
A review on Parkinson's disease treatment

Abstract
Parkinson’s disease (PD) is a neurodegenerative illness and has a common onset between the ages of 55 and 65 years. There is progressive development of both motor and non-motor symptoms, greatly affecting one’s overall quality of life. While there is no cure, various treatments have been developed to help manage the symptoms of PD. Management of PD is a growing field and targets new treatment methods, as well as improvements to old ones. Pharmacological, surgical, and therapeutic treatments have allowed physicians to treat not only the main motor symptoms of PD, but target patient-specific problems as they arise. This review discusses both the established and new possibilities for PD treatment that can provide patient-specific care and mitigate side effects for common treatments.
Keywords
Introduction
Basics of Parkinson’s disease
Parkinson’s disease (PD), or paralysis agitans, is a common neurodegenerative condition, which typically develops between the ages of 55 and 65 years[1]. This disease was first named and described by James Parkinson in 1817. The progression of this disease is gradual and prolonged. It has a plausible familial incidence, although the estimates of these occurrences are low and usually sporadic[2]. This disease is organized into two classifications: genetic and sporadic. Genetic PD follows Mendelian inheritance. Sporadic PD, which accounts for about 90% of all Parkinson’s cases, is a more complex category in which the pathogenic mechanisms that underlie it are not yet fully understood[3]. Nonetheless, it is known that the byzantine interactions of genetic and environmental influences play roles in the determination of sporadic PD[3-5]. Several subtypes of PD exist. Each has its own set of causative factors and susceptibilities, pathology, and treatment courses. General risk factors, symptoms, and pathology will be discussed first, before addressing some of the subtypes.
General risk factors
Research studies have linked theories regarding the outbreak of PD to both environmental and genetic circumstances[6]. These theories propose associations between PD and chemical reactions, neurotoxins, and genetic susceptibility or predisposition[3,7-9]. Environmental determinants positively associated with PD include factors such as injuries to the head, rural living, pesticides, anxiety and/or depression, and intake of dairy products; whereas physical inactivity, smoking, consumption of coffee and/or alcohol, and serum uric acid concentration are reported as having an inverse relationship to PD[5]. Though it is undisputed that familial factors play a role in this disease, the extent of heritability is heavily debated. As of yet, 41 different genetic loci have been linked to PD pathogenesis through the completion of 6 large meta-analysis studies[3].
General symptoms
A precedent for the clinical diagnosis of PD, according to the Movement Disorder Society, is centralized on a motor syndrome, Parkinsonism, and is based on three overriding motor symptoms (MS): bradykinesia, rigidity, and resting tremor[2]. Onset of motor manifestations usually begins unilaterally with asymmetrical effects enduring on the side of commencement[1].
Symptoms include resting tremor, bradykinesia, gait, speech difficulties, hypophonia, muscle dystrophy, postural deformities and instability[10]. Pain, stiffness or numbness in limbs, bradykinesia, tremors, a decline in facial expressions, and hypophonia are motor symptoms seen in the early stages of this disease’s onset[1,10]. Late-stage motor features may include motor fluctuations, dyskinesia, gait freezing, and falling. Initial diagnosis may be made based on evaluation of clinical features of patient history and examination[11]. Positive or negative responses to dopamine agents may also be used in the diagnosis of PD over time[1].
Since motor symptoms are the traditional primary identifiers of PD, common non-motor symptoms have been under-reported, often being overlooked or untreated. While non-motor symptoms are more heavily focused on during advanced stages, they occur during all stages of PD[12,13]. They have the potential to be early biomarkers for PD, with symptoms such as olfactory dysfunction, sleep problems, constipation, and erectile dysfunction often predating the diagnosis of PD by years[14,15]. These symptoms can greatly compromise quality of life (QoL) and daily activities, so typical non-motor treatments are based on improving QoL, with new treatments being developed, but still needing more research because PD treatments heavily focus on motor symptoms[16].
General pathology
Biochemical studies show a decrease of dopamine (DA) in the caudate nucleus and putamen; PD is therefore considered to be a disease of the neuronal system, which largely involves the nigrostriatal dopaminergic system[2]. This disease is identified on the premise of two considerable pathological processes: early selective loss of dopamine neurons, and the buildup of Lewy bodies (LBs) made up of α-synuclein that become misfolded and accumulate in a number of body systems of Parkinson’s patients[1].
The central nervous system is composed of groups of various nerve cells which form complex interactions that allow for skillful movement. The substantia nigra located in the midbrain is of essential importance to PD, as nigral neurons give rise to an extensive axonal network which innervates the basal ganglia. Liberation of dopamine, a neurotransmitter, by neurons of the substantia nigra allows for communication with neurons of the basal ganglia. Fine tuning of an organism’s movements is possible due to this biochemical interaction. Substantia nigra neurons degenerate progressively, leading to lowered levels of dopamine available for neurotransmission in the corpus striatum. This makes Parkinson’s a neurological disorder in which movement is affected[17]. Resting tremor, rigidity, declining balance and motor coordination, and bradykinesia, which is characterized by a creeping slowness of voluntary movement, are all movement-related symptoms of PD[17].
Dopamine has indirect roles in the striatum, which decreases cortical excitation of striatal neurons[18]. Boosts in the physiological state of corticostriatal glutamatergic transmission may possibly be an effect of Parkinsonism impairment of dopaminergic neurotransmission[19]. Furthermore, this may consequently emphasize the imbalance between subsets of striata neuronal systems that regulate the basal ganglia’s functional output[17,19].
LBs are fibrillar aggregates composed majorly of α-synuclein. LBs and Lewy neurites are pathologically important to Parkinson’s as they serve to be a prominent indication of the disease, being actively associated with Parkinson’s pathogenesis[4]. The formation of LBs has been considered an explanation to the neuronal degeneration that occurs in PD patients since neuronal loss has been found in predilection sites for LBs. Of the 70+ molecules that have been identified in LBs, α-synuclein is the most prominent[4,20]. Immunochemistry of this constituent has uncovered that diffuse cytoplasmic staining cultivates into pale bodies, which are anti-ubiquitin antibodies, by compaction[20]. The peripheral portion of pale bodies give rise to LBs. This abnormality has been identified in 10% of pigmented neurons in the substantia nigra and over 50% in the locus coeruleus in PD[20]. Six genetic PD-associated mutations of α-synuclein have been discovered[21]. Three mutations related to PD have demonstrated acceleration of α-synuclein aggregation, while an additional three show delay of aggregation kinetics. It is therefore troublesome to provide a unifying mechanism describing how familial PD-associated mutations affect the structure of α-synuclein, and how their accumulation and function link with PD, as there have been several suggestions pointing in this direction[21]. For instance, membrane binding studies propose that a minute number of these PD mutants strongly bind to synthetic membrane vesicles, while others show a weakened ability to bind to the membrane[21]. PD mutations have not been shown to drastically alter the toxicity of α-synuclein oligomers or fibrils, although more recent studies have indicated that the most potent toxic species responsible for PD are oligomers that are formed early on in the disease process[21]. p.A30P is the only mutant that has been found to form faster oligomers and slowed the conversion from oligomers to fibrils. It is plausible that every mutation associated with PD alters α-synuclein biology in various ways, which is possibly responsible for the pathogenesis of this disease[21].
