
Inflammation in the anterior visual pathway in multiple sclerosis: what do the animal models teach us?
 0
0Abstract
A provocative and overly reductive mantra is that “the back of the eye is the front of the brain”. Retinal imaging techniques that take advantage of this “window” to the central nervous system can provide valuable information regarding injury to the nervous system with relative ease and with a limited burden to patients. The retina develops embryonically as part of the neuroectoderm, is made up principally of neurons and their supporting cells, and is synaptically tied to the central nervous system (CNS). This has led to significant interest in using retinal health as a biomarker for brain health - given the relatively limited accessibility of brain tissue in chronic neurodegenerative diseases that progress over decades. The retina is not truly part of the CNS, and as with much of brain imaging - the grounds for asserting the pathological specificity of retinal imaging is limited. Biophotonics-based methods such as optical coherence tomography indirectly provide an opportunity to evaluate retinal neurodegeneration, while autopsy studies, histology and immunohistochemistry predominate as the methods that collect direct pathological data. Our understanding of pathological retinal lesions characteristic of demyelinating diseases, specifically diseases showing anterior visual pathway involvement, has grown significantly in recent years. However, much of the underlying pathobiology of injury remain unexplored. This review aims to highlight the major pathological features of the retina in multiple sclerosis, and its most used animal models (experimental autoimmune encephalomyelitis and cuprizone), with a particular focus on n the role of inflammation.
Keywords
Introduction
Multiple sclerosis is an inflammatory and neurodegenerative disease of the central nervous system (CNS) involving the retina, the anterior visual pathway (AVP) and less frequently the posterior visual pathway, that in most cases leads to an inexorable neurological disability[1,2]. The AVP can be impacted both directly, as a consequence of acute demyelination of the optic nerve [acute demyelinating optic neuritis (ON)]/chronic inflammation[3], and potentially indirectly, as a consequence of trans-synaptic processes affecting the posterior visual pathway[4]. The retina is a unique structure in the nervous system in that it is the only part of the nervous system where axons can be directly viewed[5]. Furthermore, given that the retina is generally completely devoid of myelin, and that the axons visible in the retina are myelinated after exiting the globe, the retina represents a great model to evaluate the effect of distal demyelination on axonal health and survival. Moreover, the retina exhibits all the pathological hallmarks of multiple sclerosis (MS) (except for demyelination), such as gliosis/fibrosis, perivascular infiltration of immune cells, innate immune activation and blood-brain/retinal barrier disruption[6]. Here, we review retinal pathology and in particular the role of inflammation in ON, in MS patients’ eyes without previous ON, and in the most used animal models of demyelination (experimental autoimmune encephalomyelitis, EAE, and cuprizone); we discuss briefly how optical coherence tomography (OCT) and other imaging tools have improved our ability to directly, longitudinally monitor retinal pathology in brain diseases in the absence of samples for “routine” pathological analysis. We summarize what is understood (and what is assumed) about the underlying mechanisms.
Optic neuritis in MS
ON is an episode of optic nerve inflammation, resulting in demyelination, conduction block, and consequent subacute vision loss. It is frequently encountered in relapsing-remitting multiple sclerosis (RRMS) (around 70% of patients experience it) and is the presenting symptom in up to 25% of patients[3]. ON is also a central feature of the less common neuromyelitis optica (NMO) and myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease, and it can develop in conjunction with acute disseminated encephalomyelitis, sarcoidosis, Lyme disease, syphilis and vasculitis. ON can also be provoked by a viral illness or immunization. Axonal damage is a less prominent but well-recognized feature of demyelinating lesions, with a direct relation between transected axons and degree of inflammation within the lesion[7]. The timing and causes of axonal loss following a demyelinating injury are unclear. Direct damage via immune/glial cells[7-9], secondary ischemic injury and loss of trophic support from myelin have all been implicated, and each may be operative to a different degree. With acute transection, animal studies have established that dying back to the retinal ganglion cell (RGC) soma may take several months[10]. The extent and retinal manifestations of retrobulbar demyelinating injury are highly dependent on the location of the main lesion. Indeed in the acute stage, an ophthalmologic examination reveals that up to two-thirds of patients present with a normal-appearing optic disc (retrobulbar ON), while a third show a swollen optic disk (papillitis, bulbar ON)[11]. Visual evoked potentials (VEPs) p100 delay, asymmetry and wave morphology alteration, together with associated pain with eye movement, relative afferent pupillary defect, magnetic resonance imaging (MRI) of the optic nerve, deficits on static and kinetic visual field testing (which usually follows the topography of the retinal nerve fiber layer (RNFL), with consequent central, cecocentral, altitudinal, and arcuate deficits) and performance on low contrast visual sensitivity and color discrimination, are methods that can determine if a patient with a normal-appearing funduscopic examination is experiencing an acute inflammatory ocular event[12-14].
Optic nerve injury is more common than clinically identifiable events with nearly 100% of patients exhibiting AVP demyelination at the end of life[15].
OCT, a non-invasive imaging technology that has been used to develop methods for quantitative assessment of the inner retina, has produced significant improvements in the strength of our knowledge regarding the timing of inflammation and neurodegeneration in MS optic neuritis (MSON) eyes[16]. Almost twenty years ago, using time-domain OCT, it was established that the peripapillary RNFL (pRNFL) and macular volume (MV) are abnormal in ON[17,18]. The pRNFL, which represents the intraocular, non-demyelinated axons of RGCs around the optic nerve head, is significantly reduced three months after the acute event, and the thinning terminates approximately after six months[19,20]. The temporal quadrant of the retina, containing the papillomacular bundle, which subserves central vision, is the most affected by ON and MS[21]. The papillomacular bundle is composed primarily of parvocellular axons. A plausible hypothesis suggests that size-selective loss is a consequence of the failure of small axons to remyelinate. RNFL thinning after a single ON episode is approximately 5 to 40 μm (averaging at 10-20 μm)[22,23]. In contrast, eyes with more than one previous ON show a significant thinning compared with the single ON ones[22]. During acute retrobulbar ON, OCT reveals more RNFL swelling than can be appreciated on routine fundoscopy with up to 82% demonstrating swelling[24], possibly due to axoplasmic flow stasis. Recently, the introduction of segmentation algorithms for MV has enabled the quantification of the RGCs (macular ganglion cell-inner plexiform layer, GCIPL) integrity[25]. Being less affected by axonal swelling due to inflammation and edema in acute ON, GCIPL evaluation is a more promising tool to track axonal loss and potentially the effect of neuroprotective/remyelinating drugs in the acute stages compared to pRNFL. pRNFL edema is present at clinical onset (being almost absent in the macula) and disappears after one month[26]. Volumetric evaluation of macula and retinal layers reveals a biphasic trend, characterized by atrophy of inner retinal layers (macular RNFL or mRNFL and combined ganglion cell and inner plexiform layer (GCIP) and swelling of the outer layers in the first two months, followed by edema reduction in the outer layers and progressive neurodegeneration of mRNFL and GCIP in the subsequent four months. As expected, GCIP thinning was confirmed to be the best predictor of the short-term visual deficit outcomes, such as low-contrast vision, visual fields, and color vision recovery[26].
Despite the findings described above, we must, however, keep in mind that all this information is to some extent the result of speculations, due to unavailability of tissue samples for pathological retinal evaluations. Therefore, there is a lack of certainties regarding the real meaning (cell infiltration/edema and axonal loss rather than neuronal atrophy) of the described acute thickening and subsequent subacute thinning of the retinal layers. Despite these aspects, monitoring retinal changes due to ON remains a useful way to assess therapeutic strategies targeting neurodegeneration during demyelinating CNS disease.
