
Postnatal toxicant exposure in 3xTgAD mice promotes gene x environment-related early alterations to neuroimmune epigenetic profiles
 0
0Abstract
Aim: The purpose of this study was to evaluate sex-biased, maladaptive changes to epigenetic regulation critical for development of neuroimmune crosstalk resulting from an early-life toxicant exposure previously associated with increased susceptibility to later-life neurodegeneration.
Methods: An evaluation of early-life gene x environment (GxE) interactions was performed in a mouse model of Alzheimer’s disease (Tg) orally exposed to lead acetate (Pb) from postnatal day (PND) 5-9. Following exposure, immunohistochemical analysis was used to evaluate hippocampal expression of DAP12, a marker for perinatal microglia related to microglial-mediated postnatal synaptic pruning of neurons. Altered profiles of three microRNAs critical to homeostatic microglia: neuron signaling (miR-34a, miR-124, miR-132) were measured by qRT-PCR.
Results: Atypical and deleterious expression patterns in Pb-exposed Tg mice were detected with significant female bias by PND 10. Early exposure to Pb resulted in the upregulation of miR-124, a microRNA involved in microglial quiescence, as well as miR-34a, involved in p53-dependent apoptosis and decreased phagocytosis, by PND 21 and during a period of microglial-mediated synaptic pruning specific to females. In addition, we observed a sustained, imbalanced upregulation of miR-132 in Pb-exposed Tg females as well as decreased expression of DAP12.
Conclusion: This study demonstrates the exacerbating effects and early manifestation of GxE interactions in this model. Furthermore, these findings underscore a period of female-specific vulnerability to epigenetic maladaptation during postnatal development, with implications on the faulty later-life adaptability of neuroimmune signaling. Further investigation is warranted to evaluate the persistence and relative contribution of these early influences on the etiopathology of Alzheimer’s disease.
Keywords
Introduction
Numerous studies examining the developmental origins of adult disease (DOAD) hypothesis have shown that environmental perturbations to neuroimmune development may contribute to later-life neurodegenerative diseases, like Alzheimer’s disease (AD)[1-3]. The consequence of gene x environment (GxE) interactions is much more profound during development due to critical windows in which phenotypic plasticity is instructed by, and extremely sensitive to, exogenous signaling. In our previous studies using a DOAD model for AD, we reported that the interaction of GxE exacerbated AD susceptibility later in life in a transgenic AD mouse model postnatally exposed to lead acetate (Pb)[4,5]. This GxE-related vulnerability was concurrent with dysfunctional microglial phenotypes and synaptic defects indicative of atypical microglia-neuron interactions, and significantly biased towards females. In agreement with the literature, microglia in aging brains can become senescent, dystrophic, and mount ineffective or inappropriate responses to neuronal signaling[6-8]. Importantly, the spatially- and temporally-synchronized development of microglia and neurons during the perinatal period fine-tunes this crosstalk necessary for homeostatic signaling in the adult[9]. Thus, imbalances in neuroimmune signaling and function may be developmentally promoted.
Healthy homeostatic interactions between microglia and neurons in adulthood depend on the tightly-controlled developmental programming of microglia in response to cues from neurons[9], clearly demonstrated in studies of early immune insults resulting in cognitive decline, impaired memory, and even neurodegeneration in adulthood[2,10-13]. Mutations to the transmembrane adaptor protein DAP12 or its ligand TREM2 are associated with a form of presenile dementia called Nasu-Hakola disease in humans[14], and patients with heterozygous variants of TREM2 have a significantly higher susceptibility to AD[15-17]. Furthermore, the typical expression of DAP12 is limited to perinatal microglia and is critical for healthy developmental synaptic pruning by microglia[18,19]. DAP12-deficient rodent models exhibit persistent synaptic defects to glutamatergic synapses in adulthood[20,21]. Therefore, DAP12 may highlight a particular junction of the neuroimmune interface during development by which microglia and neurons establish and promote healthy synapse dynamics throughout life, with implications for the promotion of early AD-like phenotypes in its absence. However, little is known about the molecular mechanisms through which early-life toxicant exposure alters this junction to promote atypical signaling between neurons and microglia that then persist beyond development.
As environmentally sensitive genomic regulators, microRNAs represent a form of long-term epigenetic regulation relevant to GxE studies to fine-tune cellular phenotypes. Nearly 60% of all protein-encoding genes are thought to be regulated by miRNAs[22], and, with half-lives nearly 10x longer than mRNA[23], numerous studies have already begun exploring the potential of these molecules to act as biomarkers of disease[24]. In the healthy adult brain, transient changes to miRNAs are often the result of the immediate microenvironment, promoting functional changes to parenchymal targets as a cellular adaption to exogenous influences - temporarily shifting the requirements for homeostasis. However, altered miRNAs during critical windows of neuroimmune development have direct consequences on the promotion of homeostatic baselines, immune maturation and learning, and even patterns of functional response phenotypes. MicroRNA expression patterns are both evidentiary of past exogenous influences and predictive of future cellular response, such that altered miRNAs promoting the disjointed development of microglia and neurons early in life may impact the intertwined functionality of neuroimmune cells throughout life. MicroRNAs are highly involved in central nervous system development[25] and, more pertinently, the promotion of varied microglial states of activation and quiescence and the dynamic transitions therein[26]. Notably, a cluster of microRNAs referred to as “NeurimmiRs” have been described in modulating both neuronal and immune processes, such a miR-124 and miR-132, that act as negotiators at the neuroimmune interface[27]. The most abundant microRNA expressed in the adult brain, miR-124 exhibits highly conserved expression patterns consistent with a critical role in neurodevelopment and neurogenesis[25,28]. The expression of miR-124 has also been shown to promote microglial quiescence transitioning from an activated amoeboid state through the downregulation of M1-associated markers[26,29]. Likewise, miR-132 is involved in the regulation of neurotransmission and synaptogenesis and is upregulated during postnatal development[27]. Correspondingly, both miR-124 and miR-132 are downregulated in the brains of patients with AD[30,31]. Although a recent report by Gillet et al.[32] underscored how certain neurodevelopmental disorders were correlated with altered microRNA expression from toxicant exposures, there is still a disconnect with etiopathological relevance in DOAD models for aging-related neurodegenerative diseases like AD. Furthermore, while microRNAs are persistently modified due to early-life Pb exposure[33], there is a little characterization of the sexually dimorphic effects in Pb-altered epigenetic profiles related to neuroimmune function.
