safety and tolerability of IDO inhibitor and TMZ combination with or without bevacizumab or radiation in patients with malignant brain tumors with no published results. Checkmate 498 is an active phase 3 clinical trial investigating whether the combination of nivolumab and radiotherapy provides an advantage to adult patients with newly diagnosed unmethylated MGMT glioblastoma in comparison with standard therapy of TMZ and radiation (NCT02617589). The addition of nivolumab to standard TMZ plus radiation therapy is also under active investigation in a phase 3 study of newly diagnosed adult subjects with MGMT-methylated glioblastoma (NCT02667587). The combination of nivolumab with another checkpoint inhibitor known as anti-lymphocyte-activation gene 3 is planned in a phase 1 study (NCT02658981). Another recruiting phase 1 trial aims to evaluate the safety and tolerability of pembrolizumab and vorinostat (a histone deacetylase inhibitor) in combination with TMZ and radiotherapy in adult glioblastoma cohorts (NCT03426891). The combination of TMZ-based therapy with IDO immune checkpoint inhibition is currently under investigation in a phase 1 study to treat pediatric patients (NCT02502708).
Completed clinical trials of checkpoint inhibitors in combination with other treatments in human brain cancer patients
| Drug name or therapy | Intervention or therapeutic target | Patient population | Study phase | Tumor type | Median OS (months) | Median PFS (months) | Study identifier |
|---|---|---|---|---|---|---|---|
| Pembrolizumab ± bevacizumab | Anti-PD-1 ± anti-VEGF-A | Adult, Older Adult | 2 | Recurrent glioblastoma | +bevacizumab:8.8-bevacizumab:10.3 | N/A | NCT02337491 |
| Indoximod + TMZ with or without bevacizumab or radiation | Anti-IDO + chemotherapy with or without anti-VEGF-A or radiation | Child, Adult, Older Adult | 1/2 | TMZ-Refractory Primary Malignant Brain Tumors | N/A | N/A | NCT02052648 |
| Durvalumab + radiotherapy | Anti-PD-L1 + Radiotherapy | Adult, Older Adult | 2 | Newly diagnosed unmethylated MGMT glioblastoma | 15.1 | N/A | NCT02336165 |
| Avelumab + axitinib | Anti-PD-L1 + tyrosine kinase inhibitor | Adult, Older Adult | 2 | Recurrent glioblastoma | 6.5 | N/A | NCT03291314 |
The synergistic effects of cancer vaccines and other immunotherapeutic approaches or standard-of-care treatment have been reported in clinical trials [Table 3]. The integration of autologous dendritic cell-based vaccination with the standard glioblastoma treatment regimen of surgical resection, radiotherapy, and chemotherapy was evaluated in a phase 2 study. This combination proved to be feasible, safe, and clinically beneficial[127]. In a large phase 3 clinical trial, the concomitant use of autologous tumor lysate-pulsed dendritic cell vaccine and standard chemoradiotherapy with TMZ in newly diagnosed glioblastoma patients demonstrated feasibility, safety, and survival-improving potential[32]. The results of other clinical trials investigating the efficacy of combining first-in-line brain cancer treatment with DCs loaded with autologous tumor lysates are not published yet (NCT01213407; NCT02496520). Another phase 3 study evaluated the addition of autologous cytokine-induced killer cells to radiotherapy-TMZ in newly diagnosed glioblastoma patients. The trial significantly improved PFS, but not OS[128].