Multiple sclerosis
In patients with MS, retinal periphlebitis, optic disc pallor, and disruption of the inner retina (slits) can be visualized on funduscopic examination[27,28]. A pale optic disc reflects a pattern of atrophy in the pRNFL[28]. Disruptions of the RNFL will manifest functionally in decreases in visual function. As already pointed out, it is upon histological and immunohistochemical examination that the cellular and molecular processes underlying retinal changes in demyelinating disease can be better understood. Unfortunately, the assessment of demyelinating injury in human disease is encumbered by the limited availability of relevant tissue for evaluation. Most tissue samples in MS come from autopsy specimens at the end of life (and end of the disease course) or from brain biopsies for acute fulminant episodes that are characterized by severe inflammation. These samples, therefore, do not provide unimpeachable data regarding the temporal course of the demyelinating process. Furthermore, frequently the works regarding retinal pathology in MS describe a small number of cases and do not provide an in-depth clinical history of the patients described. In 1983, Toussaint et al.[15] described in a pathological study 32 cases of MS, including 15 cases in which the retina was investigated. Abnormal fibrillar material along the retinal veins was described in 6 of these 15 patients, with no correlation with the duration of the disease. This regularly thickened material, staining green with Masson’s trichrome and unstained with glial fibrillary acidic protein, was distributed around all the retinal veins, from the peripapillary region to the equator. Rarefaction of macular RGCs, as well as a reduction in the number of RNFL axons, were described as obvious in most of the analyzed eyes. Interestingly, in two of these specimens, they found some nodular lymphoplasmacytoid infiltrates surrounding retinal vessels, with one invading the vitreous. Kerrison et al.[29] presented a pathological retinal evaluation of 29 patients (26 MS, 3 NMO), mainly focused on the retinal vasculature. Initial vessel evaluation in histopathological sections in the horizontal plane through the pupil and optic nerve showed only one case of inflammation (among 26) while studying the trypsinized vascular network of the caps, inflammation was evident in 20% of cases (4 of 20), 3 of them, bilateral. Atrophy of RNFL and ganglion cell layer was present in 73% of cases. There was no evidence of venous sclerosis, while choroiditis was present in 11.5% of the cases[29]. In 2001, Nikos Evangelou et al.[30] published a work showing a pattern of axonal loss and a cell-specific decrease in neuronal density that offered insight to a pathological mechanism of MS through the examination of autopsy-collected tissue of the lateral geniculate nucleus, optic nerve, and optic tract. Through imaging tissue preparations via a pixel intensity cluster program to count axons, it was found that patients with MS had smaller mean cross-sectional areas in the optic nerve and optic tract and significantly reduced axonal density. Neuronal density was also significantly reduced in the parvocellular layer of the lateral geniculate nucleus of affected patients, demonstrating a size susceptibility of axons in the anterior visual pathway to damage. In patients with MS with and without ON, pathological alteration of tissues has been described in the iris, retina (peripapillary and macular), optic disc, and optic nerve head[18,22,31]. Green et al.[6] completed a systematic pathological evaluation of retinas from 82 patients with MS in which they demonstrated that retinal inflammation is pervasive and prominent in the disease (especially perivascularly). The involved vessels lie in the nerve fiber layer of the retina (the primary layer of interest in MS). Mononuclear lymphocytes may be found near retinal vasculature while HLA-DR-positive cells can be found in the iris, RGC layer, inner nuclear layer, optic disc and optic nerve. Astrocyte proliferation could be seen both around vessels and in the optic nerve head. Furthermore, they found that both tissue atrophy and inflammation could be detected in deeper layers of the retina (including the inner nuclear layer). Additionally, optic nerve and optic disc astrocytic gliosis and evidence of gliomesodermal reaction can also be expected to be seen. Finally, Confocal microscopy of Z01 and occludin protein stained preparations of retinal and optic nerve vasculature reveals tight junction opening that may be associated with glial cell reactions. Morphological changes are also found in the eyes of individuals with MS - optic discs appear indented or “cupped”[6]. Although their work presents some limitation, such as the reduced group of eyes (eight) examined with immunohistochemistry and the paucity of previous clinical history, the description of inner nuclear layer atrophy together with the reduced presence of mononuclear cells represented strong evidence in favor of retrograde trans-synaptic degeneration, confirming previous results suggesting this phenomena[32,33].
A comparison between brain and retinal pathology is complicated by the fact that the human retina is usually devoid of myelin. Despite this limitation, the analysis of different patterns of inflammation and neuronal damage among different anatomical structures has considerable importance to achieve a complete understanding of the pathophysiology of the disease. Active white matter demyelinating lesions are characterized by the presence of macrophages containing cytoplasmic early myelin degradation products both at the lesion edge and throughout the lesion[34]. These lesions are extremely rare in chronic MS and frequent in the active forms of MS (RRMS, SPMS with relapses), probably representing the pathologic substrate of the attacks[35]. Perivascular inflammation, broadly described in brain lesions as containing T lymphocytes and fewer B lymphocytes, plasma cells and granulocytes[36], is a common feature with MS retina, where the vessels running through the RNFL are described as showing a perivascular inflammation involving lymphocytes and other mononuclear cells, apparently phagocytic. Localized perivascular infiltrate in the retina is more frequent in RRMS and SPMS (29%) than PPMS (5%)[6], as well as acute demyelinating lesions. As suggested by Calabresi et al.[37], these findings underline the hypothesis that immune response could be directed against other antigens, unrelated to myelin.
Neuronal damage in the MS brain has been described as a consequence of acute local inflammation, decreasing with lesion chronicity[38,39]. Although axonal loss is most severe in active MS plaques followed by smoldering lesions, inactive plaques and normal-appearing white matter[7,40], a greater extent, at a lower level, of cell loss is present in SPMS and PPMS[38] than RRMS. Similarly, reduction in the population of RGCs was described in 79% of chronic MS retinal samples, while when the cases were classified as acute, RGC loss was evident in 55% of eyes[6]. These data suggest that the already described pronounced diffused inflammatory reaction during the progressive disease course in the normal-appearing white matter, (perivascular cuffs of mononuclear cells, diffuse infiltration of the tissue by T lymphocytes and profound microglial activation)[41], may have the same consequences of the chronic retinal inflammation described in the progressive cases of MS.
OCT imaging of the RNFL is qualitatively comparable to histological measurement in healthy populations[6,31]. As previously mentioned, it is well documented that regardless of a diagnosis of ON, people with MS develop RNFL loss. Utilizing OCT, various groups published about RNFL thinning in the eyes of MS patients with no previous ON[42-45]. The integration of OCT into cutting-edge MS research now allows a more accurate view of structure-function relations in the understanding of the pathophysiology of this enigmatic disease[46]. Loss of myelin in diseases such as MS results in the disruption of nerve signal transmission, damage to the axon, and finally neurodegeneration. This chronic process appears to underlie and be able to predict short- and medium-term disability in MS[47,48]. In chronic disease, some subtypes of neurons/axons show increased relative vulnerability to loss - especially those with a smaller axonal diameter and soma size[49]. In a metanalysis, RNFL loss due to neurodegeneration was quantified as 7 μm (average)[46], while the RNFL loss per year, obtained studying 299 patients for up to 4 years, was described to be 2 μm. Interesting works combining MRI and OCT tried to investigate the processes behind the described chronic axonal loss. Gabilondo et al.[4] evaluated the association between the damage to the anterior and posterior visual pathways, showing the presence of retrograde and anterograde trans-synaptic degeneration, throw volumetric measurement of the synaptic relay between anterior and posterior pathway (thalamus), optic radiation lesions and OCT. Petracca et al.[50] assessed the presence of relations between OCT and cortical lesions in MRI (double inversion recovery and phase-sensitive inversion recovery sequences) in a cohort of primary progressive MS patients and showed an association of cortical lesions with GCIPL and MV, but not with RNFL. More recently, Caverzasi et al.[51] described models based on OCT and MRI metrics able to predict visual disability measured with low contrast letter acuity.
Vacuolar macular changes, predominantly in the inner nuclear layer (INL) of the perimacular rim[52], are found in 0.8%-6% of MS patients[52-54], especially in eyes with a history of acute ON, and in 20% of patients with neuromyelitis optica, where microcystic macular edema (MME) is found exclusively in eyes with prior ON[55]. However, MME is not unique to demyelinating disease and has also been described in prominent vitreous traction, compressive optic neuropathy, glaucoma, age-related macular edema, neurofibromatosis 1-associated optic nerve glioma, dominant optic atrophy, group 2A idiopathic juxtafoveolar retinal telangiectasis, Leber’s hereditary optic atrophy, tamoxifen retinopathy, trauma and hydrocephalus[56-59]. While it has been attributed to INL microglial activation leading to blood-retinal barrier breakdown[53], others have postulated that MME could be a result of Müller cell dysfunction[54] or trans-synaptic retrograde degeneration[57,60]. All these explanations could certainly be compatible with different contributory mechanisms acting on patient-specific factors[55]. In MS patients, the presence of MME has been associated with worse disability and disease progression, as well as lower visual acuity[52,53], an increased INL thickness[52], and a thinner retinal nerve fiber layer[53]. INL volume reflects response to immunotherapy[61] and might be a useful tool in stratification of patients with progressive MS for treatment choice[62]. INL swelling seems to be most prominent during early disease, while later in the disease course, the rate of thickening declines until such a time that atrophy begins to supersede the swelling[63]. In addition to MME, macular edema can be found in eyes from patients with MS when accompanied by comorbid clinical uveitis or as a consequence of treatment with fingolimod, a sphingosine-1-phosphate receptor (S1PR) modulator, which was the first FDA-approved oral drug for the treatment of relapsing forms of MS. Inflammation of the uveal tract can be found in 1-3% of patients with MS, and it is ten times more common in MS patients than in the general population. It is typically intermediate (pars planitis), chronic and bilateral, and it can complicate with retinal neovascularization and/or detachment, epiretinal membrane formation and cystoid ME[64]. Interestingly, approximately 10% of patients with intermediate uveitis will develop MS. Cystoid ME can also be the consequence of some MS therapies, such as fingolimod. ME is found in 0.5% of MS patients treated with fingolimod, with a dose-dependent incidence, and normally ceasing upon discontinuation of the drug[65]. Since the S1PR acts on the cell cytoskeleton and on intercellular junctions[66], it regulates endothelial barrier integrity. Fingolimod-associated macular edema may then be caused by inner blood-retinal barrier breakdown through receptor internalization[67].
Worthy of mention in this context are the recent works showing that the visuomotor system could be used to track motor dysfunction in MS through the monitoring of fixational microsaccades[68,69].
Neuromyelitis optica
NMO is an inflammatory demyelinating disease of the CNS caused by antibodies against aquaporin-4 expressed on astrocytes, which causes vision loss and paralysis. ON is the cause of visual disability in NMO. Fundoscopy shows disc atrophy and vascular changes with “frosting”[70]. Eighty percent of NMO eyes experience severely reduced visual acuity (< 20/200) during an acute attack, and most suffer substantial permanent vision loss due to incomplete recovery with greater axonal degeneration (average 31.1 μm) than MS, as measured by OCT[71]. Interestingly the OCT pattern of the RNFL thinning following ON is different if compared with MS, presenting a global pattern of loss with less predilection for the temporal quadrant[70,72]. Small-diameter axons are spared likely because of their dimensions[46]. Both the fundoscopy vascular changes, (attenuation of the peripapillary vascular tree and focal arteriolar narrowing) and the OCT pattern in NMO outline the possibility of a vascularly mediated damage in NMO[71]. Fellow eyes without previous history of ON do not exhibit RNFL thinning[70,73]. Recent works have shown morphological foveal and perifoveal alterations in NMO patients, explained by high-level expression of aquaporin-4 channels in Müller cells[74,75].