Both rodent and human brains undergo sexually dimorphic neuroimmune development, specifically within the postnatal period, during which PGE2 secreted by microglia critically regulates the masculinization of the male brain[34]. Blocking PGE2 with the NSAID indomethacin (indo) resulted in a “feminization” of male microglia, significantly reducing the number of amoeboid, but not total, microglia in the preoptic area of 2-day-old male mice[35]. Importantly, the number and phenotype of microglia at varying time points and brain regions varies dramatically by sex throughout development; a spike in the number of amoeboid microglia in males parallels a testosterone surge at PND 4, whereas amoeboid microglia numbers within specific brain regions don’t peak in female brains until PND 30[36]. Thus, the transition from DAP12-positive immature amoeboid phenotypes to fully mature, ramified microglia occurs earlier in males than females. Previously, the first two postnatal weeks were generally considered a period of male-specific microglial vulnerability to later-life immune-related priming[37], but, here, we report a comparable postnatal window of female-specific microglial and neuronal vulnerability to epigenetic regulation in a GxE model for AD. Nearly 2/3rds of the 250 miRNAs surveyed in neonatal mouse brains by Morgan and Bale[38] were shown to be differentially expressed in males and females, implicating miRNAs as major epigenetic regulators of sex differences in the developing brain[39,40]. In the current study, miR-34a was also evaluated alongside the neurimmiRs miR-124 and miR-132, given reports that the upregulation of miR-34a decreased TREM2 expression by targeting parts of its mRNA[41]. Furthermore, expression of miR-34a both strongly promotes and is itself promoted by, p53 as a cellular stress response leading to apoptosis and senescence[42], and modifications to this microRNA may suggest the presence of cellular stress signals that are readily detectable by microglia. Interestingly, mutations to presenilin2, one of the genes implicated in familial AD, has been shown to trigger neuronal apoptosis via the miR-34a/p53 axis[43]. Likewise, TP53, the “apoptosis gene” that encodes for p53, is mutated in some cases of AD[42].
The present study aimed to delineate the early epigenetic regulation of neuroimmune phenotypes related to the promotion of lifelong homeostatic microglia: neuron signaling as a consequence of toxicant exposure. The adaptive response of immune cells to fine-tune signaling in homeostatic pathways is not only critical in adulthood but defined during development. Thus, we hypothesized that the combination of a genetic proclivity to AD and postnatal exposure to Pb would result in persistent, differential changes in interrelated neurimmiRs known to regulate microglial quiescence and neuronal maturation and sprouting, thereby promoting lifelong neuroimmune imbalances associated with increased vulnerability to AD. By including indo exposure, we aimed to discriminate sex-biased effects derived from sexually dimorphic microglial signaling during this postnatal period related to PGE2. Here, we confirm the hypothesis that postnatal Pb exposure in a genetic mouse model of AD significantly alters early epigenetic regulation related to the neuroimmune interface in a sexually dimorphic manner, highlighting the exacerbating effect of the GxE model in the formation of a developmental phenotype for later-life female-biased susceptibility to neurodegeneration.
Methods
Animal handling
All handling and experimental manipulations were carried out in accordance with procedures approved by the East Carolina University Institutional Animal Care and Use Committee (IACUC). Pregnant wildtype (WT) and transgenic dams [3xTgAD; B6; 129Psen1tm1MpmTg (APPSwe, tauP301L) 1Lfa/Mmjax] were obtained from the seed colony in the ECU Department of Comparative Medicine and kept on a 12:12 hour light/dark cycle, with access to food and water ad libitum. Litters were culled to eight after birth (postnatal day, PND 1), if needed, and monitored for overt signs of toxicity.
Dosing and tissue preparation
Exposure to lead acetate was based on our previous findings that an identical exposure concentration and timing in the 3xTgAD mouse model resulted in significantly altered microglia and exacerbated AD pathology in adult females[4,5]. Indomethacin concentration (1 mg/kg/day) was chosen to recapitulate previous reports of efficacious concentrations for PGE2 inhibition in rodent microglia[35,44,45]. Dosing solutions were dissolved in sterile water and prepared fresh weekly for lead acetate (100 ppm) and indomethacin (1 mg/kg/day) (Sigma-Aldrich, Milwaukee, WI, USA). From PND 5-9 neonates were dosed once per day (10 µL/g body weight/day) with a vehicle, indomethacin (1 mg/kg), lead acetate (100 ppm), or indomethacin followed by lead acetate 30 min later using a modified gavage technique[46]. One mouse per sex, litter, and treatment group were randomly assigned and euthanized at PND 10, 15, or 21, with n = 3 mice/sex/age/treatment/strain for each assay. As per ethical use protocol, animals were euthanized with inhaled isofluorane followed by immediate decapitation. The brain was then carefully removed and placed in ice-cold PBS. For histochemical analysis, the left hippocampus was dissected and fixed for 24 h in 10% neutral buffered formalin followed by 70% ethyl alcohol before paraffin fixation. The right hemisphere sans cerebellum was flash-frozen whole and stored at -80 °C.
Immunohistochemistry
Formalin-fixed, paraffin-embedded hippocampi were sliced on a rotary microtome at 10 μm and mounted on Superfrost Plus slides (Azer Scientific, Germany). Briefly, slides were dewaxed in Histo-Clear II (Electron Microscopy Sciences, Halfield, PA, USA), followed by washes in 100% and 95% ethyl alcohol and phosphate-buffered saline (PBS). Antigen unmasking was accomplished using a heat-mediated citrate buffer, followed by incubation in 0.3% hydrogen peroxide for 30 min. All subsequent staining was performed using Sequenza-Coverplate racks (Thermo Scientific, Waldorf, Germany). Sections were permeabilized with PBS with Tween-20 and blocked with diluted normal serum (ABC Vectastain; Vector Laboratories, Burlingame, CA, USA). Slides were then incubated with anti-DAP12 primary antibody (4 µg/mL; unconjugated rabbit polyclonal IgG, Cat#orb156537, Biorbyt, Cambridge, UK), for 60 min at room temperature or overnight at 4 °C. Indirect labeling was then performed using biotinylated anti-rabbit IgG secondary antibodies and reagents from a high-sensitivity avidin-biotin kit with peroxidase-based detection (Vectastain Elite ABC-HRP kit, Peroxidase (Rabbit IgG), Cat# PK-6101; Vector Laboratories). Slides were visualized with diaminobenzidine (DAB) (DAB Substrate Kit, Peroxidase (HRP), Cat# SK-4100; Vector Laboratories), with a consistent DAB development time of 60 s for all slides, and counterstained with Harris’ Alum Hematoxylin. After sequential washing in ethyl alcohol and Histo-Clear, slides were coverslipped with Permount (Fisher Scientific, Fair Lawn, NJ, USA) and cured overnight before visualization.