PD pathology affects more than just the dopaminergic nigrostriatal system. Non-motor symptoms can be better explained when examining the effects of multi-system neurodegeneration. Decades of research point to altered cholinergic neurotransmissions. LBs have been found in the neurons of the nucleus basalis of Meynert, the source of cholinergic innervation of the cerebral cortex[22]. The basal forebrain complex, which provides the principal cholinergic input of the entire cortical mantle, degenerates in PD and can lead to symptoms such as dementia, depression, or apathy[22]. Anosmia and hyposmia are common side effects of PD. While the pathophysiology is not fully understood, it could be related to α-synuclein deposits in the olfactory bulb, medulla oblongata, anterior olfactory nucleus, and limbic rhinencephalon[22,23]. Progressive, non-linear loss also occurs in serotonergic terminals, although slower than the progressive loss seen in dopaminergic terminals. It can lead to both motor and non-motor symptoms such as depression, tremors, weight loss, and visual hallucinations.[24]. Reduced levels of serotonin and its metabolite are found in the caudate nucleus, hippocampus, brainstem, and frontal cortex[25,26]. Additionally, adrenergic neurons are impacted. One study has shown an increase in α1 and β1 receptors, especially in demented PD patients, and a decrease in α2 receptors within the pre-frontal cortex[27]. Disruptions in adrenergic pathways may lead to or worsen dementia and depression[27].
GBA-associated PD
Common risk factors for PD are mutations in the glucocerebrosidase (GBA) gene, which is a gene that encodes for the lysosomal enzyme. During a clinical study on patients with Gaucher’s disease (GD), a rare lysosomal storage disorder, this risk factor was identified. It has been discovered that mutations in the GBA gene are more prominent than in any other implicated genes, including dardarin (LRKK2), α-synuclein (SNCA), and parkin (PARKIN2), in the majority of the PD population.
GD is a recessive disorder in which there is a deficiency in the GBA enzyme[28]. It typically involves the mononuclear phagocyte system in which the lysosomes within macrophage lineage cells become excessively stored with lipids[28]. This disease is induced by bi-allelic variants in the GBA gene, which encodes acid beta-glucosidase (glucocerebrosidase)[29] Manifestations such as hepatosplenomegaly, anemia, thrombocytopenia, and bony involvement are common with this disorder. As of yet, 300 distinct mutations have been pinpointed on this gene. These mutations include point mutations, frameshift mutations, splice-site alterations, and recombinant alleles that embody segments of the pseudogene sequence[28,30]. Gaucher patients who develop parkinsonian features, dementia, or both, are amid the more atypical and uncommon Gaucher phenotypes. A group of 17 GD and Parkinsonism patients were described in 2003. The parkinsonian manifestations were aligned with those of sporadic PD. Many of these patients were in their 40s when they experienced the disease onset, and most responded positively to levodopa (L-DOPA)[28,31]. The GBA gene in these patients was sequenced and it was discovered that there were 12 different genotypes; the most prominent type being 1 N370S allele, which was found in 14 of the 17 patients, including five N370S homozygotes. Four patients had an autopsy performed on them, which revealed LB inclusions, predominantly in the cerebral cortex and the hippocampus[28].
Research has shown that the onset of motor impairments among GBA mutation carriers occurs 1.7-6.0 years sooner than in those without mutations. Screening showed that GBA mutations were twice as common in PD patients with an early onset (< 50 years) than those with a late onset[28]. 951 patients were screened for N370S and L444P that had an onset of PD before 51 years of age and it was discovered that 6.7% were carriers for the GBA mutation, which is equivalent to the prevalence of patients with homozygous or heterozygous PARK2 mutations[28,32]. It was also found that GBA mutation carriers developed clinical symptoms earlier than non-mutation carriers when comparing with patients who developed PD before 50 years of age[28].
Analyses from a study screening for GBA mutations found that GBA carriers were primarily male, had greater occurrences of cognitive dysfunction or dementia, and experienced hallucinations more frequently (not associated with drug treatment) when compared to patients without GBA mutations[28]. A second study recorded that GBA mutation carriers with PD more frequently suffer dementia than non-carriers[28,33]. The UPDRS, mini-metal state examination, and Hoehn and Yahr staging scales did not detect any noteworthy differences in the magnitude of PD manifestations or the rate of disease progression between GBA carriers and non-carriers[28,34]. However, several other studies have documented greater rates of cognitive decline, bradykinesia, olfactory dysfunction, and less rigidity correlated with GBA mutations[28]. A German PD study found that GBA mutation carriers have higher frequencies of dementia, neuropsychological disturbances, and autonomic dysfunction, when they compared 20 GBA mutation patients with 20 patients without the mutation[28,35].
The exact mechanisms that contribute to GBA-associated Parkinsonism are not fully known. Further studies are necessary to identify these mechanisms and to discover related risk factors that work in adjunction with GBA and favor the progression of Parkinsonism. Aging has a role in the onset and development of PD. Factors that change during the aging process such as an increase in α-synuclein cellular concentrations, and declining numbers of lysosomes and poorer lysosomal function may play a part. Much is known about the GBA protein, especially when compared to other genes associated with PD. Considerable therapeutic strategies for the treatment of Gaucher’s disease may lead to stabilization of GBA or improvement of enzymatic activity which could influence the development of parkinsonian neurodegeneration. A deeper understanding of this enigmatic protein and its role in the progression of Parkinsonism could improve genetic counseling for those who are GBA mutation carriers and enhance therapeutic approaches for GBA-associated Parkinsonism, possibly altering the treatment of all PD patients[28].
Early-onset
The incidence of PD increases with age, with an incidence of 0.5 per 100,000 for people below 40 years old compared to the overall incidence of 13.4 per 100,000 for all ages[36]. Early-onset PD is the development of symptoms in those below 60 years old. It can be divided further into juvenile PD in those younger than 21, and young-onset PD if between the ages of 21-40[36]. Early-onset PD is attributed to genetic factors, rather than environmental[17]. Juvenile PD is rare, but worth mentioning. Its genetic mutations can cause further distinct types of PD. For example, the mutation of ATP13A2 causes Kufor-Rekab syndrome, which is a specific juvenile PD that has less than 50 cases reported to date[36]. Early-onset PD differs in presentation of symptoms and responsiveness to treatment than late-onset PD[1]. With early-onset PD, bradykinesia and rigidity are more likely to be the presenting symptoms, rather than gait disturbance, when initially treated[1,37]. Early-onset has a slower disease progression and is more likely to develop L-DOPA-related dyskinesia, so dopamine agonists are frequently chosen as the initial treatment options over L-DOPA[1]. However, new evidence suggests that L-DOPA does not necessarily need to be avoided in treatment of early-onset PD[38]. This will be discussed in the dopaminergic medications section below.
Tremor-dominant vs. non-tremor dominant responsiveness to L-DOPA
PD is a disorder with various motor subtypes, but few neuroimaging studies have examined the variable effects of L-DOPA treatment on the different motor sub-classifications of Parkinson’s. One study by Mohl and colleagues investigated L-DOPA-induced differences in neural activation between tremor dominant (TD) PD patients and posterior instability/gait difficulty (PIGD) PD patients. They utilized neuroimaging by the use of fMRI to investigate task-induced activation and connectivity in the cortico-striatal-thalamo-cortical (CSTC) motor circuit before and after L-DOPA deliverance in 21 age- and sex-matched healthy controls, fourteen TD patients, and twelve PIGD patients. Each subject executed a right-handed, paced, finger-tapping task. There was greater activity within the left putamen in the PIGD patients on L-DOPA compared to the TD patients on L-DOPA and the healthy control group. This hyperactivation within the left putamen induced by L-DOPA during the finger-tapping task, however, was not accompanied by an upsurge in effective connectivity between the left posterior putamen and CSTC motor network. This finding implies a rooted pathophysiological discrepancy between motor subtypes of PD that can be evoked using a dopaminergic medication during a motor activity. Relative to the healthy control subjects, PD patients not taking medication displayed hypoactivation of the left primary motor cortex when performing the tapping task. To further investigate whether this data is applicable to one or both of the subtypes examined in the study, researchers separately compared TD patients off of medication and PIGD patients off of medication to healthy controls. It was discovered that hypoactivation of the motor cortex was greatly driven by the group of TD patients off of medication. Furthermore, PD patients on medication did not show any changes in activation within the primary or secondary motor cortices when contrasted with the control group[39].
