
Intestinal inflammation and compromised barrier function in idiopathic parkinsonism: scenario captured by systematic review

Abstract
Aim: To address how common are intestinal inflammation and compromised barrier function in idiopathic parkinsonism (IP), any potential treatment benefits, outcome of not treating, and whether screening is worthwhile. This may provide the missing link between systemic/brain inflammation in IP and implicated gastrointestinal microbiota/specific pathogens.
Methods: Search strategy was based on PRISMA guidelines. Fifteen of the 1395 articles (1995-2020) identified met the inclusion criteria. Seven gave results on more than one intestinal modality: inflammation, permeability, integrity, and bacterial translocation.
Results: The inter-relationship of IP with intestinal inflammation and bacterial translocation is firmly established, lacking only random sample surveys to meet Level-1 Oxford Centre for Evidence-Based Medicine evidence. Evidence for reduced integrity is limited to 2 small studies of tight-junction proteins in colonic biopsies. No overall conclusion can be drawn from studies of faecal and circulating markers of integrity: evidence based on an assay that recognizes wider zonulin family, not the specific peptide exclusively, was censored. Evidence for increased permeability is insubstantial: further work is needed in IP with and without small-intestinal-bacterial-overgrowth and including a non-fermentable sugar absorption test. Concentrations of markers of intestinal inflammation (faecal calprotectin) and bacterial translocation (circulating lipopolysaccharide-binding protein) appear not to change with time-since-diagnosis or IP severity. This is compatible with a pre-presentation insult. There are no longitudinal studies on inflammation or translocation to guide design of interventional studies. Neither are cut-points discriminant for IP-facets, or gradients prognostic for its evolution, defined.
Conclusion: Intestinal inflammation and barrier function is a strategic junctional point in the hypothesis for the aetipathogenesis of IP.
Keywords
Introduction
The gastrointestinal tract is involved in idiopathic parkinsonism (IP), which encompasses Parkinson’s disease, from the early symptom of constipation to shared histological findings between enteric nervous system and basal ganglia in established disease[1]. Constipation doubles the risk of diagnosis of IP 10 years on, severity increasing that risk[2]. Chronic colitis, with CD4+ cell mucosal infiltration, was found in five of six IP-patients with constipation[3].
The risk ratio for IP in Crohn’s disease and ulcerative colitis is similar at 1.4[4]. Two genetic risk factors for inflammation, leucine-rich repeat kinase 2 (LRRK2) risk alleles[5] and mutation in nucleotide-binding oligomerization domain-containing protein 2 (NOD2)[6,7] are shared between Crohn’s disease and IP. LRRK2 deficiency leads to aberrant macrophage activation and increased susceptibility to experimentally-induced colitis[8]. The role of NOD2 in controlling inflammation by maintaining equilibrium between intestinal microbiota, mucosa, and host immune responses is lost with mutations[9]. Furthermore, many patients with inborn errors of immunity due to single gene defects suffer from chronic intestinal inflammation[10]. Genes which are defective in these rare disorders also harbour IP-associated polymorphisms [e.g., interleukin (IL)-10, IL-10RA, IL-10RB][11]. These observations provide further support for the inflammatory aetiopathogenesis hypothesis of IP[1] and highlight the need for the present systematic review of intestinal inflammation and compromised barrier function therein.
There is indicative evidence that the alimentary microbiota is a major aetiopathogenic driver of IP[1]. However, it is unlikely that one set of drivers and mediators will explain every aspect of the syndrome directly. More likely, there are multiple junctions at which individual or sets of disease-facets can be modified, exacerbated, or ameliorated. One such scenario involves Helicobacter pylori. There is Oxford Centre for Evidence-Based Medicine (OCEBM)[12] level-1 evidence that successful H. pylori eradication in IP is disease-modifying (even in anti-parkinsonian treatment-naïve patients) but not preventive[13]: hypokinesia regresses with eradication, and overall motor severity lessens. However, H. pylori eradication may influence gastrointestinal microbiota adversely, unlocking the next stage in the natural history, the development of rigidity[1]. Indeed, a cumulative increase in rigidity is reported with successive antimicrobial courses for intercurrent infection in IP[14]. On the other hand, maintenance laxative therapy is associated with plateauing of the year-on-year 5% increase in rigidity, as though improved gastrointestinal motility promotes a less harmful intestinal microbiota[15].
Alimentary microbial imbalance or misplacement in IP has its own complex scenario. Small-intestinal-bacterial-overgrowth (SIBO) is found in between a quarter and two-thirds of IP-probands, using a lactulose-hydrogen breath-test[16-18]. It is likely to be due to stagnation in, and reflux from, the proximal colon, in turn a consequence of the distinctive pathology, namely segmental delay in the transverse colon, of IP[2]. In IP, SIBO is associated with higher circulating natural-killer (NK) and CD4+ lymphocyte counts, and lower neutrophils[16]. Facets of IP have directionally similar associations with these leucocyte subsets. Rigidity and hypokinesia are characterised by higher NK and CD4+ counts, the latter apparently modulating the NK-effect on rigidity. It should be noted that CD4+ includes regulatory T-cells which inhibit NK effector-mechanisms. Tremor is characterised by a lower neutrophil count, perhaps reflecting gastrointestinal sequestration in response to a particular microbial signature.
We adopt the core concept that neuro-inflammation in IP is mediated by systemic immunoinflammatory processes, and is not just reactive to aberrant protein deposition or degenerating neurons[1]. The position of a patient within the disease spectrum is determined by current and previous exposome (e.g., intestinal microbiota, modifying factors such as tobacco smoking), immunome, and faecal metabolome and their interaction with host genetics (risk and inflammatory). Intervention against drivers and mediators, and their perpetuators, would allow disease modification.
Here we attempt, by systematic review, to capture the third scenario, the connection between systemic/brain inflammation and gut microbiota/specific pathogens in IP. The missing link may be intestinal inflammation and compromised barrier function, caused by a pro-inflammatory alteration in the microbiota, a specific pathogen or lack of microbial products critical to epithelial maintenance and immune regulation[19]. We seek evidence for intestinal inflammation, increased permeability, disrupted integrity and translocation of microbes or their products in IP by comparison with normality.
Methods
Search strategy and study selection
This systematic review was conducted and reported in line with the “Preferred Reporting Items for Systematic Reviews and Meta-Analyses” (PRISMA) guidelines[20]. The search strategy was based on the “Population, Intervention, Comparison, Outcome” (PICO) framework: P = people with or without IP; I = any with respect to intestinal inflammation and barrier function; C = comparison of IP-status, and severity/manifestations according to inflammation and barrier function; O = frequency of pathology according to disease-status, severity and manifestations.
The literature search was conducted using the Ovid EMBASE online database, with restriction to full original articles published between 1995 and 2020 in peer-reviewed scientific journals, in, or with translation to, English. The systematic search combined two groups of keywords, one for the IP disease target and the other for the intestinal modality [Table 1]. Here, we opt to use the term “idiopathic parkinsonism” to refer to the target syndrome, with its variable combination of defined signs, in the absence of a recognised cause. The second group included keywords relating to the four intestinal modalities, inflammation, permeability, integrity and bacterial translocation, and targets, markers and tests used to assess these. The aim was to include observational (cohort or case-control) studies and any intervention trials. Excluded from the systematic review (but not from its framing narrative review) were case reports, reviews, and meta-analyses.