Experimental autoimmune encephalomyelitis
Experimental autoimmune encephalomyelitis (EAE) is the most commonly used experimental model comprising all three key clinical features of MS: inflammation, demyelination and axonal loss - with a diminished demyelinating component.
The methods of EAE induction are highly heterogeneous and have a major influence on disease pathology and response to diverse interventions. Traditionally an adjuvant, which contains bacterial components such as attenuated mycobacterium tuberculosis and activates the innate immune response, sensitizes EAE animals to administered myelin antigens such as proteolipid protein (PLP), myelin basic protein (MBP), oligodendrocyte-specific protein or myelin oligodendrocyte glycoprotein[76-80]. For example, after immunization with PLP, the first attack of a relapsing-remitting disease course starts as early as nine days post-induction (dpi) and peaks around twelve to fifteen dpi. From day eleven, 43.9% show unilateral ON and 41.5% bilateral ON; only 14.6% of animals do not show inflammatory infiltrate consistent with ON[76,77]. MOG-induced EAE can have a similar time of onset, but in contrast, the disease is usually monophasic, with the peak of disease (ca. 15-25 dpi) being later and the optic nerve inflammation mostly bilateral[78]. However, the onset of neurodegeneration and demyelination can similarly be as soon as eleven dpi. In all EAE models, the inflammation severity in the afferent visual system is correlated with subsequent demyelination and RGC loss[76-78], the time frame and distribution of disease components; however, it seems to be also influenced by multiple other factors such as the concentrations of antigens or the mouse strain[81,82]. Especially the comparability of EAE induction in transgenic mouse strains might be limited due to changed expression of the target protein or distinct myelin composition[83]. The spectrum of EAE models was further extended by spontaneous severely affected transgenic EAE models carrying myelin-specific T cell receptors or by passive transfer of encephalitogenic T cells[84-88]. Whereas most of these models are based on MOG-specific T cell reactivity[70-73], the immunization with myelin-associated oligodendrocytic basic protein elicits CD4+ T cells recently likewise showed a severe and chronic EAE associated with intense perivascular and parenchymal inflammation, demyelination and neurodegeneration [Table 1]. The following sections describe the inflammatory changes that occur in the afferent visual system in the most used and described MOG-induced EAE model.
EAE models presenting ON
| Strain | Immunization | Characteristics |
|---|---|---|
| C57BL/6J | myelin oligodendrocyte glycoprotein MOG35-55 | Usually monophasic, peak of disease ca. 15-25 dpi, optic nerve inflammation mostly bilateral |
| C57BL/6J | passive transfer of encephalitogenic T cells | Highly inflammatory phenotype. ON most frequently bilateral |
| SJL/J | proteolipid protein (PLP) | First attack of a relapsing-remitting disease course starts from 9 dpi, peaks ca. 12-15 dpi. From day eleven, 43.9% show unilateral ON and 41.5% bilateral ON; only 14.6% of animals do not show ON |
| SJL/J | oligodendrocyte-specific protein (OSP) | Chronic relapsing EAE with intense perivascular and parenchymal inflammatory infiltrates, widespread demyelination, axonal loss, and remarkable ON |
| SJL/J | myelin-associated oligodendrocytic basic protein (MOBP) | Severe/chronic EAE associated with intense perivascular and parenchymal infiltrations, widespread demyelination, axonal loss, and remarkable ON |
| TCR2D2 | Pertussis toxin | TCR2D2 mice can develop spontaneous ON more frequently than they develop spontaneous EAE. Injection of pertussis toxin promotes higher incidence of ON and EAE |
Inflammation in EAE
Inflammation in EAE is driven by microglia and CD4+ T cells, with Th1 and Th17 cells playing a crucial role[89]. In healthy mice the continuously layered RGCs in the retina and myelinated axons in the optic nerve are well organized with resting microglia and astrocytes [Figure 1]. In the early stage of EAE, the blood-brain barrier disrupts, and the activation of microglia and infiltration of CD4+ T-cells build an inflammatory CNS environment leading to a swelling of the inner retinal layers and demyelination of the optic nerve[90,91]. It is still challenging to entirely separate the peripheral immune cell infiltration from glial cell response, especially since both reactions show a major temporal overlap, and the activated glia facilitates the secondary recruitment of peripheral immune cells through the release of chemokines[76]. In later stages of EAE, the combined inflammatory reaction leads to severe demyelination, and up to half of the RGCs undergo apoptosis and depletion without a distinct retinal pattern[76]. An increasing number of active microglia is visible from seven dpi, especially in the ganglion cell and inner plexiform layer of the retina, and persists until late stages of the disease[89-93]. The greatest increase of microglia at eleven - fifteen dpi seem to coincide with an immense general cellular infiltrate and activation of astrocytes and macrophages by the excretion of pro-inflammatory markers such as IL-6[88-90]. During the peak of disease, microglia are also the major producer of inducible nitric oxide synthase, further fueling the inflammatory state[90]. A massive T cell infiltration starts circa nine to sixteen dpi around the optic nerve head and then slowly diminishes over the course of the disease[90-92,94]. Knier et al.[94] suggested that the inflammatory reaction in EAE might be region-specific with a lower frequency of macrophages and a higher frequency of centripetally infiltrating granulocytes and IlL17-producing CD4+ Th cells in the optic nerve compared to the brain and spinal cord. Apart from Th17 cells, IL-17 seems to be produced by a considerably increased subset of γδ T cells in the optic nerve, unconventional T cells possessing features of both innate and adaptive immune cells which are suggested to play a critical role in promoting and maintaining inflammation in EAE[95]. The neutralization of IL-17 completely prevented inner retinal layer thinning and immune cell infiltration, further underlining the importance of the IL-17-mediated pathway in EAE-ON[94,96]. In the early inflammatory phase of EAE, macrophages polarize towards a pro-inflammatory phenotype expressing increasing levels of pro-inflammatory markers such as CXCL-10, CXCL-11, IL-12 and IL-23 and thereby contributing to the influx of further inflammatory immune cells and cellular stress, whereas the percentage of anti-inflammatory macrophages is decreased[97]. A recent report suggested that fatty acids including omega-3 and omega-6 can promote a shift of macrophages towards the anti-inflammatory type by enhancing the phosphorylation of STAT3 and decreasing the phosphorylation of NF-κB[97]. The anti-inflammatory phenotype is mainly characterized by the expression of the anti-inflammatory cytokine IL-10, which also independently showed the prevention of further microglial infiltration, MHC II expression and optic nerve demyelination[98]. Several studies have further analyzed the contribution of CD8+ T cells, B cells and monocytes to EAE pathology, but specific descriptions in the afferent visual system are still warranted[99-101].
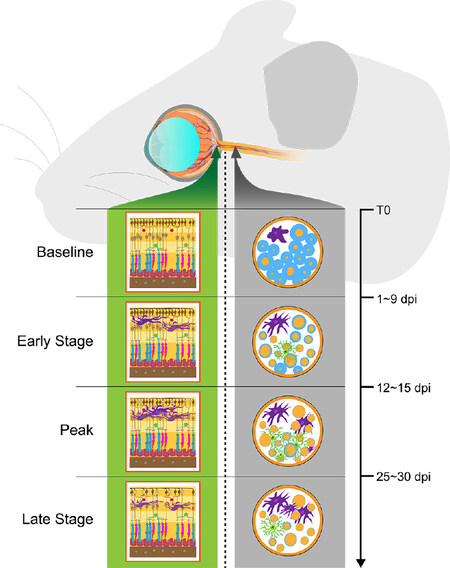
Figure 1. Retinal and optic nerve inflammation during the course of experimental autoimmune encephalomyelitis. Baseline: continuously layered retinal ganglion cells in the retina, well-structured myelinated axons in the optic nerve, resting microglia, and astrocytes. Early-stage and peak of disease: microglia and astrocyte activation, macrophage polarization, T-cell infiltration. Axonal demyelination, swelling, early degeneration. Late-stage: microgliosis, astrogliosis, T cell infiltration, axonal loss, and demyelination
Astrocyte reactivity
Astrocyte reactivity is upregulated from circa eight to nine dpi and is maintained at elevated levels with a slight decrease until a late to chronic disease stage, developing into an astroglial scar[90,91,93]. Especially in early EAE, the astrocyte-dependent inflammatory cascade is a key feature of EAE pathology, including the NF-κB associated upregulation of IL1ß, CXCL10, CCL5, ICAM-1 and TNF with a significant influence on neurotoxicity, demyelination and RGC death[102]. Suppression of this NF-κB-associated upregulation was suggested to lead to an almost absent inflammatory cell infiltrate with less microglia and astrocyte activation and the following subsistence of myelin and axonal content as well as functional output[103]. A recent transcriptomics analysis in optic nerve astrocytes revealed an upregulation of MHC II in early EAE, a reduced cholesterol synthesis in late EAE, and a constant increase in the antigen presentation and complement cascade pathways[104,105]. The latter was characterized by an upregulation of C3 in the optic nerve but not in the retina, which is associated with a neurotoxic phenotype and an aberrant phagocytic activity[90,104,105]. This was balanced only in early EAE by upregulation of Thbs1, which is associated with neuroprotection and synaptic plasticity[104,105]. Interestingly, the C3 and Thbs1 differences were more pronounced in female and male mice, respectively, without inducing a subsequent difference in inflammatory state or demyelination[104,105]. Changes of astrocyte-like Müller cells in the retina were investigated only in a very few and conflicting studies so far[89-91,105].