Putamen changes are associated with the symptomatology that defines motor subtypes of PD[39]. SPECT studies discovered a link between putaminal uptake of dopamine transporter (DaT) tracer and poorer rigidity, in addition to more noticeable dopaminergic loss in the posterior putamen in non-TD patients and mixed subtype patients relative to TD patients[39,40]. This SPECT study, together with the study conducted by Mohl and colleagues that evidenced an increase in left putamen activity by L-DOPA during finger-tapping task in PIGD patients only, implies plausible roles of the putamen in phenotypic PD subtype manifestations[39].
Furthermore, L-DOPA resulted in divergent tapping-induced coupling responses within the motor network in PIGD and TD PD patients. These findings could not be attributed to variation in the stage or duration of the disease. When comparing TD patients on medication to TD patients not on medication, there was a noteworthy increase in effective connectivity between the left posterior putamen and other motor network areas, whereas PIGD patients on and off medication did not show any compelling changes. Underlying pathophysiological disparity between recognizable PD motor subtypes may explain the differential responses of these subtypes to L-DOPA[39].
Treatment Approaches
Though the exact cause of Parkinson’s disease has not been identified, treatment discoveries have been progressive. There is no known cure for the disease, so treatments seek to manage symptoms rather than prevent or slow the progression of the disease. Treatments can vary from drugs, surgeries, therapy, or a combination of different treatments [Table 1]. They must also be adjusted throughout the course of the disease, as some common treatments, such as L-DOPA lose effectiveness over time[1]. Treatment of PD using available drugs has positive symptomatic effects; however, there are no disease-modifying or neuroprotective therapies available to slow the progression of the disease[1,41]. Therefore, treatment begins at the discretion of the patient and the physician when symptoms begin to impair function or provide social embarrassment. No one drug is more beneficial than the other for initial treatment, but instead the disease itself must be looked at in terms of severity and time of onset[42]. PD is a disease that affects multiple neural pathways in the brain. While L-DOPA may treat motor problems caused by low dopamine levels, it will not treat motor problems caused by low acetylcholine levels in other pathways. Additionally, each subtype responds differently to drugs. It is up to the discretion of the doctor to choose a plan that works for an individual patient based on responsiveness and symptoms.
Summary of different treatment options with their pros and cons
| Treatment Option | Summary | Pros | Cons |
|---|---|---|---|
| Drug Treatments | Limited to no invasive treatment required | May lose effectiveness over time
Various side effects for each drug | |
| Dopaminergic Medications | Dopamine agonists taken orally or through injection | • Current “gold standard for treatment”
• High responsiveness (80%) in idiopathic PD • Favored treatment for late-onset PD • May affect both motor and non-motor side effects • Most patients ultimately take dopamine agonists as the disease progresses and other treatments stop being effective | • Less favored for early-onset PD treatment due to levodopa-related dyskinesia
• Levodopa requires increasing dosage over time “on-off” fluctuations with oral medication • Irritation or problems at pump site for injection method |
| MAOIs, inhibitors and antagonists | Inhibit breakdown of levodopa and dopamine through oral drugs | • Do not need increased dosage over time
• May help with cognitive decline in later stages of PD • Milder side effects than dopaminergic drugs • May treat L-DOPA-related dyskinesia | • Lack extensive effects on symptoms for late stage PD
• Must be supplemented with a group of drugs for some treatments, such as COMT inhibitors |
| Anticholinergics | Reduce acetylcholine activity at choline receptors through oral drugs | • Rapid absorption, used for tremor-predominant PD
• Can be a monotherapy in early stages | • Less tolerance in elderly patients
• Lack of testing in elderly patients • New testing may show cholinergics, rather than anticholinergics may improve PD symptoms • Largely replaced by dopamine agonists in current treatment regimes • Little pharmacokinetic information |
| Neurotrophic Factors: GDNF | Small natural proteins, specifically glial cell line-derived neurotrophic factor (GDNF), delivered through an implantation in the brain | • Improvement of parkinsonian symptoms
• Increased striatal dopamine levels • Prevention from neurotoxic damage of dopaminergic neurons in the midbrain and noradrenergic neurons in the locus coeruleus | • Difficult delivering this NTF to brain cells across the BBB
• Studies show differing outcomes on the effectiveness of GDNF on symptomatic relief • Requires more research |
| Nanomedicine | Modification of existing drugs to improve delivery method | • Greater delivery through the blood brain barrier or circumventing it
• Less drug lost in peripheral tissue, increasing effectiveness and decreasing side effects • Growing field with investigations into protective factors and imaging improvements | • Relatively new field that requires more testing before being a standard practice |
| Surgical Treatments | Can have high effectiveness with long-term results | Most invasive of the treatment methods
Some methods and side effects are irreversible | |
| Ablative Surgery | Incision into the global pallidus, thalamus, or subthalamic nucleus either bilaterally or unilaterally | • High-effectiveness, relieving rigidity, hypokinesia, and tremors. | • Irreversible procedure, may have permanent side effects |
| Deep Brain Stimulation | Implant that stimulates the basal ganglia through high frequency electrical stimulation | • Bilateral DBS is likely safer than bilateral ablative surgeries
• Established treatment for advanced PD • Can improve both motor and non-motor symptoms • Equal improvement compared to L-DOPA for tremor, bradykinesia and rigidity • Closed-loop DBS is being investigated to reduce side effects and allow greater feedback • Effective at minimizing “off” episodes • Benefits can last 10+ years in some individuals | • Conventional DBS is open-loop, not allowing for feedback and patient adjustment
• Less effective for reducing gait, balance, and speech symptoms • May include dysarthria, imbalance, and dyskinesia • Limited battery life |
| Transplantation and Gene Therapy | Transplantation of stem cells into the striatum, insertion of non-disease and disease modifying transgenes | • Can generate long-lasting improvement and replace diseased neurons
• Can treat PD at a genetic level, correcting dopamine pathways | • Must consider ethical bounds for embryonic stem cell use
• Developing fields that require more research, especially human testing |
| Therapy | Can address multiple symptoms at once, both motor and nonmotor
Noninvasive treatment | Usually requires other forms of treatment in addition to therapy | |
| Physical Therapy | Addresses mobility and motor-related symptoms through physical training | • Can greatly increase quality of life
• Treat multiple symptoms at once • Multiple forms of PT exist, allowing for patient-specific care that adapts | • May require other treatments, especially as the disease progresses
• Limited by patient involvement and motivation |
| Occupational Therapy | Help patients maintain work, leisure, and self-care activities for as long as possible | • Promotes independence
• Allows one to engage meaningfully • Addresses social and environmental side effects and factors | • Usually requires PT with it, as well as additional treatments
• Does not address PD at a physiological level • Limited by patient involvement and motivation |
| Speech Therapy | Improves speech through voice exercises | • Focuses on a single parameter but improves multiple voice symptoms
• Improves quality of life | • Helpful for only speech disorders |
| Cognitive Behavioral Therapy | Management of a symptom through behavioral training methods | • Alternative for symptoms commonly treated with drugs, helping to reduce complications
• Few side-effects • As effective as drugs in treating insomnia • May be done remotely | • Cannot be double-blinded testing
• Manages only one symptom • Needs more testing • Dependent on patient motivation |
Drug treatments
Dopaminergic medications
Dopaminergic medications are the typical treatment method for motor symptoms in PD, and L-DOPA, the most potent anti-parkinsonian drug currently available, remains the “gold standard” for PD therapy. Because the lack of dopamine available for communication in the nigrostratial pathway is at the root of many PD symptoms, specifically movement-based ones, dopaminergic medications are used to help increase or mimic dopamine levels. Dopamine does not easily pass through the blood brain barrier, but its precursor, L-DOPA can[43]. It is metabolized in the small intestine and is converted to dopamine by aromatic-L-amino-acid decarboxylase (AADC) and catechol-O-methyltransferase (COMT), which can then be stored in the nigrostratial terminals. In contrast, dopamine agonists act directly on postsynaptic receptors, mitigating the need for dopamine production[43].