Select completed clinical trials of cancer vaccines in combination with other immunotherapeutic or standard-of-care approaches
| Vaccine/therapy name | Therapeutic target | Patient population | Study phase | Tumor type | Median OS (months) | Median PFS (months) | Study identifier |
|---|---|---|---|---|---|---|---|
| Immuncell-LC + TMZ | Autologous CIK cells + chemotherapy | Adult
Older adult | 3 | Newly diagnosed glioblastoma | 22.5 | 8.1 | NCT00807027 |
| Autologous whole-tumor cell vaccines + GM-CSF+ adoptive transfer of T lymphocytes | Adult, Older Adult | 2 | Primary or Recurrent Astrocytoma or Oligodendroglioma | 12 | N/A | NCT00004024 | |
| - | Autologous tumor cell vaccine + GM-CSF + ex vivo activated T-cell adoptive therapy | Adult, Older Adult | 2 | Glioblastoma | N/A | N/A | NCT00003185 |
| TVAX | TVI-Brain-1 (autologous whole-tumor cell vaccine) + adoptive transfer of T lymphocytes | Adult, Older Adult | 1/2 | Recurrent grade IV glioma | N/A | N/A | NCT01081223 |
| TVAX | TVI-Brain-1 (autologous whole-tumor vaccine) + GM-CSF + adoptive transfer of anti-CD3-activated lymphocytes | Adult, Older Adult | 2 | Recurrent grade IV glioma | N/A | N/A | NCT01290692 |
| Autologous tumor-derived heat shock protein peptide-complex (HSPPC-96) + chemotherapy | Adult, Older Adult | 2 | Newly diagnosed glioblastoma | N/A | N/A | NCT00905060 | |
| PEP-III-KLH | Anti- EGFRvIII vaccine + GM-CSF | Adult, Older Adult | 2 | Newly diagnosed EGFRvIII-expressing glioblastoma | 23.6 | 15.2 | NCT00643097 |
| Rindopepimut (CDX-110) + bevacizumab | Anti-EGFRvIII vaccine + anti-VEGF-A | Adult, Older Adult | 2 | Relapsed, EGFRvIII-positive glioblastoma | N/A | 3.7 | NCT01498328 |
| Rindopepimut (CDX-110) + sargramostim + TMZ | Anti-EGFRvIII vaccine + GM-CSF + chemotherapy | Adult, Older Adult | 2 | Glioblastoma | 21.8 | 9.2 | NCT00458601 |
| Rindopepimut (CDX-110) + adjuvant TMZ | Anti-EGFRvIII vaccine + GM-CSF + chemotherapy | Adult, Older Adult | 3 | Newly diagnosed glioblastoma | 20.1 | N/A | NCT01480479 |
| Autologous tumor cell vaccine + sargramostim | Autologous tumor cell vaccine + GM-CSF | Child, Adult, Older Adult | 2 | Recurrent or refractory brain tumor | N/A | N/A | NCT00014573 |
| Autologous DCs pulsed with tumor lysates + radiotherapy + chemotherapy | Adult, Older Adult | 2 | Glioblastoma | 23.4 | 12.7 | NCT01006044 | |
| Trivax + TMZ + Surgery + Radiotherapy | Tumour-lysate Charged DCs + Chemotherapy + Surgery + Radiotherapy | Child, Adult, Older Adult | 2 | High-grade glioma | N/A | N/A | NCT01213407 |
| IMA950 + Hiltonol + TMZ | Multi tumour-associated peptides (TUMAPs) vaccine+ poly-ICLC + chemotherapy | Adult, Older Adult | 1/2 | Glioblastoma | 19 | N/A | NCT01920191 |
| Type-1 αDCs (αDC1) loaded with glioma-associated antigen (GAA) epitopes + poly-ICLC | Adult, Older Adult | 1/2 | Recurrent malignant gliomas | 32.88 | N/A | NCT00766753 | |
| SL-701 + poly-ICLC | Synthetic peptides + poly-ICLC | Adult, Older Adult | 1/2 | Recurrent glioblastoma | 11.7 | N/A | NCT02078648 |
| Autologous DCs pulsed with tumor lysate + standard therapy | Child, Adult | 2 | Advanced solid tumors (including CNS tumors) | N/A | N/A | NCT02496520 |
Multiple phase 2 trials have assessed the efficacy of adoptive cell therapy after the use of autologous whole-tumor vaccines, such as TVAX, in combination with granulocyte-macrophage colony-stimulating factor (NCT00004024; NCT01081223; NCT01290692; NCT00014573), but the final results are unavailable to date. Results of the combination of chemotherapy with autologous tumor-derived HSPPC-96 are also expected from a completed phase 2 trial in adult patients with newly diagnosed glioblastoma (NCT00905060). Vaccines integrating multiple tumor-associated peptides have been reported and include IMA950 multipeptide which in combination with adjuvant poly-ICLC ensured almost 20 months median OS and induced both CD8 and CD4 T-cell responses. This was reported in newly diagnosed glioblastoma and grade III astrocytoma patients treated with radiochemotherapy[129]. SL-701 is another vaccine under investigation in a completed but unpublished phase 1/2 study examining the effect of administering survivin, IL-13Rα2; and EphA2 in combination with poly-ICLC in recurrent glioblastoma patients (NCT02078648). Poly-ICLC has also been administered in combination with Type-1 αDCs (αDC1) loaded with synthetic peptides associated with brain tumors, such as glioma-associated antigen epitopes, ensuring up to 32.88 median OS with relative tolerability (NCT00766753).