Search terms used in Ovid EMBASE online database
| Keywords: disease target | Keywords: intestinal modalities | |||
|---|---|---|---|---|
| Parkinson’s disease
idiopathic parkinsonism Parkinsons Parkinson* | intestinal inflammation
enteritis calprotectin lactoferrin calgranulin | intestinal permeability
intestinal mucosa permeability sugar test: - lactulose, mannitol, (L-) rhamnose, sucrose, sucralose, (D-) xylose | Intestinal
Integrity† tight junction, cingulin, claudin, junctional adhesion molecule, occludin, zonula occludens [ZO-1 (2, 3)] zonulin alpha-1-antitypsin, (AAT) (intestinal) fatty acid binding protein (2), (FABP2) diamine oxidase | bacterial translocation
bacterial DNA (bactDNA) lipopolysaccharide (LPS), endotoxin lipopolysaccharide binding protein (LBP) |
Figure 1 summarises the article selection process from search results, through screening and compliance with eligibility criteria, to inclusion. Two reviewers (C.U. and A.D.A) scanned the search results independently. First, sources, titles, and abstracts of the search results were checked to eliminate conference abstracts and original articles ineligible by language or nature (i.e., animal and cell studies). Full texts of possibly relevant articles were then retrieved and examined for eligibility. After each step, the reviewers held a consensus meeting to discuss any discrepancies in selection. A third reviewer (R.J.D.) was available to assess any equivocal selections. Several articles found in the search under one of the four modalities contributed to other modalities and so appear in more than one table of results.
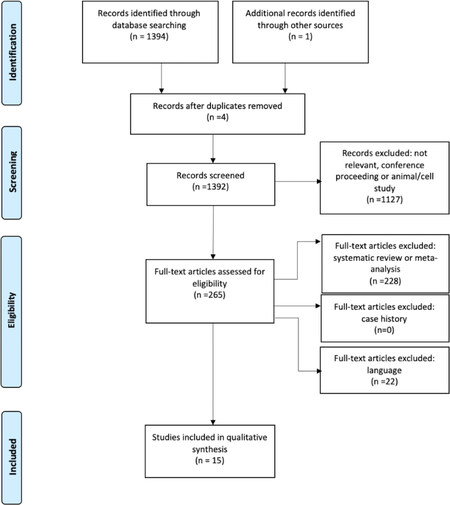
Figure 1. Preferred reporting for systematic review and meta-analysis (PRISMA) flow chart
Data extraction and questions addressed
The aim was to extract the following information from each included article: (1) citation; (2) type of study: cross-sectional observational comparison with or without retrospective/prospective data or interventional trial; (3) size of cohorts; (4) definition of disease-status of cohort(s) studied; (5) methodology for assessing intestinal modalities; (6) clinical outcome(s) in terms of association of intestinal modalities with disease-status or results of intervention on those modalities; (7) association between modalities; (8) association of a modality with disease duration and severity; and (9) country where the study was performed.
The OCEBM questions[12] to be addressed using the cumulative stratified evidence were: (1) How common is the problem (intestinal inflammation and/or compromised barrier function in IP)? (2) Does this intervention help (treatment benefits)? (3) What will happen if we do not add a therapy (prognosis)? And (4) is this (early detection) test worthwhile (screening)?
Results
Study characteristics
Search results
Of the 1,394 articles screened, 265 were assessed for eligibility, but only 15 met the inclusion criteria. However, 7 gave results on more than one intestinal modality. Results are split by modality into inflammation [Table 2][21-26], permeability [Table 3][26-30], integrity [Table 4][21,22,26,30-33], and bacterial translocation [Table 5][26,28,31,33-35]. Articles were from Europe (10/15), East Asia (1/15), and North America (4/15).
Cross-sectional observational studies of markers of intestinal inflammation in idiopathic parkinsonism
| Study | Number of participants | Marker of intestinal inflammation | Findings |
|---|---|---|---|
| Faecal calprotectin and lactoferrin | |||
| Schwiertz et al.[21] (2018)* | 34 with IP and 28 controls | Faecal calprotectina and lactoferrinb concentrations | Mean difference (95%CI) in concentration, IP minus controls, for calprotectin was 62 (17, 107) µg/g and for lactoferrin 2.8 (-4.4, 9.9) µg/g
Abnormal values (> 50 µg/g) for calprotectin found in 47 and 4%, respectively, and for lactoferrin (> 3 µg/g) 25 and 14% |
| Mulak et al.[22]* (2019) | 35 with IP and 20 controls | Faecal calprotectinc concentration | Median (25%-75% quartiles) calprotectin concentration was higher (P < 0.0001) in IP [55 (29, 138) µg/g] than in controls [10 (5, 23) µg/g]
Abnormal values (≥ 50 µg/g for < 60 y; ≥ 112 µg/g for > 60 y) for calprotectin found in 43% and 0%, respectively |
| Weis et al.[23]* (2019) | 34 with IP and 25 controls | Faecal calprotectina concentration | Abnormal values (> 50 µg/g) for calprotectin found in 41% of IP and 12% of controls |
| Faecal immune and angiogenic analytes | |||
| Houser et al.[24]* (2018) | 156 with IP and 110 controls (49 of whom IP-probands’ spouses) | 37 (immune and angiogenesis-related) analytes in faeces | Univariate analysis showed elevated concentrations of vascular endothelial growth factor receptor, IL-1α and IL-8 in IP compared with controls
Multivariate analysis showed elevated IL-1β, IL-1α, C-reactive protein and IL-8 in IP (threshold for significance P < 0.05) |
| Colonic biopsy for inflammatory cells and expression of cytokines and glial markers | |||
| Devos et al.[25]† (2013) | 19 with IP and 14 controls (requiring colonoscopy for colorectal cancer screening) | Expression of 4 pro-inflammatory cytokines and 3 enteric glial markers in ascending colon biopsiesd | Significantly greater mRNA expression of TNFα, IFNγ and IL-6 (2.5-fold), and of IL-1β (1.5-fold) in IP compared to controls
2 glial markers (GFAP and Sox-10 mRNA) up-regulated in IP compared to controls, and correlated with mRNA expression of pro-inflammatory cytokines |
| Perez-Pardo et al.[26] (2018) | 6 with IP and 6 controls | Expression of 2 cellular markers and 50 gene targets in distal sigmoid colon biopsiese | In lamina propria, significantly more TLR4+ cells in IP (mean ± SE 128 ± 30) than in controls (41 ± 10) and more CD3+ T cells in IP (89 ± 14) than controls (29 ± 6)
Significantly higher mRNA expression of TLR4 and CD3, cytokines (IFN-γ, IL-1β, IFN-β, IL-17A, IL-8) and IL-7R, and chemokines (CCL2, CCL5, CCR5) in IP than in controls |
Study design
There are no trials of intervention on intestinal inflammation or impaired barrier function in IP. All the studies were cross-sectional cohort comparisons, usually with unrelated controls, except for two studies where the comparator was reference values[29,32]. Four studies specified consecutive eligible IP-probands[29,31-33]. In one study, all controls were spouses of IP-probands, claimed to have no disease[33]. In another, half the controls were probands’ spouses, but this was taken into account in the analysis[24]: comparison between cohabiting and non-cohabiting controls showed no difference in stool faecal analytes. No sample size calculation was provided for any study. Cohort sizes are shown in Tables 2-5. Only one study declared that laboratory analysis was conducted blind to clinical data and diagnosis[21].