Responsiveness to L-DOPA occurs in 80% of patients with idiopathic PD, reducing bradykinesia and rigidity. However, it is ineffective or unsatisfactory at treating several prominent PD symptoms: posture and gait problems, speech problems, freezing, autonomic dysfunction, cognitive disorders, affective disorders, and sleep problems[1].
As previously mentioned about early-onset PD, there are concerns about L-DOPA-related dyskinesia. While dyskinesia is correlated with L-DOPA usage, the disease itself (PD) and a pulsatile drug delivery are both required for dyskinesia to develop. L-DOPA does not usually induce dyskinesia in Parkinsonism with post-synaptic involvement, and intestinal gel delivery has shown to reduce pre-existing dyskinesia. Therefore, it may be more appropriate to view dyskinesia as a result of the method of administering levodopa rather than an intrinsic effect of L-DOPA itself[38].
With late-onset PD, there is a greater risk of adverse neuropsychiatric effects from dopamine agonists, so L-DOPA is favored as an initial treatment. Both types of PD require adjustments to medication as the disease progresses, as those patients who use dopamine agonists initially often need to start taking L-DOPA after two to five years; L-DOPA too loses its effectiveness over time[1].
Along with managing motor symptoms of PD, non-motor symptoms such as depression can also be managed with dopamine agonists. Drugs like Pramiprexole are effective and clinically useful for managing depression and depressive symptoms for those with PD. However, not all dopamine agonists are effective; Rotigotine, for example, is ineffective and investigational in practical application[16]. Most drugs that treat non-motor symptoms use other neurotransmitters than dopamine[44]. Treatment of depression with dopamine agonists should not be universal, but specific to the drug and the patient’s needs[16].
Despite being the “gold standard” for PD treatment, L-DOPA does not come without a series of problems. It is unknown whether the symptomatic benefits of Levodopa have an association with neurotoxic effects and long-term deterioration. It, however, has long term complications, the most problematic being: wearing-off, dyskinesias, freezing episodes, and “on-off” fluctuations that are impossible to predict[45]. Within five years of chronic L-DOPA treatment, 40%-50% of patients will develop dyskinesias and motor fluctuations. After 10 years, that number increases to 70%-80% of patients[1]. L-DOPA’s effectiveness also declines in advanced PD due to fluctuating medication periods and levodopa’s short half-life[1]. Patients are commonly required to increase dosage amounts as well as the frequency of doses, requiring a dose every 2-3 h, for example[44]. With the resulting increase medication dosage, presentation of dyskinesias also increases.
Alleviation of PD symptoms from L-DOPA can also make it difficult to accurately assess the patient’s honest condition, making the progression of the disease troublesome to supervise. In order to make assessments of the disease progression, or deterioration, administration of L-DOPA must be withdrawn for at least two weeks. This option is not practical, especially in advanced stages of PD. The search for biological surrogate markers is underway, and there have been developments of positron emission tomography (PET) and single-photon emission computed tomography (SPECT) techniques, which have shown compelling correlations with PD’s global severity[45]. In addition to finding new biological surrogate markers, alternatives and better management of L-DOPA use are being investigated. Continuous dopaminergic stimulation is needed for those with advanced PD, which can be provided through levodopa-carbidopa intestinal gel and deep brain stimulation as an alternative for oral L-DOPA[1].
Levodopa-carbidopa intestinal gel, also referred to as Duodopa, is an option for those with advanced PD, compared to oral L-DOPA, with the intestinal gel showing less motor fluctuations and dyskinesias, though they are not completely eliminated. The gel is injected into the jejunum, with the gel delivering a continuous dosage of levodopa-carbidopa equivalent to the oral L-DOPA dose. There is faster absorption without the concern of reduced absorption[1]. Because the drugs are released continuously, “off” periods are reduced and “on” periods increased[46]. In one study, 90% of patients had an improved QoL[47]. Nocturnal sleep habits improved in all patients who switched to levodopa-carbidopa gel injections, with improvements in motor symptoms at night, PD symptoms at night, and disturbances during sleep. Daytime drowsiness also improved[48]. Improvements in other non-motor symptoms have been observed including attention, memory, gastrointestinal, urological, and pain[49]. A benefit of levodopa-carbidopa intestinal gel compared to alternatives for advanced PD is that there is no age limit for its administration. Adverse effects are mostly due to technical problems such as dislocation, kinking, obstruction and breakage of the duodenal catheter[46].
Apomorphine is a non-selective dopamine agonist that activates both D1 and D2 receptors, is used as a drug for PD patients suffering from motor fluctuations, and possibly has positive effects on non-motor symptoms. Unlike other antiparkinsonian agents, it is the only one comparable to L-DOPA in its capacity to control motor symptoms[50]. Use of apomorphine is not typically age-limited, but it is limited by a need for confirmed levodopa-responsive PD. Apomorphine is an option for patients with “off” periods including delays in effects when taking orally administered medications like L-DOPA, or “off” periods when waking up. Intermittent or continuous infusion administered subcutaneously have shown to be the most effective and bearable application processes[50]. Intermittent infusion is administered by injection via pen for rapid relief of “off” fluctuations and end-of-dose biphasic dyskinesia. Continuous infusion is administered through a modified insulin pump, most commonly dispensing in a 12-16 h regimen, although it can be given throughout the 24 h and need not be discontinued at night. This can reduce the “off” times at night[46]. Adverse side effects may include nodules at the injection side, which can be seen in up to 70% of patients, bruising, itching, or pain. An advantage of apomorphine for those with advanced PD is that it is less invasive than deep-brain stimulation or levodopa-carbidopa intestinal gel[50].
It is suspected that the frequent pain experienced by patients with PD could be partly due to central modification of nociception. A randomized, controlled, double-blind study of 25 PD patients by Dellapina et al.[51], compared apomorphine versus placebo to test their effects on pain thresholds and pain-induced cerebral activity. Apomorphine was not found to have any determinable change on subjective or objective pain thresholds, or pain activation profiles when compared with placebo. In conclusion, apomorphine has no effect on pain processing in patients with PD, suggesting thereby that other monoamine systems may be involved[51].
Monoamine oxidase inhibitors, Catechol O-methyltransferase inhibitors, and N-methyl D-aspartate receptor antagonists
Monoamine oxidase inhibitors (MAOIs), like rasagiline or selegiline, and COMT inhibitors, like entacapone, work to inhibit the breakdown of dopamine and L-DOPA to prolong their effects. MAOIs reduce the amount of dopamine broken down in the synapse. COMT inhibitors prevent COMT from prematurely converting L-DOPA into dopamine. It reduces peripheral loss of L-DOPA before it can reach the brain[1,43]. Patients with mild symptoms may be treated with MAOIs first, due to their milder side effects, less frequent doses and to delay the treatment of dopaminergic drugs. They may also be used in later stages to reduce specific symptoms such as tremor or dyskinesias[1,42]. MAOIs are dosed 1-3x daily throughout the course of the disease, not needing to increase the dosage over time, like L-DOPA[44]. Treatment with cholinesterase inhibitors, such as rivastigmine, may help with cognitive decline or hallucinations/delusions associated with advanced or later stages of PD[45]. Putative N-methyl-D-aspartate (NMDA) receptor antagonists, like amantadine and memantine, are also drugs taken as adjuncts to other treatment[1]. They block acetylcholine and NMDA receptors, which has two effects: the first is that they synergize with dopaminergic agents, enhancing release and turnover of dopamine in the striatial neurons; second, PD disrupts glutamatergic transmissions causing an overactivation of NMDA receptors, which can cause worsening dyskinesias[52]. Agonists reduce the amounts of abnormal glutamate signaling in the subthalamic nucleus[52]. In one study, amantadine was able to reduce dyskinesia severity by 60% in advanced stage PD, when taken with regular doses of L-DOPA[53]. Amantadine extended release is currently the only FDA-approved medication for the treatment of levodopa-related dyskinesia, allowing for greater sustainability of L-DOPA therapy and reducing the “off” time[54]. No existing drugs have extensive effects on symptoms that develop in the later stages of this disease, such as postural instability, dysphagia, dysphonia, dyskinesias, as well as constipation[45]. NMDA receptor agonist may possibly be neuroprotective. They can prevent disease progression by inhibiting glutamatergic-mediated excitotoxicity and stimulate synaptogenesis/neurotrophic release[52].