The combination of autologous whole-cell tumor vaccines and adoptive T-cell therapy seems promising for the treatment of grade IV glioma and primary or recurrent astrocytoma or oligodendroglioma (NCT01081223; NCT00004024), while its effect on glioblastoma in a phase 2 clinical trial setting remains unpublished (NCT00003185). In fact, adoptive transfer of in vitro activated T lymphocytes after vaccination induced a radiologically-detectable tumor response and prolonged survival in patients with recurrent gliomas[130].
Treatment of glioblastoma patients with intensified doses of chemotherapy (TMZ) and EGFRvIII-targeted vaccines potentiated vaccine-induced immune responses, eliminated EGFRvIII-expressing tumor cells and extended PFS and OS[131]. EGFRvIII-targeted vaccination using rindopepimut in a phase 2 trial confirmed promising clinical results for glioblastoma patients once rindopepimut was combined with chemotherapy (TMZ)[132]. However, the concurrent administration of rindopepimut vaccine and TMZ in patients with glioblastoma failed to prolong the survival in a phase 3 clinical trial[133]. Most recently, published results from a phase 2 study support the combination of rindopepimut and bevacizumab which confer survival advantage as well as effective antitumor responses[134]. Collectively, these results warrant further investigations into combinatorial approaches that provide the most efficient immunotherapeutic strategy in patients with brain tumors.
Conclusions and future perspectives
Clinical investigations into immunotherapeutic strategies for the treatment of brain cancer lag behind similar efforts in other cancer types due to the heterogeneity and low mutational burden of brain tumors, and the immunosuppressive and evolving nature of the tumor microenvironment. Other challenges involve T-cell sequestration in bone marrow[135] and disease-specific immunological niches[53,54]. Intracranial associated T-cell sequestration is indicative of T-cell dysfunction that contributes to tumor escape from immune surveillance[135]. The diversity of immune landscapes sculpted by different brain tumors highlights the need for better understanding of the immune cell phenotypes and functional states in each tumor type to facilitate the design of targeted immunotherapeutic approaches. Despite these hurdles, harnessing the immune system to restore antitumor immunity and to alleviate brain tumor-induced immunosuppression remains a window-of-hope for brain tumor patients. In a cancer population with a traditionally dismal prognosis, immunotherapeutic agents alone or in combination with current standard-of-care can afford a much-needed improvement in patient survival while delaying disease progression. The preliminary efficacy and tolerability of immunotherapies is evident and warrants further investigation to optimally integrate them in brain cancer management. Significant questions remain unanswered. How does the brain cancer cell affect each distinct type of immune cells starting from T-cell sequestration in bone marrow to their specific enrichment based on tumor type? Do treatment strategies influence the cross-talk between the brain tumor cells and immune cells? What about ECM as an immunotherapeutic target? Would investment in innate immunity like NK cells or even neutrophils be promising? Collectively, in-depth knowledge of the immunological signatures and their interaction with brain tumor cells is needed to develop novel immunotherapeutic agents that can be the future first-in-line curative treatments for brain tumors.
Declarations
AcknowledgmentsThe authors thank Nicholas D. Yaeger, Junji Chen, and Zachary A. Blashinsky for the initial scientific literature search and summary.