Slides were visualized using a Leica DM1000 light microscope at 20× magnification (image resolution at 1.35 µm/pixel) with a SPOTTM Idea camera attachment and Advanced Imaging software. Regions of interest (ROI) were chosen at random along the dentate gyrus with the viewing frame containing as much tissue as possible and averaged for each of the 3-4 serially-sliced tissue sections per sample for a total of n = 6-8 ROIs/animal. Negative and positive controls were routinely employed to determine immunopositive DAP12 reactivity and to instruct background thresholding to minimize batch effects. Immunopositive DAP12 was determined over a manually predetermined background threshold via FIJI[47] analysis, blinded to sample grouping. Use of a Color Deconvolution plugin using predetermined vectors for DAB and hematoxylin [Supplementary Figure 1], and the % area DAP12 positive/ROI was determined along the dentate gyrus hippocampal subregion. Mean % area DAP12+ was averaged from n = 6-8 ROIs (technical replicates) per animal, for each of the n = 3 animals/exposure/sex/age/strain. All data are represented as the mean % over control at PND10 ± SEM. Raw, untransformed values for % DAP12 immunopositive staining at each age, and exposure are listed in Supplementary Figure 2, stratified by sex, strain, and age.
Quantitative real-time polymerase chain reaction
RNA Isolation and cDNA synthesis
Total RNA was purified from no more than 20 mg frozen brain tissue taken from the cortex of the right hemisphere of 3xTgAD or WT mice using the miRCURYTM RNA Isolation Kit - Cell & Plant (Exiqon Inc., Woburn, MA, USA) and associated Lysis Additive (Exiqon) specific for fatty tissue. Following homogenization and cell lysis, RNA was purified against a proprietary resin spin column separation matrix, washed with the associated buffers, and eluted at 50 µL, as per manufacturer’s instructions. Purified RNA was measured on a NanoDropTM One/OneC Microvolume UV-Vis Spectrophotometer (ThermoFisher Scientific, Madison, WI, USA) for purity and concentration, which was then adjusted to 5 ng/µL for subsequent reverse transcription.
For each sample, cDNA was synthesized from 10 ng purified RNA, in duplicate, using the Universal cDNA synthesis kit (Exiqon) and the following protocol: 60 min at 42 °C, followed by heat-inactivation of the reverse transcriptase for 5 min at 95 °C. Newly synthesized cDNA was then stored at -20 °C, and thawed and diluted to 80× in RNase-free water before RT-PCR.
Real-time PCR
MicroRNA relative quantification was performed on an iQ5 Multicolor Real-Time PCR Detection System thermocycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA), using 96-well PCR plates (VWR, Radnor, PA, USA). A master mix was prepared for each pre-designed LNATM PCR Primer set (Table 1, Exiqon), in conjunction with ExiLENT SYBR® Green master mix (Exiqon), for the target primers (miR-124, miR-132, miR-34a), endogenous reference primer (SNORD110), and UniSp6 RNA Spike-in control primer included in the kit. Each well consisted of 6 μL master primer mix and 4 μL cDNA template per sample, for a final well volume of 10 μL per sample, performed in duplicate. Reaction conditions included polymerase activation/denaturation at 95 °C for 10 min, 40 amplification cycles at 95 °C for 10 sec and 60 °C for 1 min (1.6 °C/s ramp rate), and a melt curve analysis consisting of a setpoint at 60 °C and endpoint at 95 °C, incrementally increasing by 0.5 °C with a 10 s dwell time.
Primer sequences of miRCURY LNA® PCR Primer sets (Exiqon)
| MicroRNA | RT primer sequence 5’-3’ | |
|---|---|---|
| hsa-miR-124-3p | UAAGGCACGCGGUGAAUGCC | target |
| mmu-miR-132-5p | AACCGUGGCUUUCGAUUGUUAC | target |
| hsa-miR-34a-5p | UGGCAGUGUCUUAGCUGGUUGU | target |
| SNORD110 | [UGACUUAUAUAUCUGUCAAUCCCCUGAGAGAUCACUGACGACUCCAUGUGUCUGAGCAA] | reference |
| UniSp6 | CUAGUCCGAUCUAAGUCUUCGA | control |
Automatically generated threshold cycle values (Ct) were evaluated using LinRegPCR[48] quality assessment to account for amplicon and assay efficiency. Fold change in relative miRNA expression compared to sex-, strain-, and age-matched controls was determined by the Pfaffl method, accounting for differential amplicon efficiencies calculated by LinRegPCR.
Statistical analysis
All statistics were carried out using the Statistical Analysis System (SAS Institute, Cary, NC, USA) or GraphPad PRISM (GraphPad, La Jolla, CA, USA) software. Initially, exploratory analyses were carried out using mixed modeling (PROC MIXED, SAS) with random intercepts to evaluate possible relationships between GxE variables with sex and age. Immunohistochemical (IHC) analyses were conducted in untreated mice stratified by strain modeling the fixed effects of age and sex. The fixed effects of treatment and age were then modeled, stratified by strain and sex. Possible interactions between these variables were also evaluated. Two-way ANOVA (PROC GLM, SAS) for age and sex was performed for each strain and treatment group for quantitative real-time polymerase chain reaction (qRT-PCR). Individual pairwise comparisons were made with a t-test corrected for multiple comparisons using the Holm-Sidak method or a Tukey’s studentized range distribution method. IHC data are represented as % mean DAP12 immunopositive staining/ROI over strain- and sex-matched controls at PND10 ± SEM, and qRT-PCR data are represented as mean fold change over sex-, strain-, and age-matched controls, with respect to endogenous reference gene levels, ± SEM. Statistical significance was determined at *P < 0.05 and *P < 0.01 for interactions.