Cross-sectional observational studies of markers of intestinal permeability in idiopathic parkinsonism
| Study | Number of participants | Measure of intestinal permeability | Findings |
|---|---|---|---|
| Physiological tests | |||
| Davies et al.[27] (1995) | 15 with IP and 15 controls | Lactulose/mannitol differential sugar absorption test with 5- hour urine collection | Significantly reduced mannitol absorption in IP [mean 12 (range 2, 20) %] compared with controls [16 (7, 51) %] but no difference in lactulose absorption
Ratio lactulose/mannitol absorption significantly higher in IP 0.024 (0.007, 0.071) than in controls 0.012 (0.000, 0.038) |
| Forsyth et al.[28] (2011) | 9 with IP and 10 controls | Lactulose/mannitol and sucralose differential sugar absorption test with two sequential 12-hour urine collections | No significant difference in mannitol or lactulose absorption, or in ratio lactulose/mannitol, between IP and controls
Sucralose absorption in IP (mean ± SE 1.12% ± 0.1 % of oral dose) significantly greater than in controls (0.58% ± 0.1%) |
| Salat-Foix et al.[29] (2012) | 12 IP patients compared with pre-defined age-adjusted reference values | Lactulose/mannitol and sucrose differential sugar absorption test ‘before bed’ with ‘overnight urine’ collection | Lactulose/mannitol ratio abnormal in three patients and sucrose absorption in three, both being abnormal in two, compared with reference values |
| Perez-Pardo et al.[26] (2018) | 6 with IP and 6 healthy controls | Lactulose/mannitol and sucralose differential sugar absorption test with 24 h urine collection, including separate initial 5 h sample | No difference in recovery of lactulose, mannitol, or the lactulose/mannitol ratio.
Significantly higher mean (SE) 24 h sucralose excretion in IP (2.33% ± 0.30 %) compared with controls (0.97% ± 0.28%) |
| Direct measurement in colonic biopsies | |||
| Clairembault et al.[30] (2015) | 31 with IP and 11 controls | Three sigmoid/descending colon biopsies per individual for assessment of para-cellular permeability by sulfonic acid flux and trans-cellar by horseradish peroxidase flux | No significant difference in para- or trans-cellular permeability between IP and controls |
Cross-sectional observational studies of markers of intestinal integrity in idiopathic parkinsonism
| Study | Number of participants | Marker of intestinal integrity | Findings |
|---|---|---|---|
| ZO-1 and occludin | |||
| Clairembault et al.[30] (2015) | 31 with IP and 11 controls | Sigmoid and descending colon biopsy ZO-1 and occludin by Western blot and immunofluorescence | Significantly lower expression of occludin in IP, but not of ZO-1, compared with controls Normal reticular pattern of both occludin and ZO-1 staining in 6/8 controls but only 9/31 IP (P = 0.02) |
| Perez-Pardo et al. [26] (2018) | 6 with IP and 6 healthy controls | Distal sigmoid colon biopsy for ZO-1 by immunocytochemistry | Significant disruption in ZO-1 expression in IP (mean ± SE 1.58 ± 0.05) compared with controls (2.32 ± 0.18) (P = 0.003) |
| Zonulin and a-1-antitrypsin | |||
| Schwiertz et al.[21] (2018) | 34 with IP and 28 controls | Faecal zonulina Concentration Faecal a-1-antitrypsinb concentration | Mean difference (95%CI) between zonulin concentration in IP and controls of 57 (26, 89) ng/ml. Abnormal values (> 78 ng/ml) in 44 and 14%, respectively Mean difference (95%CI) between a-1-antitrypsin concentration in IP and controls of 25 (11, 39) mg/dL, Abnormal values (> 56 mg/dL) in 75 and 29%, respectively |
| Mulak et al.[22] (2019) | 35 with IP and 20 controls | Faecal zonulina concentration | No significant difference in zonulin concentration between IP [median (25%-75% quartiles) 162 (66, 276) ng/ml] and controls [128 (67, 264) ng/ml] |
| Loffredo et al.[31] (2020) | 8 with IP and 64 controls | Serum zonulinc concentration | Significantly higher zonulin concentration in IP (mean ± SE 3.4 ± 0.7 ng/ml) than in controls (1.6 ± 0.6 ng/ml) |
| Dufek et al.[32] (2009) | 29 with IP compared with healthy blood donor reference values | Serum a-1-antitrypsind concentration | No abnormality in a-1-antitrypsin Concentration |
| Diamine oxidase | |||
| Hasegawa et al.[33] (2015) | 52 with IP and 36 probands’ spouses | Serum diamine oxidasee | Diamine oxidase concentration not significantly different in IP [mean ± SD 19.5 ± 13.0) ng/ml] than in controls (16.8 ± 7.3 ng/ml) |
Cross-sectional observational studies of markers of bacterial translocation in idiopathic parkinsonism
| Study | Number Participants | Marker of bacterial translocation | Findings |
|---|---|---|---|
| Circulating lipopolysaccharide binding protein | |||
| Pal et al.[34] (2015) | 94 with IP and 97 controls | Plasma LBPa | Significantly lower LBP in IP (mean ± SD 9.3 ± 6.7 µg/ml) than in controls (11.3 ± 7.4 µg/ml) (P = 0.02) |
| Perez-Pardo et al.[26] (2019) | 6 with IP and 6 controls | Plasma LBPa | Significantly lower LBP in IP (mean ± SE 15.7 ± 3.8 µg/mL) than in controls (33.5 ± 6.2 µg/ml) |
| Hasegawa et al.[33] (2015) | 52 with IP and 36 probands’ spouses | Serum LBPa | Significantly lower LBP in IP (mean ± SD 7.8 ± 2.4 µg/ml) than in controls (10.1 ± 5.1 µg/ml) |
| Serum lipopolysaccharide | |||
| Loffredo et al.[31] (2020) | 8 with IP and 64 controls | Serum LPSa | Significantly higher LPS in IP (Mean ± SD 29 ± 5 pg/ml) than in controls (12 ± 6 pg/ml) |
| Wijeyekoon et al.[35] (2020) | 41 with IP and 41 case controls | Serum LPSa,b* | Significantly higher LPS in IP than in controls (P = 0.02) |
| Circulating lipopolysaccharide binding protein, lipopolysaccharide and colonic biopsy immunohistochemistry | |||
| Forsyth et al.[28] (2011) | 9 with IP and 10 controls | Plasma LPBc Serum LPSb Plasma LPS IgG core antibodiesc Serum co-receptor for LPS (soluble CD14)d Immunochemical staining of sigmoid biopsies for E. coli | Significantly lower LBP in IP (mean ± SD 22.9 ± 5.5 µg/ml) than in controls (84.3 ± 31.4 µg/ml)
No significant difference in LPS, LPS core antibodies, or soluble CD14 Higher score for intensity of E. coli staining, in epithelium and lamina propria, in IP than in controls |
In nine studies, IP and control groups were matched for age[21-26,31,33,35], in three for gender[27,31,35], and in one also for genotype relating to tau protein dementia[35]. Of the non-gender matched, non-reference value studies, there were more males than females with IP in all but one[33]. The reverse was true in controls, there being more females than males, except for in three studies[28,30,33].