Authors’ contributionsDesigned the outline, wrote the paper, and drew figures: Khamis ZI
Wrote the paper, drew figures, and prepared tables: Al-Akkary N
Assisted with literature search and summary, manuscript revision, and figure preparation: Hua T, Draughon SA
Revised the manuscript: Li Y
Conceived the idea, directed this work, designed the outline, and revised the manuscript: Sang QXA
Obtained the funding: Khamis ZI, Li Y, Sang QXA
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was in part supported by Florida Department of Health (FDOH) Live Like Bella award (9LA01) (to Sang QXA and Li Y). http://www.floridahealth.gov/provider-and-partner-resources/research/funding-opportunity-announcements/bellafoa.html. The authors are also funded in part by the Florida State University. This work is also funded by an Endowed Chair Professorship in Cancer Research from anonymous donors (to Sang QXA), and a Lebanese grant (to Khamis ZI). The funding agencies and funders did not play any role in the study design, collection, analysis and interpretation of data, and writing of the manuscript.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
2. Patel AP, Fisher JL, Nichols E, Abd-allah F, Abdela J, et al. Global, regional, and national burden of brain and other CNS cancer, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2019;18:376-93.
3. Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012-2016. Neuro Oncol 2019;21:v1-100.
4. Smith MA, Reaman GH. Remaining challenges in childhood cancer and newer targeted therapeutics. Pediatr Clin North Am 2015;62:301-12.
5. Perus LJM, Walsh LA. Microenvironmental Heterogeneity in Brain Malignancies. Front Immunol 2019;10:2294.
6. Gupta A, Dwivedi T. A simplified overview of world health organization classification update of central nervous system tumors 2016. J Neurosci Rural Pract 2017;8:629-41.
7. Claus EB, Walsh KM, Wiencke JK, Molinaro AM, Wiemels JL, et al. Survival and low-grade glioma: the emergence of genetic information. Neurosurg Focus 2015;38:E6.
8. Rasmussen BK, Hansen S, Laursen RJ, Kosteljanetz M, Schultz H, et al. Epidemiology of glioma: clinical characteristics, symptoms, and predictors of glioma patients grade I-IV in the the Danish Neuro-Oncology Registry. J Neurooncol 2017;135:571-9.
9. Barnholtz-Sloan JS, Ostrom QT, Cote D. Epidemiology of brain tumors. Neurol Clin 2018;36:395-419.
10. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009;10:459-66.
11. Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol 2019;16:509-20.
12. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 2016;19:20-7.
14. Gerstner ER, Fine RL. Increased permeability of the blood-brain barrier to chemotherapy in metastatic brain tumors: establishing a treatment paradigm. J Clin Oncol 2007;25:2306-12.
15. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol 2018;20:184-91.
16. Zhang F, Stephan SB, Ene CI, Smith TT, Holland EC, et al. Nanoparticles that reshape the tumor milieu create a therapeutic window for effective T-cell therapy in solid malignancies. Cancer Res 2018;78:3718-30.
17. Le Rhun E, Preusser M, Roth P, Reardon DA, van den Bent M, et al. Molecular targeted therapy of glioblastoma. Cancer Treat Rev 2019;80:101896.
18. Durand T, Bernier MO, Léger I, Taillia H, Noël G, et al. Cognitive outcome after radiotherapy in brain tumor. Curr Opin Oncol 2015;27:510-5.
19. Brinkman TM, Krasin MJ, Liu W, Armstrong GT, Ojha RP, et al. Long-term neurocognitive functioning and social attainment in adult survivors of pediatric CNS tumors: results from the St Jude lifetime cohort study. J Clin Oncol 2016;34:1358-67.
20. American Cancer Society. Cancer Facts & Figures 2019. Available from: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html. [Last accessed on 2 Sep 2020].
21. Schläger C, Körner H, Krueger M, Vidoli S, Haberl M, et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 2016;530:349-53.
22. Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci 2018;21:1380-91.
23. Weber JS, D’angelo SP, Minor D, Hodi FS, Gutzmer R, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375-84.
24. Gubens MA, Sequist LV, Stevenson JP, Powell SF, Villaruz LC, et al. Pembrolizumab in combination with ipilimumab as second-line or later therapy for advanced non-small-cell lung cancer: KEYNOTE-021 cohorts D and H. Lung Cancer 2019;130:59-66.
25. Shanafelt TD, Wang XV, Kay NE, Hanson CA, O’Brien S, et al. Ibrutinib-Rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med 2019;381:432-43.
26. Long GV, Atkinson V, Lo S, Sandhu S, Guminski AD, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. Lancet Oncol 2018;19:672-81.
27. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 2019;25:477-86.