Results
Sexually dimorphic hippocampal DAP12 expression during postnatal development is altered by early-life exposures
DAP12 expression on perinatal microglia is critical for phagocytosis of apoptotic neurons, and thus the development of healthy neuroimmune interactions[19,49]. To determine baseline expression of DAP12 during the postnatal period in this GxE mouse model immunohistochemical analysis was quantified in the hippocampus of untreated control mice, stratified by sex and genetic strain. In both WT and Tg males DAP12+ expression along the dentate gyrus was not significantly altered by age [Figure 1]. In WT females, DAP12 expression was significantly increased at both PND 15 and PND21 compared to expression levels at PND 10 [Figure 1A and C]. Similarly, untreated Tg females had increased DAP12 by PND 21 compared to PND 10 but was not significantly altered at PND 15 [Figure 1B and D]. Notably, while significant sex differences were detectable in WT mice at both PND 15 and 21 [Figure 1A and C], only PND 21 differed significantly by sex for DAP12 expression in the Tg mouse strain [Figure 1B and D]. These data suggest that DAP12 expression in the hippocampus during the postnatal period is sexually dimorphic regardless of genetic background and that this sexual dimorphism is dependent on age in WT mice (*Pinteraction < 0.0001).
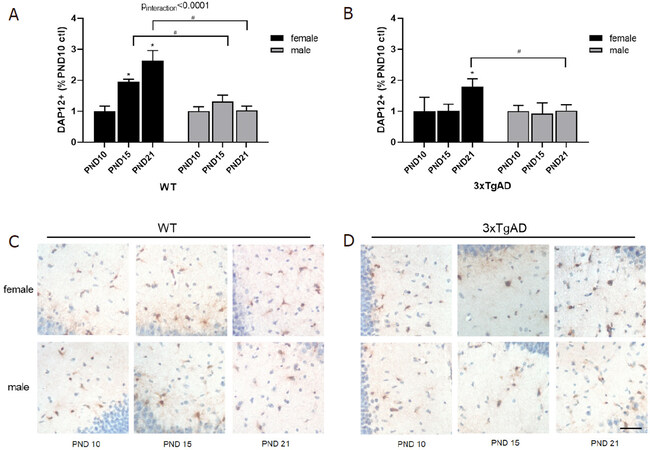
Figure 1. Quantification of hippocampal DAP12+ expression by sex over time in untreated WT (A) and 3xTgAD (B) mice. Representative images of hippocampal DAP12+ expression at PND10, 15, and 21 for females and males in WT (C) and 3xTgAD (D) hippocampus. Data expressed as mean % of sex- and strain-matched PND10 controls ± SEM. n = 3 mice/sex/age/strain. scale bar = 26 mm. Statistically significant at P < 0.05 (*-by age) (#-by sex), *P < 0.01 interaction (age*sex). PND: postnatal day; WT: wildtype; ctl: control
Postnatal Pb exposure significantly increased DAP12 expression in both WT and Tg female mice at PND 10 that then decreased over time [Figure 2A and C], with an inverse age-related trend compared to controls, suggesting a profound and persistent effect on female hippocampal DAP12 expression due to Pb. Interestingly, Tg males exposed to Pb also had significantly increased DAP12 at PND 10 that persisted with age [Figure 2D], whereas WT male expression was unaffected by Pb at all ages [Figure 2B].
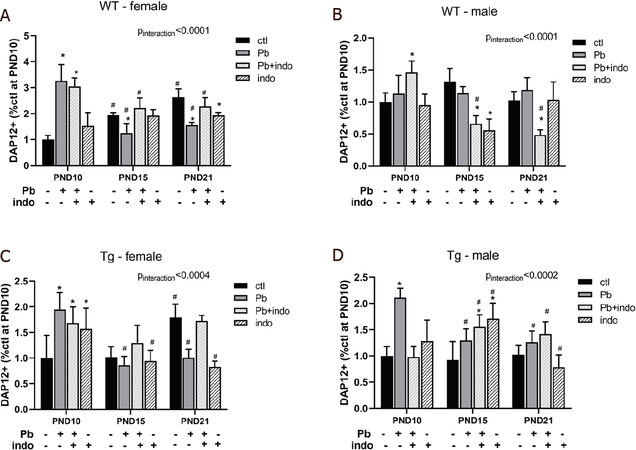
Figure 2. Quantification of hippocampal DAP12+ expression by treatment over time in WT females (A), WT males (B), 3xTgAD females (C), and 3xTgAD males (D). Data expressed as mean % of sex- and strain-matched PND10 controls ± SEM. n = 3 mice/sex/age/treatment/strain. P < 0.05 (*-by treatment, #-by age) and *P < 0.01 interaction (treatment*age) was considered statistically significant. PND: postnatal day; ROI: region of interest; WT: wildtype; Tg: 3xTgAD; Pb: lead; indo: indomethacin; ctl: control
Importantly, the addition of indomethacin before Pb exposure was able to moderate the long-term depression of DAP12 by Pb in Tg females [Figure 2C]. This is striking when contrasted with the nearly identical pattern of long-term depression by either Pb or indo alone in this group, suggesting that the combination of Pb and indo was able to rectify individual detriments over time. Although this indo+Pb rescue was similarly beneficial over time compared to Pb only exposure in WT females [Figure 2A], the individual effect of indo did not mimic that of Pb, suggesting the genetic proclivity to AD altered the female DAP12 response to postnatal indo exposure.
Due to the absence of temporal change in DAP12 expression in male mice regardless of strain [Figure 1], the significant effect of indo+Pb in differentially altering DAP12 expression over time in WT male mice [Figure 2B] would suggest that the inundation of both agents acted to modify typical developmental expression more so than either agent alone. However, male DAP12 expression at PND 15 was particularly sensitive to the effects of indo alone or in conjunction with Pb, with directionality dependent on genetic strain as expression decreased and increased in WT and Tg mice, respectively [Figure 2B and D]. This effect was not seen at PND 10 or 21, indicating a temporal vulnerability in male DAP12 expression patterning at PND 15 in response to the NSAID indo.
Postnatal toxicant exposure induced aberrant mouse brain miRNA expression profiles by PND 10
To assess early regulatory changes to neuroimmune signaling following postnatal toxicant exposure, as well as any sex bias and effect of genetic strain, we performed qRT-PCR for three distinct, yet interrelated microRNAs at the neuroimmune interface at PND 10 [Figure 3] and PND 21 [Figure 4]. At PND 10, one day after cessation of exposure, there was a significant interaction between microRNA expression levels in WT mice and treatment but not a significant effect of treatment itself [Figure 3A and B]. These data suggest, first, that atypical environmental cues due to exposure to either Pb, indo, or both affect miR-124 and miR-132 in a divergent manner than miR-34a very early during the postnatal period. Second, the inverse trends in these profile dynamics with sex would suggest that neuroimmune-related epigenetic regulation parallels the sexually dimorphic postnatal development of the neuroimmune system itself.