Contribution of oxidative stress
During the acute and chronic inflammatory process, microglia and other immune cells release cytokines, oxidative products and free radicals. The subsequent neurodegeneration includes complex overlapping processes apart from the primary apoptosis, including ion channel dysfunction, iron accumulation, mitochondriopathy and proteolytic enzyme production, which lead to further production of reactive oxygen species and therefore additional oxidative stress[106]. Multiple studies have shown that the enhancement of antioxidant pathways and mitochondrial function has a major protective effect against immune cell infiltration, optic nerve demyelination, and RGC degeneration and subsequent function[107-121] in the afferent visual system.
Testing anti-inflammatory strategies
Since EAE is the most commonly used animal model for MS with a predominant inflammatory component, it is regularly used to test the effects of new anti-inflammatory treatment strategies and to better understand the pathophysiological mechanisms of on- and off-label drugs. Recent structural and functional measurements such as OCT, VEP, optomotor response, optokinetic response, and electroretinogram (ERG), thereby allow a sensitive longitudinal evaluation of structure and function[79,82,96,106,113,114,122-126]. Advantages and disadvantages of these in vivo readouts, which are increasingly being used in animal models, are described in Table 2.
Structural and functional rodent in vivo readouts: advantages and disadvantages
| Characteristics | Advantages | Disadvantages | |
|---|---|---|---|
| Optical Coherence Tomography (OCT) | OCT uses a low coherence, infrared light source and an interferometer to obtain cross sectional images of biological tissues with extremely high axial resolution (1 microns or less) | It has been optimized for cross sectional imaging of the retina - and has been used to develop methods for quantitative assessment of inner retinal pathology in optic neuropathies. Allows for longitudinal assessment of retinal neurodegeneration | OCT imaging doesn’t allow for distinguishing between layer thickness changes due to infiltrate or cell degeneration. Moreover, OCT does not measure retinal function and being neurodegeneration a slow process it needs several months to show the consequences of even massive optic nerve damage |
| Visual Evoked Potential (VEP) | VEP is an electric potential recorded from the visual cortex in response to a repeating visual stimulus, allowing the evaluation of time needed for a signal to travel from the retina to the brain’s visual cortex while averaging out background cortical potentials | VEP has showed the efficacy of a remyelinating drug in MS patients. It could be developed as a high throughput method for pre-clinical screening of remyelinating therapies.
This model would permit assessment of the response to therapy and would allow for the correct optimization of timing and dosing for such therapies | The execution of a non-invasive, reliable and longitudinal VEP in mice presents technical issues. Histological data assessing the direct relationship between demyelination and remyelination and latency delay are lacking |
| Optokinetic Response (OKR) | OKR measures compensatory eye movements in head-fixed animals exposed to globally rotating striped patterns | evoked easily and reliably, can be used to measure visual function. It allows for a functional evaluation of a different area of the visual system | Less easy to translate to humans than OCT and VEP, due both to the limited use of it in clinic and to the anatomical differences between rodents and humans |
| Optomotor Response (OMR) | OMR measures compensatory head movements in unrestrained animals exposed to globally rotating striped patterns | It allows for a functional evaluation of a different area of the visual system | Less easy to translate to humans than OCT and VEP, due both to the limited use of it in clinic and to the anatomical differences between rodents and humans |
| Electroretinogram (ERG) | ERG measures the electrical activity of the retina in response to a light stimulus | It allows for testing the function of the outer layers of retina. RGCs function can be studied performing a pattern ERG (pERG) | The outer retina is not affected primarily by MS or ON. The execution of pERG in mice presents technical issues |
For example, intravenous immunoglobulin treatment is an established second-line treatment for acute ON but only recently showed a dose-dependent reduction of microglial infiltration, demyelination and neuroaxonal loss in EAE helping us to better understand its mechanism of action[127]. On the other hand, very promising EAE studies are often only moderately translatable into the effects we see in clinical trials. In this context, the translation from EAE to clinical trials usually works better for treatments with a mainly anti-inflammatory mechanism of action such as laquinimod[128-130], or potential neuroprotective agents such as phenytoin[131,132]. In contrast, the translatability from EAE of remyelinating therapies such as clemastine and monoclonal antibodies against anti-LINGO-1 is very limited due to the underrepresented demyelinating disease component in EAE[133-135]. In the light of upcoming regenerative therapy approaches, MS animal models with a predominant demyelinating component gain further importance.
Magnetic resonance imaging of the optic nerve in the EAE model of MS
MRI has proven useful to assess structural and functional alterations of the AVP and optic nerve portion, which is hidden from clinical evaluation by the bones of the orbit and skull base. The diameter of the optic nerve varies between 1.4 and 4 mm in humans, and 0.06-0.18 mm in mice. Due to its small size, there are challenges associated with imaging of the optic nerve. Specifically, acquisition of high spatial resolution images is required to minimize partial volume effects from surrounding tissue, cerebral spinal fluid, and/or orbital fat. Furthermore, the optic nerve is located next to air-filled sinuses and skull base, which may induce inhomogeneities in the magnetic field, resulting in signal distortions and drop-out. Nonetheless, several studies have highlighted the use of MRI for small animal optic nerve imaging following EAE induction.
Conventional T2 MRI detected the appearance of hyperintensities and swelling of the optic nerve in MOG-induced EAE in marmoset and rodent models, corresponding to areas of demyelination, inflammation and axonal loss determined by post-mortem histopathological analyses[136-139]. However, slight inflammatory infiltration in the optic nerve could not be detected with T2 MRI[136]. In the MOG-induced EAE mouse model, the signal increase on T2-weighted images was observed around the optic nerve at disease onset, peak and chronic time points[92]. Conventional T1-weighted MRI displayed a marked decrease of T1 signal intensity in the optic nerve in a rat model of EAE, which correlated with demyelination, inflammation and axon density[138]. T1-weighted MRI performed post-injection of a gadolinium-based contrast agent enables the detection of alterations in blood-brain barrier integrity. Local gadolinium signal enhancement was detected in the optic nerve of both marmoset and rodent EAE models, as early as two weeks post-immunization, and remained detectable at a later stage of the disease[137,140,141]. Diffusion tensor imaging (DTI) has been the most widely used method to investigate optic nerve abnormalities. DTI makes it possible to measure the directional diffusivities of water molecules, allowing the identification of fiber orientation as well as fiber structure integrity[142]. Several studies have shown that white matter axonal injury results in reduced axial diffusivity (λ||), while demyelination results in increased radial diffusivity (λ⊥)[139,140,143-145]. Sun et al.[143] showed a 19% decrease in λ|| and a 156% increase in λ⊥ in optic nerve at a chronic time point (3 months post-immunization) and reported significant correlations between phosphorylated neurofilaments, indicative of axonal damage, and λ||, and between myelin basic protein and λ⊥. DTI-derived λ|| and diffusion basis spectrum imaging have both shown high potential to detect axonal damage in the acute phase of EAE[144,146], while changes in λ⊥ are usually detectable at a later stage of the disease (4 weeks onward)[92,140,145]. Recently, DTI metrics of the optic nerve have been associated with OCT measures. The ganglion cell complex thickness derived from OCT was positively correlated with λ|| and negatively correlated with λ⊥ at chronic time points[92,145]. In a longitudinal study, λ⊥ was associated with VEP latency in the early stage of EAE (2 weeks), and to VEP amplitude at a later stage (8-12 weeks)[140]. Manganese-enhanced MRI can be used to visualize axonal transport in the optic nerve. As a paramagnetic agent, manganese reduces T1 relaxation time, and its presence can be detected as hyper-intensities in T1-weighted images. When injected directly into the eye, manganese ions (Mn2+) are taken up by RGCs and transported along microtubules in the axons. Lin et al.[147] showed that axonal accumulation rate and axonal transport rate are significantly reduced in optic nerve of mice at the onset of EAE and correlated with impaired visual acuity, as well as histopathological markers of inflammation, demyelination, axonal damage and tubulins. When injected intraperitoneally, manganese was shown to accumulate in the optic nerve of a rat model of EAE and correlated with a reduction of axons[138]. As Mn2+ has been shown to enter neurons through Ca2+ voltage-gated channels, increased detection of Mn2+ might reflect increased calcium influx caused by axon damage. Interestingly, increased Mn2+ signal could be detected as early as 10 min post manganese injection and last up to 7 days, suggesting an increased Ca2+ uptake into the affected nerve and storage within the cells. Besides neurons, macrophages and activated microglia also possess Ca2+ voltage-gated channels and thus might contribute to the increased signal detected by manganese-enhanced MRI[148]. Diffusion functional MRI has been show to detect changes in water diffusion during brain activation following visual or sensory stimuli and has been suggested to identify functional integrity of neurons and axons[149,150]. Following visual stimulation, the apparent diffusion coefficient perpendicular to the axonal fibers (ADC⊥) decreased in the optic nerve of normal mice (~27% ADC⊥ decrease)[151]. In contrast, at the onset of EAE, this decrease in ADC⊥ was reduced (~7% non-significant reduction) and correlated with impaired visual acuity and histological markers of inflammation, demyelination and axonal injury[152]. MR imaging of the retina, including anatomical T1-weighted, T2-weighted, contrast-enhanced, diffusion MRI, as well as blood-flow and functional MRI have been described in health and diseased states, and we refer the reader to a very comprehensive review by Duong for more details[153].