Anticholinergics for early on
The first pharmacological agents used in PD therapy were anticholinergic drugs[55]. They reduce the activity of acetylcholine by acting as antagonists at choline receptors, hoping to restore the balance between dopamine and acetylcholine levels that was disturbed by PD[56]. These drugs have largely been replaced by L-DOPA and other centrally acting dopaminergic agonists, but they still remain available for use in the treatment of PD. Benztropine, biperiden, diphenhydramine, ethopropazine, orphenadrine, procyclidine, and trihexyphenidyl are included in this therapeutic class of drugs, though there is little pharmacokinetic information available on them because of their low plasma drug concentrations[55,57]. Typically, anticholinergic drugs have a greater role in tremor-predominant PD and can be a monotherapy in early stages, but are usually done in adjunct with L-DOPA or other prescribed medications[56].
Oral administration of this drug class has rapid absorption in humans. There is variability in the oral bioavailability among the various drugs of this class, ranging anywhere from 30% to 70%. Volume of distribution in humans and animals of these drugs is large, and tissue distribution is rapid. A characterization of these drugs is their relatively low clearance when compared to hepatic blood flow. These drugs appear to be broadly metabolized, chiefly to N-dealkylated and hydroxylated metabolites[55]. There is less tolerance of these drugs in the elderly than in younger patients. Additionally, these drugs may be avoided in the elderly because of an increased risk of confusion[56]. Since the majority of studies have been restricted to young healthy volunteers who are given single doses, there is a lack of pharmacokinetic information regarding these drugs in the elderly. Furthermore, the shortcoming of information for multiple dose administration and administration in the elderly may be a possible deterrent in the safe and competent use of these pharmaceuticals in PD patients[55].
It should be noted that while anticholinergics remain a viable drug treatment, there are studies that point to the possibility of cholinergic therapy helping to manage mobility problems in PD by looking at the pedunculopontine nucleus-laterodorsal tegmental complex (PPN). A key role of the PPN is maintenance of balance, and PPN dysfunction can cause impaired postural control and gait in PD[22]. Anticholinergic medicine can increase the incidence of falling in the elderly. Additionally, the loss of CNS cholinergic neurons can worsen balance. Preliminary data of a controlled, small study shows that PD patients that are given donepezil, a central cholinesterase inhibitor, fell half as often as the control group[58].
Neurotrophic factors
Nerve cells require neurotrophic factors (NTFs), which are small natural proteins, for their development and continued survival. In addition, NTFs help maintain the morphological and functional phenotype of nerve cells. Neuronal networks require the release of NTFs at the target structures. NTFs are taken up by the nerve terminals and moved retrogradely to the soma of the projecting neurons for their formation and preservation. This is described by the “neurotrophic hypothesis”. Neuronal survival and phenotype specification is stimulated by the induction of a gene upon the arrival of NTFs at the nucleus. There have been several proteins that have been classified as NTFs based on the effects they have on neuronal survival, differentiation, maturation of electrophysical properties, and plasticity. The functional roles of NTFs provide promising possible benefits in the treatment of neurodegenerative diseases, including PD[59]. Glial cell line-derived neurotrophic factor (GDNF) is one of the more closely related NTFs associated with PD as a result of the potent trophic action it has on cultured dopaminergic neurons[59,60]. Production of GDNF is done by striatal neurons and is required for the sustenance of adult nigrostriatal dopaminergic neurons and other central and peripheral nuclei affected in PD[59,61]. Exogenous administration of GDNF has also shown neurotoxic damage preventative abilities of dopaminergic neurons in the midbrain, and noradrenergic neurons in the locus coeruleus[59,61,62]. Evidence of GDNF and its effect on the latter neurons implies that the stimulation of endogenous GDNF generation and/or administration of exogenous GDNF could serve as competent therapeutic strategies for PD[59,63,64].
Different human studies testing the effects of striatal delivery of GDNF through the permanent implantation of a cannula have demonstrated varying degrees of symptomatic relief[62]. Some subjects experienced incremental improvement, such as the study conducted by Gill and colleagues, while other studies showed no evident benefits[32,62,65]. There is much variability in the outcomes of documented studies: some have recorded antibody development in response to the administration of recombinant human GDNF, a randomized trial detailed reactions to the placebo, and one study noted that subjects experienced strong adverse effects to intraventricular delivery of GDNF[33,34,62,65]. In addition, deliverance of GDNF to brain cells across the blood brain barrier poses as a hindrance to the use of this approach as a PD treatment, suggesting that the simple administration of the NTF protein may not be a plausible option[35]. Alternative studies are needed to uncover ways to sustainably exploit the benefits of the potent trophic action that GDNF has on dopaminergic neurons[62].
GDNF is necessary for the survival of adult catecholaminergic neurons. The onset of PD due to the loss of dopaminergic neurons has been linked to GDNF signaling failure. A study conducted by Hadaczek and colleagues showed weakened GDNF signaling in neurons that were deficient in ganglio-series gangliosides (glycosphingolipids that contain sialic acid), and renewal of this dampened signaling with LIGA20, which is a membrane permeable ganglioside analog of GM1[36,66]. GM1 is a commonly studied ganglioside that is shown to be associated in situ with the GDNF receptor protein components, GFRα1 and RET, which are essential for the ternary receptor complex[36]. Whole or partial deficiency of GM1 due to interruption of the B4galnt1 gene in mice resulted in the development of PD symptoms. Inadequate levels of tyrosine phosphorylated RET were observed in PD patients that had severely deficient GM1 in nigral neurons. In addition, PD patient brains exhibited significantly lower levels of GM1 in the occipital cortex when compared to age-matched controls. This implies that decreased systemic GM1 may be a risk factor in PD[66].
Nanomedicine
Nanomedicine and the manipulation of compounds on the nanometric scale is being investigated for the improvement of drug delivery systems, specifically to improve drug bioavailability, half-life, or achieve more sustained levels of circulation. The most widely used approach is to increase the ability of drugs to be transported across the blood brain barrier[67,68]. This is particularly important with L-DOPA and dopamine agonists because the process with which they are mediated through the blood brain barrier can also occur in peripheral tissues[67]. Intranasal delivery allows for drug transport through the olfactory and trigeminal pathways, circumventing the blood brain barrier[69]. Nanoparticles accelerate this method of absorption. However, this is not the only delivery system being investigated for nanotechnology. With drugs such as apomorphine, the drug is associated with a lipid-based manipulated nanomolecule, trapping the drug inside or adhering it to itself. When taken orally, it stimulates chylomicron formation and allows for better systemic absorption by the lymphatic system[67]. Promising results from a study showed 12- to 13-times higher bioavailability using solid lipid nanoparticles compared to standard oral apomorphine. However, this study and a majority of similar studies have only been performed on rats and while they have promising results, there is a need for more research before they can be standard practice[70]. This is the main limitation for nanomedicine. It is a new and developing field, so many of the studies and drug delivery methods have not reached human testing yet and will need years before being viable practices in the field.
Nanomedicine is also being looked at for more than just drug delivery systems for PD. Studies are investigating the delivery of neuroprotective factors, enzymes, antioxidants, growth factors, and inhibition of α-synuclein aggregation. Nanomedicine is also focusing on areas including imaging and detection improvements, as well as deep brain stimulation[67].