Participant characteristics
Idiopathic parkinsonism was defined by the UK PD Society Brain Bank Criteria[36] in 11 of the 15 studies[21,23-26,29,30,32-35]. Cases were diagnosed by a hospital physician in three studies [22,31,33], clinically diagnosed in one study[28], and recruited from a Parkinson’s Disease Association in one study[33]. Where information was available, mean/median duration of IP was 4-10 years[21-23,25-27,29,30,35], with an outlier of 2 years median duration (with no dopaminergic treatment except in one participant)[28]. Hoehn and Yahr[37] functional staging was 3 (i.e., balance impairment with mild to moderate disease but physically independent)[21-23], 2 (i.e., symptoms on both sides but no impairment of balance)[26,28,29,34], or ≤ 2[35]. It was declared that some IP-probands were taking levodopa in 11 studies[21-23,25-27,29,30,32,33,35], and all were taking dopaminergic medication in five studies[21,23, 27,29,32]. Two studies declared laxative usage (6%) in IP patients[21,23], another study reported no laxatives usage[26].
Cognitive deficiency in IP was an exclusion in two studies[25,32]. More anxiety, depression and sleep disorder was noted in IP than in controls in one study[24]. A study of intestinal inflammation excluded those with questionnaire-based constipation[26]. A permeability study excluded irritable bowel syndrome and anorectal dysfunction30, another used the lactulose-hydrogen breath-testing to exclude SIBO[28]. A study of bacterial translocation excluded those taking steroids or non-steroidal anti-inflammatory drugs (NSAID), those with current infection, recent surgery or a chronic inflammatory, or autoimmune disorder[35]. Diet was specified as omnivorous with no pre- or probiotics for 3 months in two studies[21,23], no probiotics specified in one[26], and no antimicrobials for 2-3 months in five[21-23,26,27]. No difference in probiotics, diet, or antimicrobial usage between IP and controls was stated in another study[24], and no medication for digestive problems[24] and no co-existing chronic disease in IP[27] in others. The potential for false positives for calprotectin was considered in those on proton pump inhibitors (PPI)[21] or NSAID[22,24,25].
Inclusion criteria for controls in two studies were being without either pre-existing medical condition or chronic medication[21,23]. In five studies controls were described as healthy[22,25,27,30,32], this being qualified in two by requiring a total colonoscopy for colorectal cancer screening[25,30]. Specified exclusions in controls were IP symptoms, cognitive deficiency, irritable bowel syndrome, or anorectal dysfunction[30]; a history of neurological disease or self-reported memory problems or depression[35]; and taking parkinsonism-inducing drugs, history of gastrointestinal malignancy, diagnosis of inflammatory bowel disease, disease that impairs gastrointestinal motility, acute illness requiring hospitalisation, or severe malnutrition[34].
Study findings
The markers and measures used for each intestinal modality are briefly set in context, before describing the results and tabulating relevant studies, in chronological order by type of test.
Inflammation
Colonic inflammation can be demonstrated by cellular infiltrates in biopsies and localised cytokine release. Elevated faecal calprotectin indicates migration of neutrophils into the intestinal mucosa[38]. Up to 60% of the soluble protein content of neutrophil cytosol is calprotectin, which is released by neutrophil activation. The amount of calprotectin released reflects the number of neutrophils participating in the inflammation, with the context of patchy intestinal inflammation and lower bacterial load in small bowel than in colon being considered in the interpretation[39]. Lactoferrin is a major component of neutrophil secondary granules and is released by degranulating during the inflammatory process[40].
Of the six eligible articles on intestinal inflammation [Table 2], three concentrated on faecal calprotectin[21-23] (plus lactoferrin in one21), one on faecal immune and angiogenesis-related analytes[24], and two on colonic biopsies for mononuclear cells and/or expression of cytokines[25,26] (plus glial markers in one[25]).
In studies with 55-62 participants, abnormal values[41] for faecal calprotectin were more frequent in IP than controls, both when counting the number of participants exceeding a single cut-point (50 µg/g)[21,23] and when referring to two age-defined cut-points (50 µg/g for < 60 years of age, and 112 µg/g for > 60 years)[22]. Faecal calprotectin concentration was significantly higher, three- to six-fold, in IP than controls[21,22]. For faecal lactoferrin, there was no significant difference between IP and controls, numerically or in the proportion exceeding a single cut-point[21]. Calprotectin concentration was not associated with disease duration[21,22] or with levodopa or equivalent dopaminergic dose[22].
In a study with 266 participants, of 37 faecal immune and angiogenesis-related analytes, interleukins IL-1α and IL-8 discriminated for IP-status in both univariate and multivariate analysis[24]. Gender, body mass index, history of smoking, and use of probiotics influenced faecal analyte concentrations strongly. Multivariate analysis accounting for these potential covariates confirmed elevated faecal concentrations of IL-1α and IL-8, and revealed elevated IL-β and C-reactive protein, in IP.
In a study of ascending colon biopsies from 33 participants, mRNA expression of all four pro-inflammatory cytokines studied [tumour necrosis factor-alpha (TNF-α), interferon-gamma (INF-γ), IL-6, and IL-1β] and two of the three glial markers [glial fibrillary acidic protein (GFAP) and SRY-Box transcription factor-10 (Sox-10)] was significantly greater in IP than in controls[25]. All, except GFAP, were negatively correlated with disease duration. However, no correlation was seen with age or disease severity with respect to axial involvement. There was no difference in marker expression between IP-probands with and without enteric Lewy pathology. In a study with distal sigmoid colonic biopsies from 12 participants[26], increased expression of Toll-like receptor-4 (TLR4+) and CD3+ T cells was seen in IP compared with controls, on immunofluorescence histopathology and confirmed by gene expression. The increased expression of IFN-γ, IL-1β[25] in IP was replicated here[26], and, four other cytokine and three chemokine targets were implicated.
Permeability
Intestinal permeability is defined as the modality by which intestinal epithelium allows molecules to pass through by non-mediated passive diffusion[28]. Passage may be para-cellular or trans-cellular. Mannitol, a readily-absorbed monomer, serves as a marker of trans-cellular uptake, whereas lactulose, a dimer, which is only slightly-absorbed, serves as a marker for para-cellular uptake[42]. That is, mannitol can traverse pores of all sizes, whereas lactulose can only pass through larger pores. An elevated lactulose to mannitol ratio is one indicator of intestinal barrier dysfunction. Increased lactulose absorption, as opposed to decreased mannitol, has been shown in 34 gastroenterology patients with “colonic-type” microbiota on culture of small intestinal aspirate compared with 34 asymptomatic volunteers[43]. (It was confirmed that none of the gastroenterology patients had villous atrophy.) This was despite fermentation of lactulose by the SIBO, reducing its bioavailability.
The first[27] of four studies using differential sugar absorption to measure permeability in IP [Table 3] is the largest (30 participants). It showed the lactulose/mannitol absorption ratio to be doubled in IP compared with controls, the increased ratio being attributable entirely to reduced mannitol absorption. Those lactulose-hydrogen breath-test positive for SIBO had been excluded to avoid a false-negative. Two subsequent studies in IP[26,28], with 19 and 12 participants respectively, found no difference by IP-status in lactulose or mannitol recovery or their ratio.
Oral sucralose is absorbed through large pores in the colon as well as the small intestine. As it is non-fermentable, increased recovery in the presence of a normal lactulose/mannitol ratio[26,28] might reflect increased colonic permeability.