28. Westphal M, Heese O, Steinbach JP, Schnell O, Schackert G, et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur J Cancer 2015;51:522-32.
29. Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of Nivolumab vs Bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro-Oncology 2017;19:iii21-2.
30. Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol 2018;20:674-86.
31. Ferreri AJM, Cwynarski K, Pulczynski E, Ponzoni M, Deckert M, et al. Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: results of the first randomisation of the International Extranodal Lymphoma Study Group-32 (IELSG32) phase 2 trial. Lancet Haematol 2016;3:e217-27.
32. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, et al. First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med 2018;16:142.
33. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19:1423-37.
34. Bejoy J, Yuan X, Song L, Hua T, Jeske R, et al. Genomics analysis of metabolic pathways of human stem cell-derived microglia-like cells and the integrated cortical spheroids. Stem Cells Int 2019;2019:2382534.
35. Griffin K, Bejoy J, Song L, Hua T, Marzano M, et al. Human stem cell-derived aggregates of forebrain astroglia respond to amyloid beta oligomers. Tissue Eng Part A 2020;26:527-42.
36. Song L, Yuan X, Jones Z, Vied C, Miao Y, et al. Functionalization of brain region-specific spheroids with isogenic microglia-like cells. Sci Rep 2019;9:11055.
37. Chen W, Wang D, Du X, He Y, Chen S, et al. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med Oncol 2015;32:43.
38. Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 2015;161:803-16.
39. Galvao RP, Kasina A, McNeill RS, Harbin JE, Foreman O, et al. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc Natl Acad Sci U S A 2014;111:E4214-23.
40. Hide T, Shibahara I, Kumabe T. Novel concept of the border niche: glioblastoma cells use oligodendrocytes progenitor cells (GAOs) and microglia to acquire stem cell-like features. Brain Tumor Pathol 2019;36:63-73.
42. Henrik Heiland D, Ravi VM, Behringer SP, Frenking JH, Wurm J, et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat Commun 2019;10:2541.
43. Hide T, Komohara Y, Miyasato Y, Nakamura H, Makino K, et al. Oligodendrocyte progenitor cells and macrophages/microglia produce glioma stem cell niches at the tumor border. EBioMedicine 2018;30:94-104.
44. Azambuja JH, da Silveira EF, de Carvalho TR, Oliveira PS, Pacheco S, et al. Glioma sensitive or chemoresistant to temozolomide differentially modulate macrophage protumor activities. Biochim Biophys Acta Gen Subj 2017;1861:2652-62.
45. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res 2016;76:5671-82.
46. Domenis R, Cesselli D, Toffoletto B, Bourkoula E, Caponnetto F, et al. Systemic T cells immunosuppression of glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLoS One 2017;12:e0169932.
47. Li Z, Liu X, Guo R, Wang P. CD4+Foxp3- type 1 regulatory T cells in glioblastoma multiforme suppress T cell responses through multiple pathways and are regulated by tumor-associated macrophages. Int J Biochem Cell Biol 2016;81:1-9.
48. Frei K, Gramatzki D, Tritschler I, Schroeder JJ, Espinoza L, et al. Transforming growth factor-β pathway activity in glioblastoma. Oncotarget 2015;6:5963-77.
49. Mostafa H, Pala A, Högel J, Hlavac M, Dietrich E, et al. Immune phenotypes predict survival in patients with glioblastoma multiforme. J Hematol Oncol 2016;9:77.
50. Tamura R, Tanaka T, Akasaki Y, Murayama Y, Yoshida K, et al. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: perspectives for therapeutic implications. Med Oncol 2019;37:2.
51. Valdor R, García-Bernal D, Riquelme D, Martinez CM, Moraleda JM, et al. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 2019;116:20655-65.
52. Guo X, Xue H, Shao Q, Wang J, Guo X, et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 2016;7:80521-42.
53. Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 2020;181:1626-42.e20.
54. Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 2020;181:1643-60.e17.
55. Drachsler M, Kleber S, Mateos A, Volk K, Mohr N, et al. CD95 maintains stem cell-like and non-classical EMT programs in primary human glioblastoma cells. Cell Death Dis 2016;7:e2209.