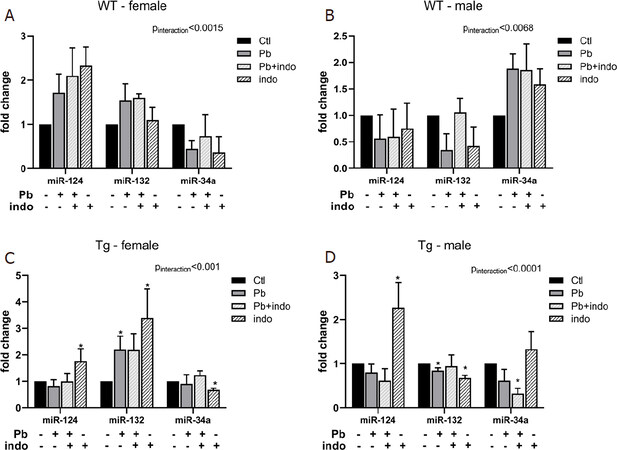
Figure 3. MicroRNA expression at PND 10 related to postnatal exposures in WT female (A), WT male (B), Tg female (C), and Tg male (D) mice. Quantification of microRNAs relative expression expressed as fold change ± SEM over sex-, strain-, and age-matched controls in WT (A, B) and Tg (C, D) mice. n = 3 mice/sex/age/treatment/strain. P < 0.05 (*-by treatment) and *P < 0.01 interaction (treatment*microRNA) was considered statistically significant. PND: postnatal day; Tg: 3xTgAD; indo: indomethacin; ctl: control; ns: not significant
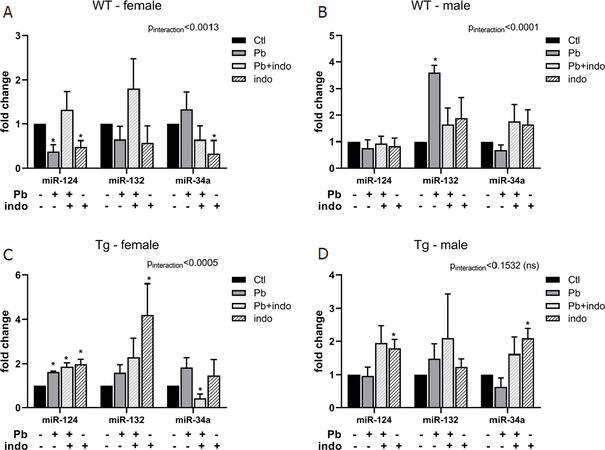
Figure 4. MicroRNA expression at PND 21 related to postnatal exposures in WT female (A), WT male (B), Tg female (C), and Tg male (D) mice. Quantification of microRNAs relative expression expressed as fold change ± SEM over sex-, strain-, and age-matched controls in WT (A, B) and Tg (C, D) mice. n = 3 mice/sex/age/treatment/strain. P < 0.05 (*-by treatment) and *P < 0.01 interaction (treatment*microRNA) was considered statistically significant. PND: postnatal day; Tg: 3xTgAD; indo: indomethacin; ctl: control; ns: not significant
While transgenic mice at PND 10 also exhibited significant interactions between treatment and microRNA expression, the effect of treatment varied significantly [Figure 3C and D]. Surprisingly, miR-124 expression levels were dramatically upregulated in both Tg females and Tg males only exposed to indo, whereas Pb and indo+Pb did not affect, suggesting that genetic proclivity to AD impacted the immediate miR-124 response to indo. On the other hand, miR-132 expression levels in Tg mice were similar in directionality by sex to that of the WT; specifically, increased in females and decreased in males by either Pb or indo. This would suggest that the directionality and altered expression of miR-132 at PND 10 consequent to either Pb or indo was a consequence of sex-related vulnerabilities to this particular epigenetic reprogramming.
The expression of miR-34a in Tg males was significantly decreased by the combination of Pb and indo but not by the agents individually [Figure 3D], in contrast to the overall trend of postnatal exposures increasing miR-34a in WT males [Figure 3B].
Postnatal toxicant exposure induced aberrant mouse brain miRNA expression profiles that persisted until PND 21
qRT-PCR analysis of microRNA expression at PND 21 revealed expression profiles in WT mice generally devoid of any carryover or long-term upregulation from PND 10, with the notable exception of dramatically increased miR-132 in WT males in response to postnatal Pb exposure [Figure 4B]. No other microRNAs were significantly affected by postnatal exposures in WT males at this time, nor was miR-132 upregulated at PND 10 in this group [Figure 3B], suggesting that Pb induced persistent epigenetic remodeling in WT males in the form of long term changes in miR-132 expression. In contrast, WT females exposed to indo exhibited decreased expression of both miR-124 and miR-34a, with miR-124 also decreased by Pb at PND 21 [Figure 4A].
Although no significant interaction between treatment and microRNA expression was detected in Tg males (Pinteraction = 0.1532), indo exposure increased the expression of both miR-124 and miR-34a at PND 21 [Figure 4D], with the increase in miR-124 persisting from PND 10 [Figure 3D]. This indo-related increase in miR-124 at both PND 10 and PND 21 was also detectable in Tg females [Figure 3C, Figure 4C], suggesting that in the transgenic strain indo exposure alone was enough to result in persistently elevated miR-124 regardless of sex. Importantly, Tg females exposed to Pb alone or in combination with indo also exhibited significantly increased miR-124 at PND 21 [Figure 4C], which was not seen in the earlier time point [Figure 3C]. These data suggest that increased miR-124 may act as a long-term response to postnatal exposures in Tg females, implicating this microRNA in the regulation of dysfunctional neuroimmune phenotypes observed in postnatal Pb-exposed Tg females with exacerbated AD pathologies[4]. Likewise, the decreased expression of miR-34a in Tg females with indo+Pb exposure at PND 21 [Figure 4C] mimicked that of Tg males at PND 10 [Figure 3D], suggesting similar epigenetic remediation of neuronal injury-related events, but more delayed.