Sucrose is rapidly degraded into glucose and fructose after leaving the stomach, so increased urinary recovery would reflect increased gastric permeability. Three studies included sucrose in the sugar mix[26,28,29]. However, only one[29] reported results, finding that 3/12 IP-probands had abnormally high sucrose recovery compared with a reference population.
Direct measurement of para- or transcellular permeability, using sulfonic acid and horseradish peroxidase flux, in sigmoid and descending colon biopsies from 42 participants, failed to show a difference by IP-status[30].
Integrity
Epithelial tight junctions are formed by transmembrane proteins, such as occludins, claudins, and junction adhesion molecule proteins, connected to the actin cytoskeleton via the high molecular weight proteins, zona occludens[30] (of which ZO-1 was the first to be cloned).
Two studies of sigmoid/descending colon biopsies [Table 4] used immunofluorescence to detect ZO-1. Staining was present in less than a third of 31 IP-probands, but in nearly three-quarters of the 11 controls, in one study[30]. Significant disruption in ZO-1 expression was seen in 6 IP-probands compared with 6 controls in the other[26]. However, on Western blotting, there was no difference in ZO-1 expression, but occludin expression was lower in IP[30].
Zonulin is the only human protein known to regulate intestinal permeability by modulating the function of tight junctions[44]. Small intestinal exposure to bacteria is a trigger for zonulin release. However, in 2019, the nature of the antigen detected in serum by two commercial “zonulin” ELISA assays was investigated using immunoprecipitation, followed by mass spectrometric analysis and sodium dodecyl sulphate-polyacrylamide gel electrophoresis, with protein staining[45]. The top match for the antigen was complement C3: the authors concluded that the “zonulin assay” was not detecting the actual protein, prehaptoglobin-2. Recent studies show a potential role for complement C3, secreted by intestinal epithelial cells, in modulating barrier integrity[46,47]. Whether complement C3 acts in concert with, or is independent of, the zonulin pathway is undetermined.
Two studies[21,22] of faecal zonulin, published in 2018 and 2019, used a commercial enzyme-linked immunosorbent assay (ELISA) assay from one of the above manufacturers [Table 4]. One claimed a significant (2-fold) greater faecal zonulin concentration in IP than in controls, and that three-times as many IP-probands had abnormal values[21]. The other showed no significant difference in concentration between IP and controls[22]. A later study, 2020, using nephelometry found a 2-fold higher serum zonulin in 8 IP-probands than in 64 controls[31].
Alpha-1-antitrypsin, a protease inhibitor, reflects loss of proteins to the intestinal lumen. The study [Table 4] with 62 participants[21] found a significant (1.5-fold) increase in faecal alpha-1-antitrypsin in IP compared with controls, which translated into a three-fold greater number of IP-probands than controls having abnormal values. However, another study with 29 IP-probands found serum concentrations were not different from reference values[31].
Diamine oxidase is a regulating enzyme in rapidly proliferating tissues, such as intestinal mucosa. Plasma concentrations of diamine oxidase fall with experimental hyperosmolar mucosal damage in rats,[48] but increase with ischaemic damage in dogs[49]. A study in 88 human participants[33] showed no difference in serum diamine oxidase between IP-probands and their controls. However, all controls were probands’ spouses, so may have shared environmental aetiopathogenic influences.
Translocation
Intestinal barrier dysfunction permits the passage of microbes and their products into the intestinal mucosa and systemic circulation. The bacterial product lipopolysaccharide (LPS) has been widely studied in this respect. Its release from gram negative bacteria does not require the destruction of the bacterial cell wall, since it is secreted as part of normal outer membrane vesicle trafficking. It binds to the soluble acute-phase protein, lipopolysaccharide-binding protein (LBP), presenting LPS to cell surface pattern receptors such as CD14 and TLR4, which are involved in consequent innate immunity. The term ‘endotoxin’ is mostly used synonymously with LPS.
The studies [Table 5] are remarkably consistent in their results, showing lower circulating LBP[26,28,33,34], and higher LPS[31,35] in IP than in controls, with one exception. In that study[28], LPS (and its core IgG antibodies and co-receptor, soluble CD14) were not significantly different between IP and controls. However, the latter study showed direct evidence of E. coli translocation into the epithelium and lamina propria of the sigmoid colon.
Discussion
Diverse study characteristics revealed by this systematic review reflect the range of avenues to be explored. However, diversity, and in some cases curtailed numerical description of results, constrains overall interpretation. Most of the information addresses whether intestinal inflammation and compromised barrier function are features of IP, but there are some insights into their role in early detection and their prognostic implications. The markers have not been targeted in interventional studies.
Four questions addressed by systematic review
How common is the problem?
Intestinal inflammation is more common in IP compared with controls, as judged by higher faecal calprotectin concentrations. The three local non-random sample studies[21-23], consolidated by this systematic review, provide level-2 OCEBM evidence. Applying the usual cut-point of 50 µg/g to distinguish those with intestinal inflammation from healthy individuals[50], nearly half of IP-probands were abnormal[21,22]. Wide ranging severity of inflammation is implied by the values for IP exceeding cut-points (100 and 188 µg/g) for differentiating irritable bowel syndrome from inflammatory bowel disease[50]. Indeed, the maximum values given[21,22] would have a high positive predictive value for inflammatory bowel disease pathology[39]. Faecal lactoferrin appears to be a relatively insensitive marker of inflammation in IP[21]. However, the level 2 evidence is further consolidated by a large study[24] finding an increase in faecal immune and angiogenesis-related analytes in IP compared with controls. Moreover, two smaller studies of colonic biopsies[25,26], together showed greater mRNA expression of proinflammatory cytokines, glial markers, TLR4, and CD3, as well as more TLR4+ cells (presumed to be lymphocytes) and CD3+ T-cells.
Bacterial translocation is more common in IP, as judged by lower circulating LBP[26,28,33,34] and higher LPS[31,35]. In the one study where LPS was not higher in IP, LBP was lower[28]. This gives level 2 OCEBM evidence, consolidated by this systematic review and corroborated by finding greater E. coli immunostaining in colonic epithelium and lamina propria in IP compared with controls[28].
There is insubstantial evidence from the lactulose/mannitol differential sugar absorption test that intestinal permeability is increased in IP. Indeed, a case-series[51] (as such excluded from the systematic review), in IP-probands with normal faecal calprotectin and circulating transglutaminase antibody, showed normal ratios of lactulose and (the efficiently absorbed) 3-O-methyl-O-glucose to the monomer, L-rhamnose. Moreover, direct measurement of marker fluxes across colonic biopsies showed no difference in para- or transcellular permeability in IP[30]. However, in two small studies, the increased urinary recovery of the non-fermentable sucralose, in the presence of a normal lactulose/mannitol ratio, points to greater colonic permeability[26,28].
The evidence for compromised integrity rests on a replicated finding of disruption in the reticular pattern of ZO-1 staining in colonic biopsies from IP-probands compared with controls[26,30]. Regarding faecal and serum markers of integrity, α-1-antitrypsin concentration was increased in faeces[21], but not in serum[32], in IP. One study reports high serum zonulin, using nephelometry, in IP[31], whilst two others[21,22] were censored because of doubts as to the validity of the ELISA assay used (which recognizes the wider zonulin family, not exclusively the tight junction modulator, zonulin).
Does this intervention help (treatment benefits)?