56. Ge H, Mu L, Jin L, Yang C, Chang YE, et al. Tumor associated CD70 expression is involved in promoting tumor migration and macrophage infiltration in GBM. Int J Cancer 2017;141:1434-44.
57. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res 2014;20:5290-301.
58. Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res 2012;18:6110-21.
59. Abou-Ghazal M, Yang DS, Qiao W, Reina-Ortiz C, Wei J, et al. The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin Cancer Res 2008;14:8228-35.
60. Piperi C, Papavassiliou KA, Papavassiliou AG. Pivotal Role of STAT3 in shaping glioblastoma immune microenvironment. Cells 2019;8:1398.
61. Yan J, Kong LY, Hu J, Gabrusiewicz K, Dibra D, et al. FGL2 as a multimodality regulator of tumor-mediated immune suppression and therapeutic target in gliomas. J Natl Cancer Inst 2015;107:djv137.
62. Yan J, Zhao Q, Gabrusiewicz K, Kong LY, Xia X, et al. FGL2 promotes tumor progression in the CNS by suppressing CD103+ dendritic cell differentiation. Nat Commun 2019;10:448.
63. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017;545:495-9.
64. Davidson TB, Lee A, Hsu M, Sedighim S, Orpilla J, et al. Expression of PD-1 by T cells in malignant glioma patients reflects exhaustion and activation. Clin Cancer Res 2019;25:1913-22.
65. Fecci PE, Sampson JH. The current state of immunotherapy for gliomas: an eye toward the future. J Neurosurg 2019;131:657-66.
66. Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, et al. Single-cell RNA-Seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep 2017;21:1399-410.
67. Zhao Y, Yang W, Huang Y, Cui R, Li X, et al. Evolving roles for targeting CTLA-4 in cancer immunotherapy. Cell Physiol Biochem 2018;47:721-34.
68. Maxwell R, Jackson CM, Lim M. Clinical trials investigating immune checkpoint blockade in glioblastoma. Curr Treat Options Oncol 2017;18:51.
70. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol 2017;8:561.
71. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987;328:267-70.
72. Romani M, Pistillo MP, Carosio R, Morabito A, Banelli B. Immune checkpoints and innovative therapies in glioblastoma. Front Oncol 2018;8:464.
73. Chen C, Rowell EA, Thomas RM, Hancock WW, Wells AD. Transcriptional regulation by Foxp3 is associated with direct promoter occupancy and modulation of histone acetylation. J Biol Chem 2006;281:36828-34.
74. Simonelli M, Persico P, Perrino M, Zucali PA, Navarria P, et al. Checkpoint inhibitors as treatment for malignant gliomas: “A long way to the top”. Cancer Treat Rev 2018;69:121-31.
75. Tsui K, Gajjar A, Li C, Srivastava D, Broniscer A, et al. Subsequent neoplasms in survivors of childhood central nervous system tumors: risk after modern multimodal therapy. Neuro Oncol 2015;17:448-56.
76. Chen RQ, Liu F, Qiu XY, Chen XQ. The prognostic and therapeutic value of PD-L1 in Glioma. Front Pharmacol 2018;9:1503.
77. Hwang K, Koh EJ, Choi EJ, Kang TH, Han JH, et al. PD-1/PD-L1 and immune-related gene expression pattern in pediatric malignant brain tumors: clinical correlation with survival data in Korean population. J Neurooncol 2018;139:281-91.
78. Liu F, Huang J, Liu X, Cheng Q, Luo C, et al. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int 2020;20:7.
79. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, et al. Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23-34.
80. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, et al. Nivolumab versus Docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 2015;373:123-35.
81. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, et al; CheckMate 025 Investigators. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803-13.
82. Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol 2016;17:976-83.
83. Reiss SN, Yerram P, Modelevsky L, Grommes C. Retrospective review of safety and efficacy of programmed cell death-1 inhibitors in refractory high grade gliomas. J Immunother Cancer 2017;5:99.
84. Chamberlain MC, Kim BT. Nivolumab for patients with recurrent glioblastoma progressing on bevacizumab: a retrospective case series. J Neurooncol 2017;133:561-9.
85. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med 2019;25:470-6.