Inflammation of the anterior visual pathway in the cuprizone model
Cuprizone (CPZ) is a copper chelating agent. When fed to adult mice, usually as a 0.2% CPZ-supplemented diet for 4-6 weeks, CPZ induces metabolic stress, myelin sheath breakdown, and oligodendrocyte apoptosis. This results in primary acute demyelination of the corpus callosum with microglial activation, phagocytosis of myelin, and astrocytosis. After three weeks of CPZ treatment, there is spontaneous repopulation of oligodendrocyte precursor cells, ultimately resulting in complete remyelination upon CPZ discontinuation. When CPZ is given for 12 weeks, lesions appear chronically demyelinated, and there is limited remyelination capacity[154]. While short-term CPZ administration leads to synaptic dysfunction and disturbed neurotransmitter concentrations with preservation of axons, long exposures to CPZ can result in axonal degeneration[155]. Classically, it had been considered that CPZ does not cause demyelination in the optic nerves or the spinal cord of C57Bl/6 mice, but later studies have described different degrees of optic nerve injury after CPZ diet. Through immunoblot analysis of CNPase and MBP-like immunoreactivity, Namekata et al.[156] demonstrated that CPZ induces a small yet significant optic nerve demyelination after 12 weeks of treatment. This was accompanied by impaired visual function as assessed by multifocal ERGs. In those experiments, demyelination was partially prevented in mice where the oligodendrocytes in the optic nerve overexpressed dedicator of cytokinesis 3 (Dock3, an activator of GTPase Rac1), possibly by increased activation of extracellular signal-regulated kinases. Interestingly, retinal morphology was normal, and there was no RGC degeneration. Optic nerve injury was not found when limiting CPZ treatment to 6 weeks. A more recent work, however, found demyelination in the optic nerve of mice treated for as short as three weeks with CPZ[157]. In that study, demyelination was reduced (showing a higher g-ratio) in mice treated from day 11 to 21 with daily intraperitoneal injections of probenecid (a pannexin-1 antagonist), suggesting a role for ATP-release channels in modulating the inflammatory response to toxic demyelination. Bagchi et al.[158] saw considerable demyelination in the optic nerve as well after 8 weeks, with axonal preservation. They found spreading of voltage-activated K+ channels (KV1.1 and 1.2 a subunits) from typical juxta-paranodal sites to nodes in demyelinated axons. This resulted in altered evoked compound action potentials (CAPs) in vitro, characterized by initial synchronous waveform followed by a dispersed component - in contrast to the monophasic CAPs seen in controls. Kojima and Hayashi[159] found no oligodendrocyte loss in the optic nerve after CPZ treatment for nine weeks but saw that CPZ induced new contacts between axons and myelin at the internode region. While the lengths of paranodes and nodes (stained with anti-Nav and anti- Caspr, respectively) in CPZ-treated optic nerve were not altered, the lengths of juxtaparanodes (stained with anti-Kv1) were significantly larger than that of the control mice. In any case, demyelination in the visual pathway, at least during shorter CPZ intoxication (i.e., 3-5 weeks), appears to be more prominent in the dorsal lateral geniculate nucleus (dLGN) and the superior colliculus, with relative sparing of the optic tract, according to Araújo et al.[160]. Furthermore, CPZ-induced demyelination is associated with inflammation and reactive astrogliosis in the dLGN. After five weeks of CPZ diet, there is a higher density of ameboid-shaped microglia, which has been linked to a repairing, pro-remyelinating phenotype. At the same time-point, there are synaptic changes toward inhibitory network activity, where excitatory synaptic contacts from glutamatergic retinal input in the dLGN are decreased, and inhibitory GABAergic input from the thalamic reticular nucleus is increased[160].
Conclusion
The dilemma regarding the main causative factors of ON and MS is still a significant puzzle for researchers. However, information regarding the pathogenesis of MS has progressed substantially over the last 20 years. Studies with animal models are one of the main tools responsible for this progress, and although experimental models of ON and MS are not a perfect fit, they make it possible to understand several pathway mechanisms underlying demyelinating pathology.
As such, the development of novel therapies that effectively target the inflammatory and neurodegenerative aspects of ON and MSON cannot inhibit the study of the disease processes and the response to different treatments via various animal models.
Various studies have demonstrated that some drugs are capable of promoting remyelination and increase the number of oligodendrocytes. These remyelinating/neuroprotective approaches aiming to prevent axonal loss as well as myelin damage are incredibly promising, even if physiologically relevant and validated preclinical methods for measuring remyelination in a longitudinal manner are still needed.
Once their beneficial properties are hopefully translated to clinical practice, this new class of drugs has relevance for delaying the progression of disease in patients with both relapsing-remitting and progressive phenotypes of MS and for treating patients with clinically isolated syndrome (like ON) to prevent progression to MS.
Of relevance, most of the studies used young adult female animals. As a consequence, there is a need for additional studies to be performed with male animals, especially using the EAE model of MS. Also, studies with aged animals are warranted, since chronic low-grade inflammation is an essential contributor to various age-related pathologies and natural processes in aging tissues, including the nervous system.
Since as we have seen, the importance of oxidative stress in MS is becoming increasingly recognized, combination therapies using immunomodulators and antioxidant drugs would be of great interest. Broadly speaking, it is conceivable that combination therapy may be more effective for MS patients, especially using agents that target neuroinflammation and remyelination/neurodegeneration. Remyelinating/neuroprotective agents, coupled with an immunomodulator, could be a viable way of enhancing the effectiveness of current drugs. Future studies should determine the effectiveness of combining agents shown to be effective singly in alleviating neuroinflammation, axonal loss and oxidative stress in animal models of chronic progressive disease, as they may exert synergistic effects and may provide more effective therapies for patients with MS.
Declarations
AcknowledgmentsWe thank Frederike Cosima Oertel for comments that greatly improved the manuscript and Jung Hyung Sin for assistance with the preparation of the figure.
Authors’ contributionsStudy concept and design, manuscript composition: Cordano C
Manuscript composition: Ramos C, Arnow S, Cruz-Herranz A, Guglielmetti C, Iester M, Bandini F
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 2008;31:247-69.
2. Tur C, Goodkin O, Altmann DR, et al. Longitudinal evidence for anterograde trans-synaptic degeneration after optic neuritis. Brain 2016;139:816-28.
4. Gabilondo I, Martínez-Lapiscina EH, Martínez-Heras E, et al. Trans-synaptic axonal degeneration in the visual pathway in multiple sclerosis. Ann Neurol 2014;75:98-107.
5. Schmidt D, Meyer JH, Brandi-Dohrn J. Wide-spread myelinated nerve fibers of the optic disc: do they influence the development of myopia? Int Ophthalmol 1996;20:263-8.
6. Green AJ, McQuaid S, Hauser SL, Allen IV, Lyness R. Ocular pathology in multiple sclerosis: retinal atrophy and inflammation irrespective of disease duration. Brain 2010;133:1591-601.
7. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998;338:278-85.
8. Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain 1997;120:393-9.
9. Kornek B, Storch M K, Weissert R, Schmidbauer i M, Lassmann H, et al. (2000). “[J]. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions.”. Am J Pathol 2000;157:267-76.
10. Quigley HA, Davis EB, Anderson DR. Descending optic nerve degeneration in primates. Invest Ophthalmol Vis Sci 1977;16:841-9.
12. Keltner JL, Johnson CA, Spurr JO, Beck RW. Baseline visual field profile of optic neuritis. The experience of the optic neuritis treatment trial. Optic Neuritis Study Group. Arch Ophthalmol 1993;111:231-4.
13. Tintoré M, Montalban X. The optic nerve should be included as one of the typical CNS regions for establishing dissemination in space when diagnosing MS - No. Mult Scler 2018;24:123-5.
14. Galetta SL, Balcer LJ. The optic nerve should be included as one of the typical CNS regions for establishing dissemination in space when diagnosing MS - Yes. Mult Scler 2018;24:121-2.
15. Toussaint D, Périer O, Verstappen A, Bervoets S. Clinicopathological study of the visual pathways, eyes, and cerebral hemispheres in 32 cases of disseminated sclerosis. J Clin Neuroophthalmol 1983;3:211-20.
16. Bischof A, Caverzasi E, Cordano C, Hauser SL, Henry RG. Advances in imaging multiple sclerosis. Semin Neurol 2017;37:538-45.
17. Parisi V. Correlation between morphological and functional retinal impairment in patients affected by ocular hypertension, glaucoma, demyelinating optic neuritis and Alzheimer’s disease. Semin Ophthalmol 2003;18:50-7.
18. Trip SA, Schlottmann PG, Jones SJ, et al. Retinal nerve fiber layer axonal loss and visual dysfunction in optic neuritis. Ann Neurol 2005;58:383-91.
19. Henderson AP, Altmann DR, Trip AS, et al. A serial study of retinal changes following optic neuritis with sample size estimates for acute neuroprotection trials. Brain 2010;133:2592-602.