Reducing intestinal inflammation and translocation are quantifiable pathophysiological study endpoints, but design of intervention studies will require longitudinal, observational information on their markers. Encouragingly, since addressing constipation is associated with plateauing of the year-on-year increment in rigidity[15], these markers may well be associated, not just with IP-status, but also with quantifiable IP-facets. However, caution is required in design of a long-term study to record or avoid where possible potential confounders such as PPIs, NSAIDs, alcohol[39], and antimicrobial courses for intercurrent infection[14].
What will happen if we do not add a therapy (prognosis)?
All four pro-inflammatory markers in one colonic biopsy study, and of one of the two glial markers, were inversely related to disease duration, but had no association with disease severity[25]. Thus, rather than intestinal inflammation increasing with time, it might become quiescent. However, the inverse association with disease duration was not echoed for faecal inflammatory markers[21,22,24]. Within-subject follow-up on faecal and serum intestinal markers will be more sensitive to change.
Is this (early detection) test worthwhile?
Markers of intestinal inflammation and translocation could contribute to detecting a pre-presentation IP state, where rescue is feasible. Longitudinal follow-up of these markers in constipation might predict the risk of IP evolving. Delay in transverse colon transit described in IP compared with controls[2] might be associated with increase in faecal calprotectin. It would be interesting to see to what extent the age-related increase in faecal calprotectin described[41] relates to undiagnosed IP or measures of a pre-presentation IP state.
Inter-relationships between intestinal modalities
More evidence is required to access whether the 4 intestinal modalities studied move in parallel and in what order. A deficient barrier, say due to short-chain fatty acid deficiency, might allow translocation, in turn setting up inflammation. Alternatively, an inflammatory response, say to microbial imbalance or misplacement, might compromise integrity. Although increased intestinal permeability in IP is not firmly established, two studies showed significant correlation of intestinal permeability (as measured by sucralose recovery) with translocation (as measured by intensity of E. coli staining[28] or decreased circulating LBP[26]). One of these studies[26], reported that increased permeability and reduced integrity (ZO-1 staining) were associated with intestinal inflammation (expression of inflammatory mediators and greater TLR4+ and CD3+ cell response).
Mechanisms
The “decades-before-diagnosis” constipation and peptic ulceration of IP fit with intestinal inflammation being an early feature in the aetiopathogenesis. Intestinal T-lymphocytes (and other immune cells, including NK) may be primed in the gut in the IP prodrome, trafficking to local mucosal lymph nodes, systemic circulation, and brain. There are biological gradients of blood leucocyte subtype counts and circulating inflammatory markers on objective measures of IP-facets[1,16]. Higher serum IL-6 is predictive of incident IP[52]. In early IP, inflammation (measured by imaging microglial activation) in the affected nigrostriatal pathway accompanies loss of presynaptic dopamine transporter[53] and becomes increasingly widespread on follow-up[54]. The simplest biologically plausible explanation is that the circulating leucocytes and immune-inflammatory markers represent mediators for neuroinflammation and neuronal damage, their driver being chronic intestinal inflammation with translocation of microbes/their products across the gut wall. Indeed, where intestinal inflammation is unremitting in IP and reflected in an aggressive neurological condition, there is an argument for adapting and applying experience gained in immunoregulatory therapies for inflammatory bowel disease[55].
Microbial imbalance and misplacement is a potential source of intestinal inflammation in IP. Lack of beneficial products of the normal microbiota may be more important than detrimental products. Integrity of the intestinal barrier is maintained by the microbiota metabolising dietary carbohydrates into short-chain fatty acids, a major source of nutrition for the epithelium[56]. Faecal short-chain fatty acids are reduced in IP compared with controls[57]. Tryptophan catabolites generated by the gut microbiota are also important to intestinal homeostasis[58]. Indole, for example, increases expression of genes involved in maintaining epithelial cell structure and function. Tryptophan deficiency is marked by reduced T-cell regulation, and specific dysregulation of tight junctions. The microbiota is also involved in maintenance of the protective mucus layer. Furthermore, it moulds the specificity of luminal IgA which limits adhesion of microbes to, and their entry into, the epithelium[56].
Toll-like receptor 4 is a transmembrane protein belonging to the pattern recognition receptor family. It is required for phagocytosis (internalisation) by enterocytes of Gram-negative bacteria, and has a potential role in bacterial translocation[59]. Its activation signals intracellularly to activate the innate immune system via the NF-κB pathway. The TLR4 ligand, LPS, promotes macrophage differentiation toward the classically activated (M1) phenotype[60]. M1 macrophages are characterized by production of pro-inflammatory cytokines (e.g., IL-1, IL-6, IL-12, TNF-α, and chemokines) and promote differentiation of inflammatory T cells (e.g., Th1 and Th17), as opposed to M2 macrophages which are important for resolution of inflammation by production of anti-inflammatory cytokines (e.g., IL-10 and IL-13). The results of a small exploratory study[26], using colonic biopsies from IP patients and controls, fit with TLR4 activation [Table 2]. The authors proceeded to give the pesticide, rotenone (which interferes with electron transport chain in mitochondria, giving an IP-like syndrome in rodents), to TLR4-knockout and wild-type mice. The knockout mice showed less intestinal inflammation, intestinal and motor dysfunction, brain neuroinflammation, and neurodegeneration. TLR4 mediated intestinal inflammation might be a target for intervention in IP. In keeping with the apparent decrease in intestinal inflammation with disease duration, blood leukocytes from younger IP patients produced more TNFα after TLR4 stimulation by LPS, than cells from age-matched controls or older IP-patients[61].
Limitations
Evidence is limited, particularly in the assessment of whether intestinal integrity and permeability are impaired in IP. Serum concentrations of intestinal fatty-acid binding protein-2, which is reduced where integrity compromised in obesity[62], may give a clearer picture. The influence of SIBO on small intestinal permeability[42], and of Helicobacter on gastric permeability[63] in IP needs further study. Further data on permeability, as measured by sucralose recovery, are needed. Circulating microbial DNA fragments (pathogen-associated molecular pattern motifs, abundant in microbial, but rare in vertebrate, genomes) may enhance the investigation of translocation in IP.
Deviation, in IP and in constipation, from age-specific reference ranges for promising intestinal markers (e.g., calprotectin and LBP) needs to be measured. This groundwork, together with longitudinal follow-up, may allow subclassification of IP into progressive, relapsing/remitting, quiescent, or burnt out, and definition of a pre-presentation state. In ulcerative colitis, for example, faecal calprotectin can identify histological inflammation in patients in clinical and endoscopic remission[64].
In conclusion, intestinal inflammation and compromised barrier function provide a strategic junctional point for intervention in the pathophysiology of IP. Whether such intervention might modify the IP syndrome is the burning question.
In particular, markers of inflammation and translocation may provide the much needed quantifiable screen for disease activity, both in established IP and a pre-clinical state. They may allow rationalization of the nature of, and optimal timing for, disparate targeted interventions[65]. They could signal the need for containing the disease by addressing gastrointestinal drivers and mediators. As outcome criteria, they might provide an early signal of the efficacy of therapeutic interventions (e.g., diet, laxatives, probiotics, or gut microbiota- or metabolome-targeted therapy) in IP. As regards safety, they could monitor adverse effects of common concurrent potentially injurious therapies on the intestinal disease process (e.g., PPIs[66] and NSAIDs[67] may elevate faecal calprotectin). Genetic risk factors for colitis[4-9] may be an aggravating factor in IP, usefully signalled by markedly elevated inflammation and translocation measures.