86. Reardon DA, Kim T, Frenel J, Santoro A, Lopez J, et al. ATIM-35. Results of the phase IB keynote-028 multi-cohort trial of pembrolizumab monotherapy in patients with recurrent PD-L1-positive glioblastoma multiforme (GBM). Neuro-Oncology 2016;18:vi25-6.
87. Ardon H, Van Gool SW, Verschuere T, Maes W, Fieuws S, et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: results of the HGG-2006 phase I/II trial. Cancer Immunol Immunother 2012;61:2033-44.
88. Chang CN, Huang YC, Yang DM, Kikuta K, Wei KJ, et al. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci 2011;18:1048-54.
89. Schmittling RJ, Archer GE, Mitchell DA, Heimberger A, Pegram C, et al. Detection of humoral response in patients with glioblastoma receiving EGFRvIII-KLH vaccines. J Immunol Methods 2008;339:74-81.
90. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 2010;28:4722-9.
91. Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science 1995;269:1585-8.
92. Ji N, Zhang Y, Liu Y, Xie J, Wang Y, et al. Heat shock protein peptide complex-96 vaccination for newly diagnosed glioblastoma: a phase I, single-arm trial. JCI Insight 2018;3:99145.
93. Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol 2014;16:274-9.
94. Yu JS, Liu G, Ying H, Yong WH, Black KL, et al. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res 2004;64:4973-9.
95. Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res 2005;11:5515-25.
96. Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tønnesen P, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother 2013;62:1499-509.
97. Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother 2011;34:382-9.
98. Frank MO, Kaufman J, Parveen S, Blachère NE, Orange DE, et al. Dendritic cell vaccines containing lymphocytes produce improved immunogenicity in patients with cancer. J Transl Med 2014;12:338.
99. Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, et al. A randomized double-blind placebo-controlled phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res 2019;25:5799-807.
100. Brown MP, Ebert LM, Gargett T. Clinical chimeric antigen receptor-T cell therapy: a new and promising treatment modality for glioblastoma. Clin Transl Immunology 2019;8:e1050.
101. Neidert MC, Kowalewski DJ, Silginer M, Kapolou K, Backert L, et al. The natural HLA ligandome of glioblastoma stem-like cells: antigen discovery for T cell-based immunotherapy. Acta Neuropathol 2018;135:923-38.
102. Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, et al. Stem-like tumor-initiating cells isolated from IL13Rα2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res 2012;18:2199-209.
103. Pituch KC, Miska J, Krenciute G, Panek WK, Li G, et al. Adoptive transfer of IL13Rα2-specific chimeric antigen receptor t cells creates a pro-inflammatory environment in glioblastoma. Mol Ther 2018;26:986-95.
104. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016;375:2561-9.
105. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016;375:2561-9.
106. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9:eaaa0984.
107. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother 2019;42:126-35.
108. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Wakefield A, et al. Autologous HER2 CMV bispecific CAR T cells are safe and demonstrate clinical benefit for glioblastoma in a Phase I trial. J Immunotherapy Cancer 2015;3.
109. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, et al. CAR T cells for brain tumors: lessons learned and road ahead. Immunol Rev 2019;290:60-84.
110. Chow VA, Gopal AK, Maloney DG, Turtle CJ, Smith SD, et al. Outcomes of patients with large B-cell lymphomas and progressive disease following CD19-specific CAR T-cell therapy. Am J Hematol 2019;94:E209-E213.
111. Majd N, Dasgupta P, de Groot J. Immunotherapy for neuro-oncology. Adv Exp Med Biol 2020;1244:183-203.
112. Wang W, Jiang J, Wu C. CAR-NK for tumor immunotherapy: clinical transformation and future prospects. Cancer Lett 2020;472:175-80.
113. Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, et al. CAR-engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front Immunol 2019;10:2683.
114. Kruse CA, Cepeda L, Owens B, Johnson SD, Stears J, et al. Treatment of recurrent glioma with intracavitary alloreactive cytotoxic T lymphocytes and interleukin-2. Cancer Immunol Immunother 1997;45:77-87.
115. Dillman RO, Duma CM, Ellis RA, Cornforth AN, Schiltz PM, et al. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. J Immunother 2009;32:914-9.
116. Plautz GE, Barnett GH, Miller DW, Cohen BH, Prayson RA, et al. Systemic T cell adoptive immunotherapy of malignant gliomas. J Neurosurg 1998;89:42-51.