Discussion
In the current study, we demonstrated a critical and previously unrecognized early alteration to epigenetic patterning related to the long-term regulation of neuroimmune phenotypes consequent to postnatal toxicant exposure in a GxE model of AD. Our results indicate that these exposure-related epigenetic changes parallel the established sexual dimorphism in microglial and neuronal postnatal development, thereby highlighting a critical period of neuroimmune vulnerability for sex bias not only in neurodevelopmental disorders but also neurodegenerative diseases like female-biased AD. Although previous studies have shown the critical role of miRNAs in sexually dimorphic brain development and AD[25,32], this study is the first to our knowledge to directly address immediate changes to the epigenetic landscape during the postnatal environment for GxE interactions with implications for chronic aging-related neurodegenerative diseases. The data presented here support our global hypothesis that disruption of neuroimmune development initiates sex-biased susceptibility to later-life neurodegeneration through GxE interactions and epigenetic regulation for a susceptible phenotype.
This GxE interaction, with a double hit from environmental toxicant exposure and genetic proclivity to AD, exacerbated and prolonged maladaptive microRNA expression profiles in response to an atypical developing environment for each sex. Contrary to the observed upregulation of DAP12 expression over time in females regardless of strain [Figure 1], postnatal Pb exposure significantly increased DAP12 immediately following cessation of exposure at PND 10, that then declined over time in both females [Figure 2A and C] as well as Tg males [Figure 2D]. These results not only corroborate reports of sexually dimorphic postnatal microglial development[36] using DAP12 as a marker for immature, phagocytic perinatal microglia[20], but also the heightened vulnerability of immature female microglia during this temporally critical maturation. Also, these data further corroborate our model of GxE vulnerability in a DOAD model of AD, as Pb-exposed Tg males also exhibited increased DAP12 expression [Figure 2D] during a postnatal window in which expression does not temporally fluctuate in healthy male mice [Figure 1]. Interpretation of these results is limited due to a lack of quantification of microglial number and phagocytosis or neuronal apoptosis, and future studies are warranted to delineate early physiological alterations to microglial-directed synaptic pruning resulting from atypical DAP12 expression, as well as the ramifications for total microglia numbers in the adult brain. Likewise, without the addition of colocalized staining with microglia-specific markers, cell-type-specific inferences cannot justifiably be made regarding DAP12 expression. However, reports of its expression are limited to perinatal microglia and its critical role in developmental phagocytosis of apoptotic neurons by microglia[19,49] lend credence to the interpretation that DAP12-related neuroimmune development was significantly altered in a sex-biased manner in this GxE model.
Importantly, this striking GxE effect on temporal DAP12 expression in Pb-exposed Tg females was accompanied by equally striking effects on epigenetic changes related to neuroimmune function. Namely, latent upregulation of miR-124 by Pb, indo, or both indo+Pb was observed in Tg females at PND 21 [Figure 4C]. The microRNA miR-124 critically regulates both microglial and neuronal development, maturation, and function[27]. Upregulation of miR-124 has been shown to promote microglial quiescence and restrict proliferation through inhibition of CEBPα, the transcription factor PU.1, and its downstream target CSF1R[29], while also promoting neuronal differentiation via repression of Sox9[28]. The female-specific expansion of amoeboid microglia populations around PND 30, preceded by a highly proliferative period to populate various brain regions[36], would suggest that a premature increase in miR-124, such as that observed in postnatally exposed Tg females at PND 21 [Figure 4C], may not only prematurely limit temporally-critical microglial maturation but also the proliferation and subsequent region-specific populations in the adult brain. While the additional investigation is needed to confirm any long-term changes to microglia and neuron population numbers, there is considerable evidence for a central role of miR-124 in our experimental DOAD model of AD. Long-term neuronal changes have been reported consequent to upregulation of miR-124, in which inhibition of REST by miR-124 delayed the maturation of NMDA receptors by retaining the prevalence of highly excitable GluN2B subunits, the ectopic form of which is associated with amyloid-β production[50,51]. Also, defects in glutamatergic synaptic function along with reduced TrkB expression have been reported in DAP12-deficient mice[21]. Given our previous research demonstrating a compensatory increase in hippocampal TrkB expression in Pb-exposed Tg female mice compared to WT at PND 120 that decreased with age and correlated with dysfunctional microglial phenotypes[4], the decrease in hippocampal DAP12 expression [Figure 2] and upregulation of miR-124 [Figure 4] reported here substantiate the hypothesis that perturbation to neuroimmune development promotes epigenetic changes for maladaptive microglia-neuron interactions involved in early AD pathologies. Furthermore, increased miR-124 at PND 21 in Tg females regardless of exposure type [Figure 4C] would suggest that this postnatal window represents a previously unseen window of vulnerability for female microglia development to any type of immune-altering exogenous insult, be it proinflammatory (Pb) or anti-inflammatory (indo).
Notably, primary cultures of immature microglia from female, but not male, whole mouse brain at PND 3 have been shown to exhibit increased expression of proinflammatory cytokines, such as IL-6 and IL-1β[52]. While indo was originally administered in the present study to inhibit the masculinizing effect of microglia-secreted PGE2 to elucidate its role in the sex-biased epigenetic modulation in developing males, the anti-inflammatory mechanism of action also inhibited pro-inflammatory cytokines like IL-1β. Thus, indo-related suppression of these pro-inflammatory signals critical for postnatal female microglial development also underscored the role of not only PGE2, but pro-inflammatory cytokines as well, in the epigenetic regulation of microglia in males and females, respectively. While suppression of PGE2 via indo exposure did not detectably “feminize” the vulnerability of male microglia to heightened neuroimmune epigenetic modification, indo exposure in females did reveal how timely modulation of microglia phenotype during a critical window of postnatal development significantly altered certain epigenetic cascades in neuroimmune development. For example, the significant and persistent upregulation of miR-132 in indo-exposed Tg females at PND 10 [Figure 3C] and PND 21 [Figure 4C] implies the presence of heightened sensitivity to cytokine-mediated miR-132 epigenetic reprogramming associated with neurite sprouting, synaptogenesis, and neurotransmission. Although miR-132 is upregulated for the first 2-4 weeks of postnatal development during a period of NMDA-dependent synaptic pruning[27], additional upregulation may disrupt the miR-132-promoted positive feedback loop in NMDA depolarization, while interfering in GABAergic interneuron maturation[53]. These data further corroborate the heightened GxE-related vulnerability and female bias for the epigenetic reprogramming of microglia during this postnatal window.