Surgical treatments
There are three major surgical treatments for PD: ablative surgery, deep brain stimulation (DBS), and grafting fetal mesencephalic cells into the striatum.
Ablative surgery
Ablative surgery is the oldest of the three major surgical treatments and holds the greatest risks. Pallidotomy, thalamotomy, and subthalamotomy are available surgeries[71]. Locations of each surgery and the pathways that are disrupted can be seen in Figure 1. Because of the loss of dopaminergic innervation, the internal globus pallidus and substantia nigra reticulata have hyperactivity leading to excessive inhibitory output[73,74]. The inhibitory output interferes with the movement-related center in the thalamus and generates hypokinetic PD symptoms[73]. Ablative surgery tries to alleviate these symptoms by creating a lesion within the internal globus pallidus, thalamus, or subthalamic nucleus to stop the overactive pathway either near the start, as in pallidotomies, or further down the line in thalamotomies [Figure 1][74]. In one study, stereotactic posteroventral pallidotomies were performed on 38 patients, giving complete or almost complete relief of rigidity and hypokinesia in 92% of patients[75]. As well, 32 patients had suffered tremors before the surgery, and 81% of them had complete or almost complete relief of tremor, post-surgery. Gait and speech volume also saw improvement. However, seven patients (roughly 18%) had complications, with six patients presenting with permanent partial homonymous hemianopsia; one of those patients also had transient dysphagia and facial weakness. The other patient developed transitory hemiparesis 1 week after the surgery[75]. Thalamotomy is an option for patients that have long-standing tremor as the main clinical manifestation, and it cannot be controlled by drugs. Thalamotomy is less common than pallidotomy, with less than 5% of patients having the thalamus as the preferable target for thalamotomy or DBS[76]. Ablative surgeries have lost prevalence because of concerns over high incidence of side-effects with bilateral ablative procedures[71]. At experienced centers, unilateral pallidotomy is equally safe as unilateral DBS, though bilateral DBS is likely safer than bilateral pallidotomy. Therefore, while DBS may be more common now, pallidotomy and other ablative surgeries are still viable options when DBS is not available nor feasible[77].
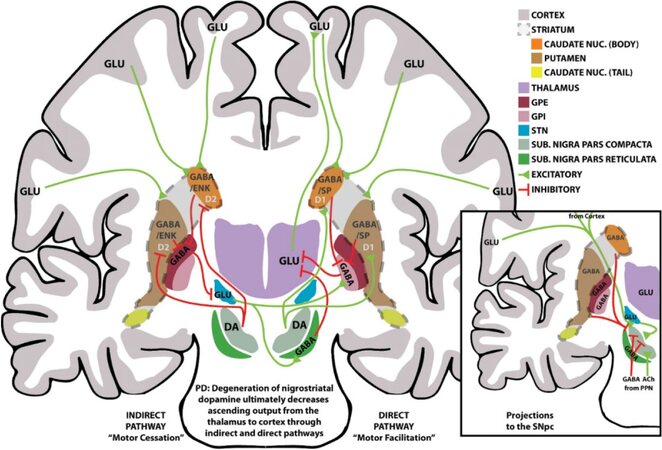
Figure 1. An overview of the basal ganglia neuroanatomy and primary motor circuits. Ablative surgery options include pallidotomy - destroying the global pallidus (GPE, GPI in maroon and pink, respectively), thalamotomy - destroying the thalamus (purple), and subthalamotomy - destroying the subthalamic nucleus (STN, blue). Procedures can either be bilateral or unilateral and are done to reduce excessive inhibitory output from the internal globus pallidus (pink) and substantia nigra reticulata (green), and their respective pathways shown in red. Image provided by Harris et al.[72] 2020
Deep brain stimulation
Deep brain stimulation (DBS) developed as an alternative to risky bilateral ablative surgeries. It is applied in the internal globus pallidus, subthalamic nucleus, and thalamus[71]. It is similar to ablative surgeries in terms of the pathway and symptoms that DBS is trying to mitigate. Conventional DBS is open-loop, with the implants delivering electrical impulses continuously and without feedback[74]. It is an established treatment for advanced stage PD, improving motor symptoms both in the long and short term[78]. DBS is effective at minimizing instances of “off” episodes that tend to occur more frequently throughout the day in patients with more progressed stages of medically treated PD. There are variable effects on motor symptoms and non-motor symptoms such as postural and gait disturbances, cognitive decline, sleep, swallowing, and speech or micturition disturbances[79]. Controlled studies using large and randomized groups have displayed comparable motor benefits between the subthalamic nucleus (STN) and the globus pallidus pars interna (GPi), which are two of the most prevalent targets for DBS. However, DA replacement pharmaceuticals can be decreased following STN DBS, whereas there are less cognitive and mood side effects from GPi DBS[80]. In addition, gait instability and freezing may be improved from targeting the pedunculopontine, as evidence suggests[81,82].
DBS involves a delivery of unceasing high frequency electrical stimulation to the basal ganglia. The mechanism of this therapy is not fully understood, but it is known that there is an interference with pathological and physiological neural circuitry[79]. High-frequency stimulation of distinct targets in the brain resembles functional effects of a lesion and has inhibitory effects on neuronal activity. Prospective mechanisms include high-frequency patterns obscuring encoded information, suppression of abnormal beta oscillations, stimulation of inhibitory gamma-aminobutyric acid (GABAergic) afferents to the nucleus in focus, efferent projections or passing fibers, and the inhibition of neurotransmitter and hormone production or release. DBS mechanisms are intricate as excitation or inhibition of neural elements may occur, dynamic equilibrium states are attained, and differing forms of neural plasticity are established[83]. Internal pulse generators (IPGs) are able to record and store local field potential and multiple targets concurrently, allowing physicians and researchers to more advantageously understand the pathophysiology of PD and the mechanism by which DBS mitigates symptoms. Subthalamic nucleus (STN) and the globus pallidus pars interna (GPi) are the most prevalent targets for DBS. Common motor benefits between these targets have been indicated by large, randomized, controlled studies. Dopamine replacement drugs can generally be decreased following STN DBS, while GPi DBS can lead to less adverse cognitive and mood effects[79].
Response to DBS is equal to the best responses of L-DOPA, improving levodopa-responsive symptoms of tremor, bradykinesia, and rigidity. Gait, balance, and speech are less likely to improve[1]. In fact, stimulation-induced side effects include dysarthria, imbalance, and dyskinesia. Another limitation is a limited battery life[78]. There is no definitive duration that DBS remains effective, but it can have benefits for at least 10 years in some[1].
While conventional DBS is a successful treatment, closed-loop DBS is being investigated as a solution around the limitations caused by open-loop DBS, by being able to adjust stimulation in real time according to the patient[78]. In contrast to open-loop DBS which embeds lasting electrodes into the basal ganglia and provides uninterrupted electrical stimulation independent from any feedback, closed-loop DBS delivers electrical stimulation as a function of neuronal activity, all of which is documented and evaluated online[74]. It allows for more efficient and long-term improvements for PD treatment and has the potential to reduce or remove adverse effects in 19% of PD patients treated with subthalamic nucleus DBS according to one study[84].
Surgical therapies with transplantation and gene therapy
Cell transplantation is regarded as a potential future PD treatment. There have been trials using autologous and non-autologous cells. Human embryonic stem cells and induced pluripotent stem cells are few of the cells that have been included in these transplantation studies. One of the concerns with cell transplantation using stem cells is the ethical bounds that must be considered[79,85].