20. Klistorner A, Arvind H, Garrick R, Graham SL, Paine M, Yiannikas C. Interrelationship of optical coherence tomography and multifocal visual-evoked potentials after optic neuritis. Invest Ophthalmol Vis Sci 2010;51:2770-7.
21. Gelfand JM, Goodin DS, Boscardin WJ, Nolan R, Cuneo A, Green AJ. Retinal axonal loss begins early in the course of multiple sclerosis and is similar between progressive phenotypes. PLoS One 2012;7:e36847.
22. Costello F, Coupland S, Hodge W, et al. Quantifying axonal loss after optic neuritis with optical coherence tomography. Ann Neurol 2006;59:963-9.
23. Costello F, Hodge W, Pan YI, Eggenberger E, Coupland S, Kardon RH. Tracking retinal nerve fiber layer loss after optic neuritis: a prospective study using optical coherence tomography. Mult Scler 2008;14:893-905.
24. Kupersmith MJ, Mandel G, Anderson S, Meltzer DE, Kardon R. Baseline, one and three month changes in the peripapillary retinal nerve fiber layer in acute optic neuritis: relation to baseline vision and MRI. J Neurol Sci 2011;308:117-23.
25. Petzold A, Balcer LJ, Calabresi PA, et al. Retinal layer segmentation in multiple sclerosis: a systematic review and meta-analysis. Lancet Neurol 2017;16:797-812.
26. Gabilondo I, Martínez-Lapiscina EH, Fraga-Pumar E, et al. Dynamics of retinal injury after acute optic neuritis. Ann Neurol 2015;77:517-28.
27. Rucker CW. Sheathing of the retinal veins in multiple sclerosis. Review of pertinent literature. Mayo Clin Proc 1972;47:335-40.
28. Frisén L, Hoyt WF. Insidious atrophy of retinal nerve fibers in multiple sclerosis. Funduscopic identification in patients with and without visual complaints. Arch Ophthalmol 1974;92:91-7.
29. Kerrison JB, Flynn T, Green WR. Retinal pathologic changes in multiple sclerosis. Retina 1994;14:445-51.
30. Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM. Size-selective neuronal changes in the anterior optic pathways suggest a differential susceptibility to injury in multiple sclerosis. Brain 2001;124:1813-20.
31. Blumenthal EZ, Parikh RS, Pe’er J, et al. Retinal nerve fibre layer imaging compared with histological measurements in a human eye. Eye (Lond) 2009;23:171-5.
32. Audoin B, Fernando KT, Swanton JK, Thompson AJ, Plant GT, Miller DH. Selective magnetization transfer ratio decrease in the visual cortex following optic neuritis. Brain 2006;129:1031-9.
33. Reich DS, Smith SA, Gordon-Lipkin EM, et al. Damage to the optic radiation in multiple sclerosis is associated with retinal injury and visual disability. Arch Neurol 2009;66:998-1006.
34. Brück W, Porada P, Poser S, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol 1995;38:788-96.
35. Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707-17.
36. Popescu BF, Lucchinetti CF. Pathology of demyelinating diseases. Annu Rev Pathol 2012;7:185-217.
37. Calabresi PA, Balcer LJ, Frohman EM. Retinal pathology in multiple sclerosis: insight into the mechanisms of neuronal pathology. Brain 2010;133:1575-7.
38. Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009;132:1175-89.
39. Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Brück W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain 2002;125:2202-12.
41. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005;128:2705-12.
42. Parisi V, Manni G, Centofanti M, et al. Correlation between optical coherence tomography, pattern electroretinogram, and visual evoked potentials in open-angle glaucoma patients. Ophthalmology 2001;108:905-12.
43. Fisher JB, Jacobs DA, Markowitz CE, et al. Relation of visual function to retinal nerve fiber layer thickness in multiple sclerosis. Ophthalmology 2006;113:324-32.
44. van Dijkman SC, de Jager NCB, Rauwé WM, Danhof M, Della Pasqua O. Effect of age-related factors on the pharmacokinetics of lamotrigine and potential implications for maintenance dose optimisation in future clinical trials. Clin Pharmacokinet 2018;57:1039-53.
45. Costello F, Hodge W, Pan YI, Freedman M, DeMeulemeester C. Differences in retinal nerve fiber layer atrophy between multiple sclerosis subtypes. J Neurol Sci 2009;281:74-9.
46. Petzold A, de Boer JF, Schippling S, et al. Optical coherence tomography in multiple sclerosis: a systematic review and meta-analysis. Lancet Neurol 2010;9:921-32.
47. Martinez-Lapiscina EH, Arnow S, Wilson JA, et al. Retinal thickness measured with optical coherence tomography and risk of disability worsening in multiple sclerosis: a cohort study. Lancet Neurol 2016;15:574-84.
48. Cordano C, Nourbakhsh B, Devereux M, et al. pRNFL as a marker of disability worsening in the medium/long term in patients with MS. Neurol Neuroimmunol Neuroinflamm 2018;6:e533.
49. DeLuca GC, Ebers GC, Esiri MM. Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain 2004;127:1009-18.
50. Petracca M, Cordano C, Cellerino M, et al. Retinal degeneration in primary-progressive multiple sclerosis: A role for cortical lesions? Mult Scler 2017;23:43-50.
51. Caverzasi E, Cordano C, Zhu AH, et al. Imaging correlates of visual function in multiple sclerosis. PLoS One 2020;15:e0235615.
52. Saidha S, Sotirchos ES, Ibrahim MA, et al. Microcystic macular oedema, thickness of the inner nuclear layer of the retina, and disease characteristics in multiple sclerosis: a retrospective study. Lancet Neurol 2012;11:963-72.
53. Gelfand JM, Nolan R, Schwartz DM, Graves J, Green AJ. Microcystic macular oedema in multiple sclerosis is associated with disease severity. Brain 2012;135:1786-93.
54. Balk LJ, Killestein J, Polman CH, Uitdehaag BM, Petzold A. Microcystic macular oedema confirmed, but not specific for multiple sclerosis. Brain 2012;135:e226-7.
55. Green AJ, Schwartz D, Gelfand J. Reply: microcysts in the inner nuclear layer from optic atrophy are caused by retrograde trans-synaptic degeneration combined with vitreous traction on the retinal surface. Brain 2013;136:e261.
56. Gaudric A, Ducos de Lahitte G, Cohen SY, Massin P, Haouchine B. Optical coherence tomography in group 2A idiopathic juxtafoveolar retinal telangiectasis. Arch Ophthalmol 2006;124:1410-9.
57. Abegg M, Dysli M, Wolf S, Kowal J, Dufour P, Zinkernagel M. Microcystic macular edema: retrograde maculopathy caused by optic neuropathy. Ophthalmology 2014;121:142-9.
58. Wolff B, Basdekidou C, Vasseur V, Mauget-Faÿsse M, Sahel JA, Vignal C. Retinal inner nuclear layer microcystic changes in optic nerve atrophy: a novel spectral-domain OCT finding. Retina 2013;33:2133-8.
59. Barboni P, Carelli V, Savini G, Carbonelli M, La Morgia C, Sadun AA. Microcystic macular degeneration from optic neuropathy: not inflammatory, not trans-synaptic degeneration. Brain 2013;136:e239.
60. vanBuren JM. Trans-synaptic retrograde degeneration in the visual system of primates. J Neurol Neurosurg Psychiatry 1963;26:402-9.
61. Knier B, Schmidt P, Aly L, et al. Retinal inner nuclear layer volume reflects response to immunotherapy in multiple sclerosis. Brain 2016;139:2855-63.
62. Cellerino M, Cordano C, Boffa G, et al. Relationship between retinal inner nuclear layer, age, and disease activity in progressive MS. Neurol Neuroimmunol Neuroinflamm 2019;6:e596.
63. Cordano C, Yiu HH, Oertel FC, et al. Retinal INL thickness in multiple sclerosis: a mere marker of neurodegeneration? Ann Neurol 2020; doi: 10.1002/ana.25933.
64. Le Scanff J, Sève P, Renoux C, Broussolle C, Confavreux C, Vukusic S. Uveitis associated with multiple sclerosis. Mult Scler 2008;14:415-7.
65. Jain N, Bhatti MT. Fingolimod-associated macular edema: incidence, detection, and management. Neurology 2012;78:672-80.
66. Dudek SM, Jacobson JR, Chiang ET, et al. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem 2004;279:24692-700.
67. Sanna MG, Wang SK, Gonzalez-Cabrera PJ, et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol 2006;2:434-41.
68. Sheehy CK, Bensinger ES, Romeo A, et al. Fixational microsaccades: a quantitative and objective measure of disability in multiple sclerosis. Mult Scler 2020;26:343-53.
69. Serra A, Petzold A. Can stability of visual fixation be a measure for disability in multiple sclerosis? Mult Scler 2020;26:264-5.
70. Bennett JL, de Seze J, Lana-Peixoto M, et al. Neuromyelitis optica and multiple sclerosis: Seeing differences through optical coherence tomography. Mult Scler 2015;21:678-88.
71. Green AJ, Cree BA. Distinctive retinal nerve fibre layer and vascular changes in neuromyelitis optica following optic neuritis. J Neurol Neurosurg Psychiatry 2009;80:1002-5.
72. Schneider E, Zimmermann H, Oberwahrenbrock T, et al. Optical coherence tomography reveals distinct patterns of retinal damage in neuromyelitis optica and multiple sclerosis. PLoS One 2013;8:e66151.