Further, given reports that the majority of microRNAs only persist for about five days without external stimulus[23], this upregulation of microRNA profiles persisting until or occurring at PND 21 would suggest the continuation of an atypical epigenetic stimulus despite the cessation of exposure. In further validation of the GxE female-specific vulnerability, postnatal exposures increased miR-124 at PND 10 in WT females [Figure 3A] but PND 21 was rectified via significant downregulation with Pb or indo exposure [Figure 4A]. This was in contrast to Tg females exposed to Pb, in which upregulation of miR-124 did not occur until PND 21 [Figure 4C], indicating that a rectifying epigenetic signal for Tg miR-124 was not present for a similarly-vulnerable population of immature female microglia as with WT females despite a parallel trend for significant reductions in hippocampal DAP12 expression [Figure 2]. The current study is limited by a hypothesis-driven bias in the neurimmiRs selected for analysis in our GxE model for AD, and future analyses might benefit from wider microarrays to further characterize this window of heightened sensitivity to epigenetic reprogramming for female neuroimmune development. While little is known regarding this subtle and life-long maladaptive neuroimmune phenotype, the data presented here demonstrate that changes to the epigenetic landscape are detectable early in postnatal development and are significantly biased by sexual dimorphism for neuroimmune development during this time.
As hypothesized, our study demonstrated a clear female bias in the exacerbating effect of GxE interactions in early epigenetic regulation promoting later life maladaptibility and neuroimmune dysfunction, which we have previously correlated with heightened susceptibility to AD[4,5]. We have also shown that, although the most detrimental phenotype occurred in Pb-exposed Tg females, there was evidence to suggest that this DOAD model may be useful in investigating toxicant- and timing-specific windows of vulnerabilities for other sex-biased adult diseases. Ultimately, investigation of these early perturbations to epigenetic regulation of cellular phenotype could reveal thresholds of adaptability consequent to atypical developmental conditions, and potential identification of biomarkers for susceptibility.
Declarations
AcknowledgmentsThe authors would like to acknowledge Ishmael Gomez for his assistance with portions of immuno-histochemistry. We would also like to acknowledge Dr. Qun Lu and the Harriet and John Wooten Laboratory for Alzheimer’s and Neurodegenerative Diseases Research, our funding source, for transgenic mouse accessibility, as well as the East Carolina University Department of Comparative Medicine for their excellent husbandry and care of our animals.
Authors’ contributionsMade substantial contributions to the conception and design of the study: vonderEmbse AN, DeWitt JC
Assisted with technical expertise and dissections: vonderEmbse AN, Hu Q
Performed immunohistochemistry: vonderEmbse AN
Performed data analysis and interpretation: vonderEmbse AN
Provided administrative, technical, and material support: DeWitt JC
Involved in the writing and editing of the manuscript: vonderEmbse AN, DeWitt JC
Discussed the results and commented on the manuscript: vonderEmbse AN, Hu Q, DeWitt JC
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Harriet and John Wooten Laboratory for Alzheimer’s and Neurodegenerative
Diseases Research (https://www.ecu.edu/cs-dhs/wootenlab/). The funding body provided access to the 3xTgAD transgenic and wildtype mice used in the study.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateAll handling and experimental manipulations were carried out in accordance with procedures approved by the East Carolina University Institutional Animal Care and Use Committee (IACUC), and were conducted in accordance with the ARRIVE guidelines and the National Institutes of Health guide for the care and use of laboratory animals (NIH publication No. 8023, revised 1978).
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Fox DA, Grandjean P, de Groot D, Paule MG. Developmental origins of adult diseases and neurotoxicity: epidemiological and experimental studies. Neurotoxicology 2012;33:810-6.
2. Dietert RR. Role of developmental immunotoxicity and immune dysfunction in chronic disease and cancer. Reprod Toxicol 2011;31:319-26.
3. Miller DB, O’Callaghan JP. Do early-life insults contribute to the late-life development of Parkinson and Alzheimer diseases? Metabolism 2008;57 Suppl 2:S44-9.
4. vonderEmbse AN, Hu Q, DeWitt JC. Dysfunctional microglia:neuron interactions with significant female bias in a developmental gene x environment rodent model of Alzheimer’s disease. Int Immunopharmacol 2019;71:241-50.
5. vonderEmbse AN, Hu Q, DeWitt JC. Developmental toxicant exposure in a mouse model of Alzheimer’s disease induces differential sex-associated microglial activation and increased susceptibility to amyloid accumulation. J Dev Orig Health Dis 2017;8:493-501.
6. Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 2009;118:475-85.
7. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci 2008;28:8354-60.
8. Lopes KO, Sparks DL, Streit WJ. Microglial dystrophy in the aged and Alzheimer’s disease brain is associated with ferritin immunoreactivity. Glia 2008;56:1048-60.
9. Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016;353:aad8670.
10. Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD. Microglia and memory: modulation by early-life infection. J Neurosci 2011;31:15511-21.
11. Bilbo SD. Early-life infection is a vulnerability factor for aging-related glial alterations and cognitive decline. Neurobiol Learn Mem 2010;94:57-64.
12. Bland ST, Beckley JT, Young S, Tsang V, Watkins LR, et al. Enduring consequences of early-life infection on glial and neural cell genesis within cognitive regions of the brain. Brain Behav Immun 2010;24:329-38.
13. Bilbo SD, Barrientos RM, Eads AS, Northcutt A, Watkins LR, et al. Early-life infection leads to altered BDNF and IL-1beta mRNA expression in rat hippocampus following learning in adulthood. Brain Behav Immun 2008;22:451-5.
14. Thrash JC, Torbett BE, Carson MJ. Developmental regulation of TREM2 and DAP12 expression in the murine CNS: implications for Nasu-Hakola disease. Neurochem Res 2009;34:38-45.
15. Colonna M, Wang Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci 2016;17:201-7.
16. Painter MM, Atagi Y, Liu CC, Rademakers R, Xu H, et al. TREM2 in CNS homeostasis and neurodegenerative disease. Mol Neurodegener 2015;10:43.
17. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, et al; Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer’s disease. N Engl J Med 2013;368:117-27.
18. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011;333:1456-8.
19. Wakselman S, Béchade C, Roumier A, Bernard D, Triller A, et al. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci 2008;28:8138-43.