Since the first clinical trial in 1987 involving the transplantation of dopaminergic- neuron-rich human fetal mesencephalic tissue into PD patients’ striatums, more research has aimed to explore whether the grafted dopaminergic neurons will live and form connections in the brain, if the patient’s brain can harmonize and make use of the grafted neurons, and if the grafts can generate significant clinical improvement[86]. Clinical trials with cell therapy intend to discover if there are long-lasting improvements following restoration of striatal DA transmission by grafted dopaminergic neurons. Experimental data from rodents and nonhuman primates show that fetal ventral mesencephalon intrastriatal grafted DA neurons demonstrate many morphological and functional characteristics of normal DA neurons. Significant improvements of PD-like symptoms in animal models have been demonstrated after successful reinnervation by the grafts. Dopaminergic grafts can reinnervate the striatum in the brain, restore regulated release of DA in the striatum, and can become functionally integrated into neural circuitries[86].
A new area that is being explored in the treatment of PD is gene therapy. Both non-disease and disease modifying transgenes are being researched. Non-disease modifying transgenes target GABA and dopamine synthesis pathways. Disease modifying transgenes have the potential to slow the progress of the disease. Both transgenes are too early in trials to be conclusive but offer promising results[87]. Gene therapies address only the motor symptoms, not the non-motor symptoms[88].
Treatment of PD by gene therapy requires an appropriate delivery method for the synthesized nucleic acid: viral or nonviral. Vector choice considerably determines the delivery technique used. A vector that is peripherally delivered must cross the blood-brain barrier (BBB) with an adequate degree of tissue specificity. Rather, DBS surgical techniques may be used to precisely administer the vector to a specific region in the brain. Because nonviral techniques have a briefer duration of achieved gene expression, they are not as suitable for treating neurodegenerative diseases, including PD, despite being technically and conceptually more clear-cut. Using nonviral vectors in experiments usually involves regimens of multiple doses, since there are low transfection rates. This can present problems for translation to human studies if there is a need for repeated intracerebral injections, as there are risks associated[88]. A novel study that utilized human glial cell-derived neurotrophic factor (GDNF) gene and a neurotensin polyplex nanoparticle vector in an animal model of PD found that a single intracerebral injection of the non-viral agent may be able to activate a biochemical and functional response, suggesting that this method may still be effective, regardless of the latter concerns[89]. Region-specific ligands using intravenous vector administration in alternative studies in animal models of PD using nonviral vectors have helped to maximize tissue specificity[90]. Derivation of viral vectors from DNA or RNA are considered a more reasonable approach, as they have the potential ability to create enduring gene expression via episome formation or DNA integration into the host genome[91].
Higher doses of exogenous levodopa are required for PD patients in order to control the progression of symptoms. The enzyme, aromatic amino acid decarboxylase (AADC), is responsible for dopamine production from endogenous or exogenous levodopa. There have been indications that activity of AADC may be diminished in PD. Increasing this enzyme’s activity using gene therapy may minimize PD symptoms and lower the amount of L-DOPA needed to maintain them[91,92].
Behavioral therapy
Even with medical and surgical management, the symptoms of PD may not be completely controlled. While pharmaceutical medications can have positive results, there can also be adverse side effects and limited effectiveness at combating PD symptoms. Furthermore, Parkinson’s drugs have little effect on postural instability, balance problems and gait disorders such as freezing of gait (FoG) and stride variability[93]. Therefore, physicians must look to alternatives to either replace or supplement medical and surgical treatments while managing PD symptoms. Different types of therapies are available to help patients manage symptoms and the daily struggles that may result from them[94]. Physical therapy (PT) and speech therapy (SP) help to manage mobility and motor-related symptoms, with SP focusing on speech abilities[95]. Occupational therapy (OT) focuses on enabling patients to participate in the community, work, and home activities[96]. These therapies focus on the behavior of the patient and management of symptoms rather than on the supplementation or modification of neural pathways affected by PD.
Physical therapy
Aside from pharmaceutical treatment, PT is the most adept aid for patients with PD. It is a necessary aspect in the treatment and maintenance of the health of Parkinson’s patients. Abnormalities in the cortical silent period (CSP duration) and other corticomotor excitability measures in PD patients have been revealed through single-pulse transcranial magnetic stimulation (TMS) studies. This indicates more outstanding corticomotor excitability in these patients. CSP is primarily mediated by gamma-aminobutyric acid (GABA)-B receptors. GABAergic transmission abnormalities are fundamental elements of the pathophysiology of movement disorders concerning the basal ganglia. Promotion of synaptogenesis and neurogenesis may improve neuronal function through voluntary exercise, which enhances brain-derived neurotrophic factor (BDNF). The level of functional inhibition in an activity-dependent manner is modulated by BDNF. This is accomplished by regulation in the number of GABAergic interneurons. It is suspected that the relationship between the lengthening of CSP and high intensity exercise may have an association to an exercise-induced increase in BDNF, though the role of BDNF in inflecting GABA-mediated inhibitory transmission is not completely known[97]. Most of the reviews published about the effectiveness of physical activity (PA) in PD have supported the positive capabilities of PA for those with the disease. A PD patient’s participation in aerobic exercise may significantly improve motor action, balance, and gait parameters including gait velocity, stride length, and walking ability[98,99]. Treadmill training, using PT external cues, dance, and yoga are some PT exercises that have been studied as they relate to PD[93].
Mehrholz et al.[100] concluded that PD patients who partake in treadmill training (TT) are more likely to see improvements in gait hypokinesia. A review by Herman et al.[101] reviewing investigations on the effects of TT on gait in PD patients found that TT had immediate and more lasting effects on gait impairments. Three studies included in this review noted that over-ground walking improved (increased speed and stride length) after just one treadmill session, with effects persisting even 15 minutes after the training session. Eleven longer-term trials included in the review found that TT had positive effects on gait speed, stride length, disease severity measured by the Unified Parkinson’s Disease Rating Scale (UPDRS) and health-related QoL. These benefits endured for weeks after the conclusion of the TT. The persistence of these beneficial effects suggests that TT may elicit positive neural plasticity changes[101]. TT works as an external cue and bypasses the defective basal ganglia, allowing PD patients to focus on the action of walking. The motor movement of walking on a treadmill can generate motor learning, and lead to improvements in Parkinsonian gait. Gait training can improve overall performance since it helps reduce gait disorders including FoG, gait variability and festination. This is important for the QoL of Parkinson’s patients as those affected by gait suffer from reduced mobility, more frequent falls and injuries[93]. The rhythmic motions accomplished by treadmill use can help regularize the PD patient’s gait, visibly improving step variability and stride length, the latter of which are the main risk indicators of falls in the elderly[102,103].
In a study conducted by Arfa-Fatollahkhani et al.[104], it was found that moderate intensity TT resulted in compelling improvements in balance, functional capacity, and QoL, thereby providing evidence that this form of physical training has positive effects in PD patients[104]. In addition, another study conducted by Herman et al.[105] found that overall, TT resulted in decreased Parkinsonian symptoms, measured by the UPDRS, improving from a score of 29 to 22. There was an overall increase in gait speed, lowered (better) swing time variability, and improvement in Short Physical Performance Battery (SPPB) scores. These results suggest that TT has the potential to minimize gait impairments, reduce the risk of falls, and increase the QoL in PD patients[105]. In conjunction to the latter studies, yet another study performed by Protas et al.[106] had similar findings when the effects of step training were assessed. It was found that the trained group had a considerable reduction in the number of falls, gait speed and stride lengths increased, and the 5-step test speed increased in the trained group, but not in the control group[106]. TT may also help achieve effects that positively impact the central nervous system (CNS), specifically dopamine availability and corticomotor excitability. These two fundamental elements are impaired by PD, causing motor disabilities[93].