73. Ratchford JN, Quigg ME, Conger A, et al. Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology 2009;73:302-8.
74. Shen T, You Y, Arunachalam S, et al. Differing structural and functional patterns of optic nerve damage in multiple sclerosis and neuromyelitis optica spectrum disorder. Ophthalmology 2019;126:445-53.
75. Oertel FC, Kuchling J, Zimmermann H, et al. Microstructural visual system changes in AQP4-antibody-seropositive NMOSD. Neurol Neuroimmunol Neuroinflamm 2017;4:e334.
76. Shindler KS, Guan Y, Ventura E, Bennett J, Rostami A. Retinal ganglion cell loss induced by acute optic neuritis in a relapsing model of multiple sclerosis. Mult Scler 2006;12:526-32.
77. Shindler KS, Ventura E, Dutt M, Rostami A. Inflammatory demyelination induces axonal injury and retinal ganglion cell apoptosis in experimental optic neuritis. Exp Eye Res 2008;87:208-13.
78. Quinn TA, Dutt M, Shindler KS. Optic neuritis and retinal ganglion cell loss in a chronic murine model of multiple sclerosis. Front Neurol 2011;2:50.
79. Smith AW, Rohrer B, Wheless L, et al. Calpain inhibition reduces structural and functional impairment of retinal ganglion cells in experimental optic neuritis. J Neurochem 2016;139:270-84.
80. Kaushansky N, Zilkha-Falb R, Hemo R, et al. Pathogenic T cells in MOBP-induced murine EAE are predominantly focused to recognition of MOBP21F and MOBP27P epitopic residues. Eur J Immunol 2007;37:3281-92.
81. Soares RM, Dias AT, De Castro SB, et al. Optical neuritis induced by different concentrations of myelin oligodendrocyte glycoprotein presents different profiles of the inflammatory process. Autoimmunity 2013;46:480-5.
82. Cruz-Herranz A, Dietrich M, Hilla AM, et al. Monitoring retinal changes with optical coherence tomography predicts neuronal loss in experimental autoimmune encephalomyelitis. J Neuroinflammation 2019;16:203.
83. Guo X, Harada C, Namekata K, et al. Delayed onset of experimental autoimmune encephalomyelitis in Olig1 deficient mice. PLoS One 2010;5:e13083.
84. Guan Y, Shindler KS, Tabuena P, Rostami AM. Retinal ganglion cell damage induced by spontaneous autoimmune optic neuritis in MOG-specific TCR transgenic mice. J Neuroimmunol 2006;178:40-8.
85. Pöllinger B, Krishnamoorthy G, Berer K, et al. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med 2009;206:1303-16.
86. Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 2003;197:1073-81.
87. Shao H, Huang Z, Sun SL, Kaplan HJ, Sun D. Myelin/oligodendrocyte glycoprotein-specific T-cells induce severe optic neuritis in the C57BL/6 mouse. Invest Ophthalmol Vis Sci 2004;45:4060-5.
88. de Rosbo NK, Kaye JF, Eisenstein M, et al. The myelin-associated oligodendrocytic basic protein region MOBP15-36 encompasses the immunodominant major encephalitogenic epitope(s) for SJL/J mice and predicted epitope(s) for multiple sclerosis-associated HLA-DRB1*1501. J Immunol 2004;173:1426-35.
89. Horstmann L, Kuehn S, Pedreiturria X, et al. Microglia response in retina and optic nerve in chronic experimental autoimmune encephalomyelitis. J Neuroimmunol 2016;298:32-41.
90. Jin J, Smith MD, Kersbergen CJ, et al. Glial pathology and retinal neurotoxicity in the anterior visual pathway in experimental autoimmune encephalomyelitis. Acta Neuropathol Commun 2019;7:125.
91. Manogaran P, Samardzija M, Schad AN, et al. Retinal pathology in experimental optic neuritis is characterized by retrograde degeneration and gliosis. Acta Neuropathol Commun 2019;7:116.
92. Manogaran P, Walker-Egger C, Samardzija M, et al. Exploring experimental autoimmune optic neuritis using multimodal imaging. Neuroimage 2018;175:327-39.
93. Horstmann L, Schmid H, Heinen AP, Kurschus FC, Dick HB, Joachim SC. Inflammatory demyelination induces glia alterations and ganglion cell loss in the retina of an experimental autoimmune encephalomyelitis model. J Neuroinflammation 2013;10:120.
94. Knier B, Rothhammer V, Heink S, et al. Neutralizing IL-17 protects the optic nerve from autoimmune pathology and prevents retinal nerve fiber layer atrophy during experimental autoimmune encephalomyelitis. J Autoimmun 2015;56:34-44.
95. Malik S, Want MY, Awasthi A. The emerging roles of gamma-delta T cells in tissue inflammation in experimental autoimmune encephalomyelitis. Front Immunol 2016;7:14.
96. Larabee CM, Hu Y, Desai S, et al. Myelin-specific Th17 cells induce severe relapsing optic neuritis with irreversible loss of retinal ganglion cells in C57BL/6 mice. Mol Vis 2016;22:332-41.
97. Locri F, Cammalleri M, Pini A, Dal Monte M, Rusciano D, Bagnoli P. Further evidence on efficacy of diet supplementation with fatty acids in ocular pathologies: insights from the EAE model of optic neuritis. Nutrients 2018;10:1447.
98. Matsuda R, Kezuka T, Nishiyama C, et al. Interleukin-10 gene-transfected mature dendritic cells suppress murine experimental autoimmune optic neuritis. Invest Ophthalmol Vis Sci 2012;53:7235-45.
99. Bullard DC, Hu X, Adams JE, Schoeb TR, Barnum SR. p150/95 (CD11c/CD18) expression is required for the development of experimental autoimmune encephalomyelitis. Am J Pathol 2007;170:2001-8.
100. Koh DR, Fung-Leung WP, Ho A, Gray D, Acha-Orbea H, Mak TW. Less mortality but more relapses in experimental allergic encephalomyelitis in CD8-/- mice. Science 1992;256:1210-3.
101. Hjelmström P, Juedes AE, Fjell J, Ruddle NH. B-cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J Immunol 1998;161:4480-3.
102. Brambilla R, Dvoriantchikova G, Barakat D, Ivanov D, Bethea JR, Shestopalov VI. Transgenic inhibition of astroglial NF-κB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J Neuroinflammation 2012;9:213.
103. Guo X, Namekata K, Kimura A, Harada C, Harada T. The renin-angiotensin system regulates neurodegeneration in a mouse model of optic neuritis. Am J Pathol 2017;187:2876-85.
104. Itoh N, Itoh Y, Tassoni A, et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: focus on astrocytes. Proc Natl Acad Sci U S A 2018;115:E302-9.
105. Tassoni A, Farkhondeh V, Itoh Y, Itoh N, Sofroniew MV, Voskuhl RR. The astrocyte transcriptome in EAE optic neuritis shows complement activation and reveals a sex difference in astrocytic C3 expression. Sci Rep 2019;9:10010.
106. Khan RS, Baumann B, Dine K, et al. Dexras1 deletion and iron chelation promote neuroprotection in experimental optic neuritis. Sci Rep 2019;9:11664.
107. Larabee CM, Desai S, Agasing A, et al. Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Mol Vis 2016;22:1503-13.
108. Palani CD, Fouda AY, Liu F, et al. Deletion of arginase 2 ameliorates retinal neurodegeneration in a mouse model of multiple sclerosis. Mol Neurobiol 2019;56:8589-602.
109. Guo X, Harada C, Namekata K, et al. Spermidine alleviates severity of murine experimental autoimmune encephalomyelitis. Invest Ophthalmol Vis Sci 2011;52:2696-703.
110. Guo X, Namekata K, Kimura A, et al. Brimonidine suppresses loss of retinal neurons and visual function in a murine model of optic neuritis. Neurosci Lett 2015;592:27-31.
111. Guy J, Qi X, Hauswirth WW. Adeno-associated viral-mediated catalase expression suppresses optic neuritis in experimental allergic encephalomyelitis. Proc Natl Acad Sci U S A 1998;95:13847-52.
112. Khan RS, Dine K, Das Sarma J, Shindler KS. SIRT1 activating compounds reduce oxidative stress mediated neuronal loss in viral induced CNS demyelinating disease. Acta Neuropathol Commun 2014;2:3.
113. Khan RS, Dine K, Geisler JG, Shindler KS. Mitochondrial uncoupler prodrug of 2,4-dinitrophenol, MP201, prevents neuronal damage and preserves vision in experimental optic neuritis. Oxid Med Cell Longev 2017;2017:7180632.
114. McDougald DS, Dine KE, Zezulin AU, Bennett J, Shindler KS. SIRT1 and NRF2 gene transfer mediate distinct neuroprotective effects upon retinal ganglion cell survival and function in experimental optic neuritis. Invest Ophthalmol Vis Sci 2018;59:1212-20.
115. Qi X, Sun L, Lewin AS, Hauswirth WW, Guy J. Long-term suppression of neurodegeneration in chronic experimental optic neuritis: antioxidant gene therapy. Invest Ophthalmol Vis Sci 2007;48:5360-70.
116. Talla V, Yu H, Chou TH, et al. NADH-dehydrogenase type-2 suppresses irreversible visual loss and neurodegeneration in the EAE animal model of MS. Mol Ther 2013;21:1876-88.
117. Talla V, Porciatti V, Chiodo V, Boye SL, Hauswirth WW, Guy J. Gene therapy with mitochondrial heat shock protein 70 suppresses visual loss and optic atrophy in experimental autoimmune encephalomyelitis. Invest Ophthalmol Vis Sci 2014;55:5214-26.