Declarations
Authors’ contributionsSignificantly participated in drafting or substantive critical revision of the manuscript and provided approval of the final submitted version: all authors
Central to design and coordination of narrative and systematic reviews, interpretation of data and first drafting of manuscript: Umamahesan C, as a PhD student, under guidance from Dobbs RJ, Dobbs SM, Weller C, Charlett A, Taylor D, Augustin AD
Scanned search results independently: Umamahesan C, Augustin AD
Judged equivocal selections: Dobbs RJ
Special expertise in interpretation of results: Hayee BH, Ibrahim MAA
Reviewed statistical analyses: Charlett A
Responsible for conceptualization: Dobbs RJ, Dobbs SM, Hayee BH, Umamahesan C
Availability of data and materialsNot applicable.
Financial support and sponsorshipOur grateful thanks go to the Nomis Foundation, Zurich, Switzerland, Psychiatry Research Trust, London, UK, the Cecil Pilkington Charitable Trust, London, UK, and the Cyril Corden Trust, Letchworth Garden City, UK. Thanks also go to Gordon Roddick, Christopher Norris, Louise Barton, Richard and Diana Gloyn, Alex and Lyn Orr, Jamie and Julia Korner, and Stella Weston for their generous donations through the Psychiatry Research Trust.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Dobbs SM, Dobbs RJ, Weller C, et al. Peripheral aetiopathogenic drivers and mediators of Parkinson’s disease and co-morbidities: role of gastrointestinal microbiota. J Neurovirol 2016;22:22-32.
2. Tucker RM, Ryan S, Hayee BH, et al. Distinctive Pathophysiology Underlying Constipation in Parkinson’s Disease: Implications for Cognitive Inefficiency. J Clin Med 2020;9:1916.
3. Chen Y, Yu M, Liu X, et al. Clinical characteristics and peripheral T cell subsets in Parkinson’s disease patients with constipation. Int J Clin Exp Pathol 2015;8:2495-504.
4. Zhu F, Li C, Gong J, Zhu W, Gu L, Li N. The risk of Parkinson’s disease in inflammatory bowel disease: A systematic review and meta-analysis. Dig Liver Dis 2019;51:38-42.
5. Hui KY, Fernandez-Hernandez H, Hu J, et al. Functional variants in the
6. Bialecka M, Kurzawski M, Klodowska-Duda G, et al. CARD15 variants in patients with sporadic Parkinson’s disease. Neurosci Res 2007;57:473-6.
7. Ma Q, An X, Li Z, et al. P268S in NOD2 associates with susceptibility to Parkinson’s disease in Chinese population. Behav Brain Funct 2013;9:19.
8. Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol 2011;12:1063-70.
9. Negroni A, Pierdomenico M, Cucchiara S, Stronati L. NOD2 and inflammation: current insights. J Inflamm Res 2018;11:49-60.
10. Tangye SG, Al-Herz W, Bousfiha A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2020;40:24-64.
11. Lill CM, Roehr JT, McQueen MB, et al; 23andMe Genetic Epidemiology of Parkinson’s Disease Consortium., International Parkinson’s Disease Genomics Consortium., Parkinson’s Disease GWAS Consortium., Wellcome Trust Case Control Consortium 2). Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database. PLoS Genet 2012;8:e1002548.
12. The Oxford Centre for Evidence-Based Medicine (CEBM). OCEBM Levels of Evidence. Available from: https://www.cebm.ox.ac.uk/resources/levels-of-evidence/ocebm-levels-of-evidence. [Last accessed on 11 Jan 2021].
13. Tucker RM, Augustin AD, Hayee BH, et al. Role of
14. Dobbs SM, Charlett A, Dobbs RJ, et al. Antimicrobial surveillance in idiopathic parkinsonism: indication-specific improvement in hypokinesia following Helicobacter pylori eradication and non-specific effect of antimicrobials for other indications in worsening rigidity. Helicobacter 2013;18:187-96.
15. Augustin AD, Charlett A, Weller C, et al. Quantifying rigidity of Parkinson’s disease in relation to laxative treatment: a service evaluation. Br J Clin Pharmacol 2016;82:441-50.
16. Dobbs RJ, Charlett A, Dobbs SM, et al. Leukocyte-subset counts in idiopathic parkinsonism provide clues to a pathogenic pathway involving small intestinal bacterial overgrowth. A surveillance study. Gut Pathog 2012;4:12.
17. Fasano A, Bove F, Gabrielli M, et al. The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord 2013;28:1241-9.
18. Tan AH, Mahadeva S, Thalha AM, et al. Small intestinal bacterial overgrowth in Parkinson’s disease. Parkinsonism Relat Disord 2014;20:535-40.
19. Parada Venegas D, De la Fuente MK, Landskron G, et al. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front Immunol 2019;10:277.
20. Moher D, Shamseer L, Clarke M, et al; PRISMA-P Group. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev 2015;4:1.
21. Schwiertz A, Spiegel J, Dillmann U, et al. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Parkinsonism Relat Disord 2018;50:104-7.
22. Mulak A, Koszewicz M, Panek-Jeziorna M, Koziorowska-Gawron E, Budrewicz S. Fecal Calprotectin as a Marker of the Gut Immune System Activation Is Elevated in Parkinson’s Disease. Front Neurosci 2019;13:992.
23. Weis S, Schwiertz A, Unger MM, et al. Effect of Parkinson’s disease and related medications on the composition of the fecal bacterial microbiota. NPJ Parkinsons Dis 2019;5:28.
24. Houser MC, Chang J, Factor SA, et al. Stool Immune Profiles Evince Gastrointestinal Inflammation in Parkinson’s Disease. Mov Disord 2018;33:793-804.
25. Devos D, Lebouvier T, Lardeux B, et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis 2013;50:42-8.
26. Perez-Pardo P, Dodiya HB, Engen PA, et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut 2019;68:829-43.
27. Davies KN, King D, Billington D, Barrett JA. Intestinal permeability and orocaecal transit time in elderly patients with Parkinson’s disease. Postgrad Med J 1996;72:164-7.
28. Forsyth CB, Shannon KM, Kordower JH, et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One 2011;6:e28032.
29. Salat-Foix D, Tran K, Ranawaya R, Meddings J, Suchowersky O. Increased intestinal permeability and Parkinson disease patients: chicken or egg? Can J Neurol Sci 2012;39:185-8.
30. Clairembault T, Leclair-Visonneau L, Coron E, et al. Structural alterations of the intestinal epithelial barrier in Parkinson’s disease. Acta Neuropathol Commun 2015;3:12.
31. Loffredo L, Ettorre E, Zicari AM, et al; Neurodegenerative Disease study group. Oxidative Stress and Gut-Derived Lipopolysaccharides in Neurodegenerative Disease: Role of NOX2. Oxid Med Cell Longev 2020;2020:8630275.
32. Dufek M, Hamanová M, Lokaj J, et al. Serum inflammatory biomarkers in Parkinson’s disease. Parkinsonism Relat Disord 2009;15:318-20.
33. Hasegawa S, Goto S, Tsuji H, et al. Intestinal Dysbiosis and Lowered Serum Lipopolysaccharide-Binding Protein in Parkinson’s Disease. PLoS One 2015;10:e0142164.