117. Mondal M, Guo J, He P, Zhou D. Recent advances of oncolytic virus in cancer therapy. Hum Vaccin Immunother 2020:1-14.
118. Maxwell R, Luksik AS, Garzon-Muvdi T, Lim M. The potential of cellular- and viral-based immunotherapies for malignant glioma-dendritic cell vaccines, adoptive cell transfer, and oncolytic viruses. Curr Neurol Neurosci Rep 2017;17:50.
119. Martínez-Vélez N, Garcia-Moure M, Marigil M, González-Huarriz M, Puigdelloses M, et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat Commun 2019;10:2235.
120. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol 2018;36:1419-27.
122. Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther 2000;7:867-74.
123. Geletneky K, Hajda J, Angelova AL, Leuchs B, Capper D, et al. Oncolytic H-1 Parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol Ther 2017;25:2620-34.
124. Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn G, et al. Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). JCO 2019;37:2032.
125. Neyns B, Ben Salama L, Awada G, De Cremer J, Schwarze JK, et al. GLIAVAX: a stratified phase II clinical trial of avelumab and axitinib in patients with recurrent glioblastoma. JCO 2019;37:2034.
126. Reardon DA, Nayak L, Peters KB, Clarke JL, Jordan JT, et al. Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. JCO 2018;36:2006.
127. Inogés S, Tejada S, de Cerio AL, Gállego Pérez-Larraya J, Espinós J, et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J Transl Med 2017;15:104.
128. Kong DS, Nam DH, Kang SH, Lee JW, Chang JH, et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in Korea. Oncotarget 2017;8:7003-13.
129. Migliorini D, Dutoit V, Allard M, Grandjean Hallez N, Marinari E, et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro Oncol 2019;21:923-33.
130. Sloan AE, Dansey R, Zamorano L, Barger G, Hamm C, et al. Adoptive immunotherapy in patients with recurrent malignant glioma: preliminary results of using autologous whole-tumor vaccine plus granulocyte-macrophage colony-stimulating factor and adoptive transfer of anti-CD3-activated lymphocytes. Neurosurg Focus 2000;9:e9.
131. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol 2011;13:324-33.
132. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol 2015;17:854-61.
133. Weller M, Butowski N, Tran DD, Recht LD, Lim M, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 2017;18:1373-85.
134. Reardon DA, Desjardins A, Vredenburgh JJ, O’Rourke DM, Tran DD, et al; ReACT trial investigators. Rindopepimut with Bevacizumab for Patients with relapsed EGFRvIII-expressing glioblastoma (ReACT): results of a double-blind randomized phase II trial. Clin Cancer Res 2020;26:1586-94.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Khamis ZI, Al-Akkary N, Hua T, Draughon SA, Li Y, Sang QX. Clinical investigations of immunotherapy for human primary brain tumors. Neurosciences 2021;8:154-73. http://dx.doi.org/10.20517/2347-8659.2020.43
AMA Style
Khamis ZI, Al-Akkary N, Hua T, Draughon SA, Li Y, Sang QX. Clinical investigations of immunotherapy for human primary brain tumors. Neuroimmunology and Neuroinflammation. 2021; 8(3): 154-73. http://dx.doi.org/10.20517/2347-8659.2020.43
Chicago/Turabian Style
Khamis, Zahraa I., Nancy Al-Akkary, Timothy Hua, Sophia A. Draughon, Yan Li, Qing-Xiang Amy Sang. 2021. "Clinical investigations of immunotherapy for human primary brain tumors" Neuroimmunology and Neuroinflammation. 8, no.3: 154-73. http://dx.doi.org/10.20517/2347-8659.2020.43
ACS Style
Khamis, ZI.; Al-Akkary N.; Hua T.; Draughon SA.; Li Y.; Sang Q.X. Clinical investigations of immunotherapy for human primary brain tumors. Neurosciences. 2021, 8, 154-73. http://dx.doi.org/10.20517/2347-8659.2020.43
About This Article
Copyright

Data & Comments
Data


 Cite This Article 15 clicks
Cite This Article 15 clicks

 Like This Article 6
likes
Like This Article 6
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.