118. Talla V, Koilkonda R, Porciatti V, et al. Complex I subunit gene therapy with NDUFA6 ameliorates neurodegeneration in EAE. Invest Ophthalmol Vis Sci 2015;56:1129-40.
119. Talla V, Koilkonda R, Guy J. Gene therapy with single-subunit yeast NADH-ubiquinone oxidoreductase (NDI1) improves the visual function in experimental autoimmune encephalomyelitis (EAE) mice model of multiple sclerosis (MS). Mol Neurobiol 2020;57:1952-65.
120. Shindler KS, Ventura E, Rex TS, Elliott P, Rostami A. SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci 2007;48:3602-9.
121. Shindler KS, Ventura E, Dutt M, Elliott P, Fitzgerald DC, Rostami A. Oral resveratrol reduces neuronal damage in a model of multiple sclerosis. J Neuroophthalmol 2010;30:328-39.
122. Dietrich M, Helling N, Hilla A, et al. Early alpha-lipoic acid therapy protects from degeneration of the inner retinal layers and vision loss in an experimental autoimmune encephalomyelitis-optic neuritis model. J Neuroinflammation 2018;15:71.
123. Fonseca-Kelly Z, Nassrallah M, Uribe J, et al. Resveratrol neuroprotection in a chronic mouse model of multiple sclerosis. Front Neurol 2012;3:84.
124. Khan RS, Dine K, Wessel H, Brown L, Shindler KS. Effects of varying intranasal treatment regimens in ST266-mediated retinal ganglion cell neuroprotection. J Neuroophthalmol 2019;39:191-9.
125. Dietrich M, Koska V, Hecker C, et al. Protective effects of 4-aminopyridine in experimental optic neuritis and multiple sclerosis. Brain 2020;143:1127-42.
126. Schirmer L, Möbius W, Zhao C, et al. Oligodendrocyte-encoded Kir4.1 function is required for axonal integrity. Elife 2018;7:e36428.
127. Takahashi H, Okuda S, Tamura M, Kamei S, Aizawa R, Kobayashi T. Prophylactic treatment with intravenous immunoglobulin attenuates experimental optic neuritis in mice. Biol Pharm Bull 2019;42:173-8.
128. Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med 2012;366:1000-9.
129. Vollmer TL, Sorensen PS, Selmaj K, et al. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J Neurol 2014;261:773-83.
130. Wilmes AT, Reinehr S, Kühn S, et al. Laquinimod protects the optic nerve and retina in an experimental autoimmune encephalomyelitis model. J Neuroinflammation 2018;15:183.
131. Lo AC, Black JA, Waxman SG. Neuroprotection of axons with phenytoin in experimental allergic encephalomyelitis. Neuroreport 2002;13:1909-12.
132. Raftopoulos R, Hickman S J, Toosy A, et al. Phenytoin for neuroprotection in patients with acute optic neuritis: a randomised, placebo-controlled, phase 2 trial. Lancet Neurol 2016;15:259-69.
133. Gresle MM, Liu Y, Kilpatrick TJ, et al. Blocking LINGO-1 in vivo reduces degeneration and enhances regeneration of the optic nerve. Mult Scler J Exp Transl Clin 2016;2:2055217316641704.
134. Cadavid D, Balcer L, Galetta S, et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol 2017;16:189-99.
135. Green AJ, Gelfand JM, Cree BA, et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. Lancet 2017;390:2481-9.
136. Diem R, Demmer I, Boretius S, et al. Autoimmune optic neuritis in the common marmoset monkey: comparison of visual evoked potentials with MRI and histopathology. Invest Ophthalmol Vis Sci 2008;49:3707-14.
137. Boretius S, Schmelting B, Watanabe T, et al. Monitoring of EAE onset and progression in the common marmoset monkey by sequential high-resolution 3D MRI. NMR Biomed 2006;19:41-9.
138. Boretius S, Gadjanski I, Demmer I, et al. MRI of optic neuritis in a rat model. Neuroimage 2008;41:323-34.
139. Wu Q, Butzkueven H, Gresle M, et al. MR diffusion changes correlate with ultra-structurally defined axonal degeneration in murine optic nerve. Neuroimage 2007;37:1138-47.
140. Nishioka C, Liang HF, Chung CF, Sun SW. Disease stage-dependent relationship between diffusion tensor imaging and electrophysiology of the visual system in a murine model of multiple sclerosis. Neuroradiology 2017;59:1241-50.
141. Wuerfel E, Infante-Duarte C, Glumm R, Wuerfel JT. Gadofluorine M-enhanced MRI shows involvement of circumventricular organs in neuroinflammation. J Neuroinflammation 2010;7:70.
142. Beaulieu C. The basis of anisotropic water diffusion in the nervous system - a technical review. NMR Biomed 2002;15:435-55.
143. Sun SW, Liang HF, Schmidt RE, Cross AH, Song SK. Selective vulnerability of cerebral white matter in a murine model of multiple sclerosis detected using diffusion tensor imaging. Neurobiol Dis 2007;28:30-8.
144. Chiang CW, Wang Y, Sun P, et al. Quantifying white matter tract diffusion parameters in the presence of increased extra-fiber cellularity and vasogenic edema. Neuroimage 2014;101:310-9.
145. Nishioka C, Liang HF, Barsamian B, Sun SW. Sequential phases of RGC axonal and somatic injury in EAE mice examined using DTI and OCT. Mult Scler Relat Disord 2019;27:315-23.
146. Lin TH, Chiang CW, Perez-Torres CJ, et al. Diffusion MRI quantifies early axonal loss in the presence of nerve swelling. J Neuroinflammation 2017;14:78.
147. Lin TH, Kim JH, Perez-Torres C, et al. Axonal transport rate decreased at the onset of optic neuritis in EAE mice. Neuroimage 2014;100:244-53.
148. Gadjanski I, Boretius S, Williams SK, et al. Role of n-type voltage-dependent calcium channels in autoimmune optic neuritis. Ann Neurol 2009;66:81-93.
149. Darquié A, Poline JB, Poupon C, Saint-Jalmes H, Le Bihan D. Transient decrease in water diffusion observed in human occipital cortex during visual stimulation. Proc Natl Acad Sci U S A 2001;98:9391-5.
150. Tsurugizawa T, Ciobanu L, Le Bihan D. Water diffusion in brain cortex closely tracks underlying neuronal activity. Proc Natl Acad Sci U S A 2013;110:11636-41.
151. Spees WM, Lin TH, Song SK. White-matter diffusion fMRI of mouse optic nerve. Neuroimage 2013;65:209-15.
152. Lin T H, Spees W M, Chiang C W, et al. Diffusion fMRI detects white-matter dysfunction in mice with acute optic neuritis. Neurobiol Dis 2014;67:1-8.
153. Duong TQ. Magnetic resonance imaging of the retina: from mice to men. Magn Reson Med 2014;71:1526-30.
154. Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol 2001;11:107-16.
155. Praet J, Guglielmetti C, Berneman Z, Van der Linden A, Ponsaerts P. Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci Biobehav Rev 2014;47:485-505.
156. Namekata K, Kimura A, Harada C, Yoshida H, Matsumoto Y, Harada T. Dock3 protects myelin in the cuprizone model for demyelination. Cell Death Dis 2014;5:e1395.
157. Hainz N, Becker P, Rapp D, et al. Probenecid-treatment reduces demyelination induced by cuprizone feeding. J Chem Neuroanat 2017;85:21-6.
158. Bagchi B, Al-Sabi A, Kaza S, et al. Disruption of myelin leads to ectopic expression of K(V)1.1 channels with abnormal conductivity of optic nerve axons in a cuprizone-induced model of demyelination. PLoS One 2014;9:e87736.
159. Kojima W, Hayashi K. Changes in the axo-glial junctions of the optic nerves of cuprizone-treated mice. Histochem Cell Biol 2018;149:529-36.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Cordano C, Ramos C, Arnow S, Cruz-Herranz A, Guglielmetti C, Iester M, Bandini F. Inflammation in the anterior visual pathway in multiple sclerosis: what do the animal models teach us?. Neurosciences 2021;8:185-202. http://dx.doi.org/10.20517/2347-8659.2020.54
AMA Style
Cordano C, Ramos C, Arnow S, Cruz-Herranz A, Guglielmetti C, Iester M, Bandini F. Inflammation in the anterior visual pathway in multiple sclerosis: what do the animal models teach us?. Neuroimmunology and Neuroinflammation. 2021; 8(3): 185-202. http://dx.doi.org/10.20517/2347-8659.2020.54
Chicago/Turabian Style
Cordano, Christian, Claudia Ramos, Sam Arnow, Andrés Cruz-Herranz, Caroline Guglielmetti, Michele Iester, Fabio Bandini. 2021. "Inflammation in the anterior visual pathway in multiple sclerosis: what do the animal models teach us?" Neuroimmunology and Neuroinflammation. 8, no.3: 185-202. http://dx.doi.org/10.20517/2347-8659.2020.54
ACS Style
Cordano, C.; Ramos C.; Arnow S.; Cruz-Herranz A.; Guglielmetti C.; Iester M.; Bandini F. Inflammation in the anterior visual pathway in multiple sclerosis: what do the animal models teach us?. Neurosciences. 2021, 8, 185-202. http://dx.doi.org/10.20517/2347-8659.2020.54
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 15
likes
Like This Article 15
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.