34. Pal GD, Shaikh M, Forsyth CB, Ouyang B, Keshavarzian A, Shannon KM. Abnormal lipopolysaccharide binding protein as marker of gastrointestinal inflammation in Parkinson disease. Front Neurosci 2015;9:306.
35. Wijeyekoon RS, Kronenberg-Versteeg D, Scott KM, et al. Peripheral innate immune and bacterial signals relate to clinical heterogeneity in Parkinson’s disease. Brain Behav Immun 2020;87:473-88.
36. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181-4.
38. Lehmann FS, Burri E, Beglinger C. The role and utility of faecal markers in inflammatory bowel disease. Therap Adv Gastroenterol 2015;8:23-36.
39. Bjarnason I. The use of fecal calprotectin in inflammatory bowel disease. Gastroenterol Hepatol 2017;13:53-6.
40. Gisbert JP, McNicholl AG, Gomollon F. Questions and answers on the role of fecal lactoferrin as a biological marker in inflammatory bowel disease. Inflamm Bowel Dis 2009;15:1746-54.
41. Joshi S, Lewis SJ, Creanor S, Ayling RM. Age-related faecal calprotectin, lactoferrin and tumour M2-PK concentrations in healthy volunteers. Ann Clin Biochem 2010;47:259-63.
42. Musa MA, Kabir M, Hossain MI, et al. Measurement of intestinal permeability using lactulose and mannitol with conventional five hours and shortened two hours urine collection by two different methods: HPAE-PAD and LC-MSMS. PLoS One 2019;14:e0220397.
43. Riordan SM, McIver CJ, Thomas DH, Duncombe VM, Bolin TD, Thomas MC. Luminal bacteria and small-intestinal permeability. Scand J Gastroenterol 1997;32:556-63.
44. Fasano A. Zonulin, regulation of tight junctions, and autoimmune diseases. Ann N Y Acad Sci 2012;1258:25-33.
45. Ajamian M, Steer D, Rosella G, Gibson PR. Serum zonulin as a marker of intestinal mucosal barrier function: may not be what it seems. PLoS One 2019;14:e0210728.
46. Sina C, Kemper C, Derer S. The intestinal complement system in inflammatory bowel disease: Shaping intestinal barrier function. Semin Immunol 2018;37:66-73.
47. Sünderhauf A, Skibbe K, Preisker S, et al. Regulation of epithelial cell expressed C3 in the intestine - Relevance for the pathophysiology of inflammatory bowel disease? Mol Immunol 2017;90:227-38.
48. Luk GD, Bayless TM, Baylin SB. Diamine oxidase (histaminase). A circulating marker for rat intestinal mucosal maturation and integrity. J Clin Invest 1980;66:66-70.
49. Wolvekamp MC, de Bruin RW. Diamine oxidase: an overview of historical, biochemical and functional aspects. Dig Dis 1994;12:2-14.
50. Jha AK, Chaudhary M, Dayal VM, et al. Optimal cut-off value of fecal calprotectin for the evaluation of ulcerative colitis: An unsolved issue? JGH Open 2018;2:207-13.
51. Dobbs RJ, Dobbs SM, Weller C, et al. Role of chronic infection and inflammation in the gastrointestinal tract in the etiology and pathogenesis of idiopathic parkinsonism. Part 1: eradication of Helicobacter in the cachexia of idiopathic parkinsonism. Helicobacter 2005;10:267-75.
52. Chen H, O’Reilly EJ, Schwarzschild MA, Ascherio A. Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am J Epidemiol 2008;167:90-5.
53. Ouchi Y, Yoshikawa E, Sekine Y, et al. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol 2005;57:168-75.
54. Ouchi Y, Yagi S, Yokokura M, Sakamoto M. Neuroinflammation in the living brain of Parkinson’s disease. Parkinsonism & Related Disorders 2009;15:S200-4.
55. Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol 2017;14:269-78.
56. Brenchley JM, Douek DC. Microbial translocation across the GI tract. Annu Rev Immunol 2012;30:149-73.
57. Unger MM, Spiegel J, Dillmann KU, et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 2016;32:66-72.
58. Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun 2018;9:3294.
59. Neal MD, Leaphart C, Levy R, et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol 2006;176:3070-9.
60. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther 2017;2:17023.
61. Rocha Sobrinho HMD, Silva DJD, Gomides LF, Dorta ML, Oliveira MAP, Ribeiro-Dias F. TLR4 and TLR2 activation is differentially associated with age during Parkinson’s disease. Immunol Invest 2018;47:71-88.
62. Lau E, Marques C, Pestana D, et al. The role of I-FABP as a biomarker of intestinal barrier dysfunction driven by gut microbiota changes in obesity. Nutr Metab (Lond) 2016;13:31.
63. Sutherland L, Verhoef M, Wallace J, van Rosendaal G, Crutcher R, Meddings J. A simple, non-invasive marker of gastric damage: sucrose permeability. The Lancet 1994;343:998-1000.
64. Guardiola J, Lobatón T, Rodríguez-Alonso L, et al. Fecal level of calprotectin identifies histologic inflammation in patients with ulcerative colitis in clinical and endoscopic remission. Clin Gastroenterol Hepatol 2014;12:1865-70.
65. Pajares M, I Rojo A, Manda G, Boscá L, Cuadrado A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020;9:1687.
66. Poullis A, Foster R, Mendall MA, Shreeve D, Wiener K. Proton pump inhibitors are associated with elevation of faecal calprotectin and may affect specificity. Eur J Gastroenterol Hepatol 2003;15:573-4. author reply 574
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Umamahesan C, Augustin AD, Hayee BH, Ibrahim MAA, Taylor D, Weller C, Charlett A, Dobbs RJ, Dobbs SM. Intestinal inflammation and compromised barrier function in idiopathic parkinsonism: scenario captured by systematic review. Neurosciences 2021;8:313. http://dx.doi.org/10.20517/2347-8659.2020.57
AMA Style
Umamahesan C, Augustin AD, Hayee BH, Ibrahim MAA, Taylor D, Weller C, Charlett A, Dobbs RJ, Dobbs SM. Intestinal inflammation and compromised barrier function in idiopathic parkinsonism: scenario captured by systematic review. Neuroimmunology and Neuroinflammation. 2021; 8: 313. http://dx.doi.org/10.20517/2347-8659.2020.57
Chicago/Turabian Style
Umamahesan, Chianna, Aisha D. Augustin, Bu’ Hussain Hayee, Mohammad AA Ibrahim, David Taylor, Clive Weller, André Charlett, R. John Dobbs, Sylvia M. Dobbs. 2021. "Intestinal inflammation and compromised barrier function in idiopathic parkinsonism: scenario captured by systematic review" Neuroimmunology and Neuroinflammation. 8: 313. http://dx.doi.org/10.20517/2347-8659.2020.57
ACS Style
Umamahesan, C.; Augustin AD.; Hayee BH.; Ibrahim MAA.; Taylor D.; Weller C.; Charlett A.; Dobbs RJ.; Dobbs SM. Intestinal inflammation and compromised barrier function in idiopathic parkinsonism: scenario captured by systematic review. Neurosciences. 2021, 8, 313. http://dx.doi.org/10.20517/2347-8659.2020.57
About This Article
Copyright

Data & Comments
Data


 Cite This Article 24 clicks
Cite This Article 24 clicks

 Like This Article 24
likes
Like This Article 24
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.