20. Roumier A, Pascual O, Béchade C, Wakselman S, Poncer JC, et al. Prenatal activation of microglia induces delayed impairment of glutamatergic synaptic function. PLoS One 2008;3:e2595.
21. Roumier A, Béchade C, Poncer JC, Smalla KH, Tomasello E, et al. Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J Neurosci 2004;24:11421-8.
22. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009;19:92-105.
23. Gantier MP, McCoy CE, Rusinova I, Saulep D, Wang D, et al. Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res 2011;39:5692-703.
24. Reid G, Kirschner MB, van Zandwijk N. Circulating microRNAs: association with disease and potential use as biomarkers. Crit Rev Oncol Hematol 2011;80:193-208.
25. Bian S, Sun T. Functions of noncoding RNAs in neural development and neurological diseases. Mol Neurobiol 2011;44:359-73.
26. Guedes J, Cardoso AL, Pedroso de Lima MC. Involvement of microRNA in microglia-mediated immune response. Clin Dev Immunol 2013;2013:186872.
27. Soreq H, Wolf Y. NeurimmiRs: microRNAs in the neuroimmune interface. Trends Mol Med 2011;17:548-55.
28. Cheng LC, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci 2009;12:399-408.
29. Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α-PU.1 pathway. Nat Med 2011;17:64-70.
30. Tan L, Yu JT, Hu N, Tan L. Non-coding RNAs in Alzheimer’s disease. Mol Neurobiol 2013;47:382-93.
31. Wang WX, Huang Q, Hu Y, Stromberg AJ, Nelson PT. Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: white matter versus gray matter. Acta Neuropathol 2011;121:193-205.
32. Gillet V, Hunting DJ, Takser L. Turing revisited: decoding the microRNA messages in brain extracellular vesicles for early detection of neurodevelopmental disorders. Curr Environ Health Rep 2016;3:188-201.
33. Masoud AM, Bihaqi SW, Machan JT, Zawia NH, Renehan WE. Early-Life exposure to Lead (Pb) alters the expression of microRNA that target proteins associated with Alzheimer’s disease. J Alzheimers Dis 2016;51:1257-64.
34. Nelson LH, Lenz KM. The immune system as a novel regulator of sex differences in brain and behavioral development. J Neurosci Res 2017;95:447-61.
35. Lenz KM, Nugent BM, Haliyur R, McCarthy MM. Microglia are essential to masculinization of brain and behavior. J Neurosci 2013;33:2761-72.
36. Schwarz JM, Sholar PW, Bilbo SD. Sex differences in microglial colonization of the developing rat brain. J Neurochem 2012;120:948-63.
37. Schwarz JM, Bilbo SD. Sex, glia, and development: interactions in health and disease. Horm Behav 2012;62:243-53.
38. Morgan CP, Bale TL. Sex differences in microRNA regulation of gene expression: no smoke, just miRs. Biol Sex Differ 2012;3:22.
39. Guo L, Zhang Q, Ma X, Wang J, Liang T. miRNA and mRNA expression analysis reveals potential sex-biased miRNA expression. Sci Rep 2017;7:39812.
40. McCarthy MM, Nugent BM. At the frontier of epigenetics of brain sex differences. Front Behav Neurosci 2015;9:221.
41. Bhattacharjee S, Zhao Y, Dua P, Rogaev EI, Lukiw WJ. microRNA-34a-Mediated Down-regulation of the microglial-enriched triggering receptor and phagocytosis-sensor TREM2 in age-related macular degeneration. PLoS One 2016;11:e0150211.
42. Rokavec M, Li H, Jiang L, Hermeking H. The p53/miR-34 axis in development and disease. J Mol Cell Biol 2014;6:214-30.
43. Li LH, Tu QY, Deng XH, Xia J, Hou DR, et al. Mutant presenilin2 promotes apoptosis through the p53/miR-34a axis in neuronal cells. Brain Res 2017;1662:57-64.
44. Ajmone-Cat MA, Bernardo A, Greco A, Minghetti L. Non-Steroidal anti-inflammatory drugs and brain inflammation: effects on microglial functions. Pharmaceuticals (Basel) 2010;3:1949-65.
45. Du ZY, Li XY. Inhibitory effects of indomethacin on interleukin-1 and nitric oxide production in rat microglia in vitro. Int J Immunopharmacol 1999;21:219-25.
46. Butchbach ME, Edwards JD, Schussler KR, Burghes AH. A novel method for oral delivery of drug compounds to the neonatal SMNDelta7 mouse model of spinal muscular atrophy. J Neurosci Methods 2007;161:285-90.
47. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676-82.
48. Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 2009;37:e45.
49. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 2005;201:647-57.
50. Rodenas-Ruano A, Chávez AE, Cossio MJ, Castillo PE, Zukin RS. REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat Neurosci 2012;15:1382-90.
51. Bordji K, Becerril-Ortega J, Nicole O, Buisson A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-ß production. J Neurosci 2010;30:15927-42.
52. Crain JM, Nikodemova M, Watters JJ. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J Neurosci Res 2013;91:1143-51.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
vonderEmbse AN, Hu Q, DeWitt JC. Postnatal toxicant exposure in 3xTgAD mice promotes gene x environment-related early alterations to neuroimmune epigenetic profiles. Neurosciences 2020;7:345-59. http://dx.doi.org/10.20517/2347-8659.2019.29
AMA Style
vonderEmbse AN, Hu Q, DeWitt JC. Postnatal toxicant exposure in 3xTgAD mice promotes gene x environment-related early alterations to neuroimmune epigenetic profiles. Neuroimmunology and Neuroinflammation. 2020; 7(4): 345-59. http://dx.doi.org/10.20517/2347-8659.2019.29
Chicago/Turabian Style
vonderEmbse, Annalise N., Qing Hu, Jamie C. DeWitt. 2020. "Postnatal toxicant exposure in 3xTgAD mice promotes gene x environment-related early alterations to neuroimmune epigenetic profiles" Neuroimmunology and Neuroinflammation. 7, no.4: 345-59. http://dx.doi.org/10.20517/2347-8659.2019.29
ACS Style
vonderEmbse AN, Hu Q.; DeWitt JC. Postnatal toxicant exposure in 3xTgAD mice promotes gene x environment-related early alterations to neuroimmune epigenetic profiles. Neurosciences. 2020, 7, 345-59. http://dx.doi.org/10.20517/2347-8659.2019.29
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.