PD patients have complications with activating the appropriate stepping response for a given situation. An increasing number of studies have described that the interaction between the basal ganglia and the supplementary motor area (SMA) is interrupted during movement performance. Normal functioning of the SMA includes preparation for a constant increase in neuronal activity before movement begins, halting abruptly as soon as the external indication to move transpires. Concise bursts of phasic activity are released by the basal ganglia at the end of sub-movements performed in a sequence. Activity of the basal ganglia suggests that an internal cue is responsible for the sudden decline in SMA neuronal activity. Alas, if this cue was absent or disturbed, as it is in PD, then it is likely that the preparatory activity of the SMA would be disrupted, resulting in abnormally executed motor activity. Dopaminergic neuronal defects result in incorrect cues from the basal ganglia for motor sequences to be communicated to the cortical motor areas, consequently leading to a PD patient’s inadequacy to prepare and maintain complex, yet well-learned movements, such as walking. It is likely that an internal absence of rhythmic cues may cause FoG, festination and other gait impairments. PT strategies aim to offset these physiologic defects that lead to PD motor impairments. Visual, auditory, and somatosensory are three external cues that are shown to be effective at improving motor performance by bypassing the affected basal ganglia[93].
Studies have linked gait in PD to an inability to generate adequate stride length, leaving cadence control as an alternative intact mechanism to compensate for the inability. Several explanations have been proposed as reasonings for the reduced stride length including deficient internal cue production or insufficient contribution to the cortical motor set by the basal ganglia. Morris et al.[107] compared the long-lasting effects in bettering stride length with that of attentional strategies by exploring these possibilities. A total of 54 PD patients in three separate studies were each trained for 20 minutes by repeated 10 minute walks fixed at control stride lengths, varying for each subject based on age, sex and height. Visual floor markers or mental pictures of the suitable stride length were used. Gait patterns were monitored and assessed subsequent to the training every 15 minutes for 2 hours, while adding in secondary tasks of intensifying complexity levels, and covertly when subjects were inattentive to measurements being taken. This study found that normal stepping patterns are attainable for PD patients and that gait hypokinesia is reflected by problems in activating the motor control system. Using attentional strategies and visual cues, PD patients can improve stride length. The mechanism of concentrating on stride length appears to be shared between the two strategies but requires consistent diligence to avoid regression back to more automatic control mechanisms[107].
Emerging studies on the effects of therapeutic dance in addition to conventional PT in PD patients have aimed to find if this form of exercise is safe, feasible and effective. Aguiar et al.[108] included in their review that there is emerging evidence suggesting that therapeutic dance can be used as a safe PA exercise for those with mild-moderately severe PD, leading to improvements in walking, FoG, and health related QoL. Hackney and Earhart performed a study examining the effects of partnered and non-partnered dance movement on gait and balance in patients with PD. There were 39 participants with mild-moderate PD randomly assigned to partnered or non-partnered tango classes, which were held for 1 hour, twice a week for 10 weeks. In the weeks immediately before and after the study, and 1 month after the trial, gait and balance were assessed. Both partnered and non-partnered groups showed noteworthy improvements on the Berg Balance Scale. At the post one-month follow-up, progress was maintained. Though the non-partnered group showed as much improvement as the partnered group, participants in the partnered voiced more enjoyment and interest in continuing the dance[109].
The effects of yoga on oxidative stress, motor function and non-motor symptoms in patients with PD have been studied and assessed. In a randomized controlled trial of an immediate treatment group and a wait-list control group, two 60 minute group-based classes, which lasted for 12 weeks were held for the treatment group. Baseline, 12 week, and 6 months post-intervention assessments were conducted to measure oxidative stress, motor and cognitive function, physical activity, sleep quality and overall QoL. The mean age of participants in this study was 63 years (SD 8, range 49-75), with a disease duration of 4.8 years (SD 2.9, range 1-13), and all had a disease severity from mild-moderate. 90% of all participants (18) were on dopaminergic medications, 85% (17) of them attended 75% or more of the yoga classes and 20% (4) attended 100% of classes. At the end of the 12-week study, no major differences in blood oxidative stress markers between the treatment and control groups were found. The treatment group had better motor function scores based on the UPDRS, but worse sleep scores and physical activity levels than the control group according to the Parkinson’s Disease Quality of Life (PDQUALIF) Scale and the Longitudinal Aging Study Amsterdam Physical Activity Questionnaire, respectively. Motor-function, cognitive function, and catalase levels improved according to within-group comparisons, but social and role function, sleep and outlook domains from the PDQUALIF and physical activity levels were reported poorer at 12 weeks than at baseline. Yoga may be an acceptable complementary practice to improve motor function in PD patients, but more studies using larger sample sizes are needed in order to determine the influence of yoga on oxidative stress and non-motor symptoms[110].
Occupational therapy
There is a progressive development of both motor and non-motor disabilities in patients with PD, despite the effective drug and surgical therapies available. Occupational therapy aims to help patients maintain their self-care, work and leisure activities for as long as possible, and later to adapt to their physical and social environment as needed, as the disease progresses[96]. Occupational therapy goes hand in hand with PT to overcome the physical limitations that hinder patients including walking, manual activities, and self-transfers such as rising from a chair, all of which impact the lifestyle and activities within one’s community and home. While PT focuses on the manual effects, OT focuses on enabling one to perform and engage meaningfully. Structuring schedules, planning activities, and adapting one’s physical environment are all possible strategies adopted by occupational therapists[96]. Home-based individual OT sessions can lead to self-perceived performance improvement within daily tasks[111].
It should be noted with both PT and OT that efficacy is affected by personal and environmental factors. Limitations within an individual’s environment such as the height of furniture at home, the quality of lighting, and availability of space can impact results. Poorly illuminated areas and cramped spaces have been associated with FoG[112]. Environmental factors as well as anxiety has been associated with FoG. This anxiety can be exacerbated by environmental stressors[113]. Motivation, coping style, and adequate social support can also impact results and a patient’s active participation in therapy[96].
Speech therapy
Speech disorders are a common symptom among PD patients, affecting up to 89% of those with the disease, but only 3%-4% of patients receive speech treatment[114]. This can hurt communication skills as well as one’s QoL. Use of the Lee Silverman Voice Treatment (LSVT) LOUD has been used to improve vocal loudness and communication skills. Studies show that it has the possibility to improve not only loudness, but speech rate, intelligibility, quality, monotony, vocal fold adduction, swallowing, facial expression, and neural activation[95]. Training focuses on a single parameter, in this case, loudness and self-perception of vocal loudness, but results in improving the aforementioned symptoms. LSVT/LOUD gives the potential of treatments focused on a single target, encouraging cross-system improvement and delivery systems in line with principles of neural plasticity[115]. While other versions of speech and language therapy have been researched, LSVT/LOUD remains the main form of speech therapy. Modified versions, focusing on parameters such as articulation have been examined; novel versions too have been tried, but with the limited number of trials and patient numbers, there is not sufficient evidence to conclude if these new versions are any more effective than LSVT/LOUD[116].
Cognitive behavioral therapy (CBT)
CBT is being looked at as a possible solution to treat many symptoms such as depression, insomnia, and impulse control for PD patients. CBT studies are disadvantaged in that they cannot be double-blinded. More testing and replication are needed to prove efficacy[1]. Furthermore, CBT holds the disadvantage that each type of therapy tends to manage only one symptom rather than the management of multiple symptoms as in PT or OT, or cross-system improvement as seen in SP. For example, CBT for insomnia is different in its core therapies from other forms of CBT, so it is its own treatment and would not impact other symptoms[117].
Up to 80% of patients report sleep fragmentation and early morning wake ups, forms of sleep maintenance insomnia[118]. Even with the treatment of ‘primary’ conditions, insomnia can persist and will not spontaneously resolve if not treated[119]. The most common approach to treat insomnia is drugs, including benzodiazepine receptor agonists. However, there could be concerns over side-effects or complications with other drugs[120]. CBT is an alternative to drug therapy, focusing on factors that sustain insomnia, namely, sleep-related anxiety, sleep-interfering behaviors, etc. The advantage of CBT is that there are fewer side-effects, it focuses on the factors that cause insomnia rather than managing the effects after insomnia has set in and can have more lasting results. Indeed, CBT for insomnia was found to be at least as effective in treating insomnia as sleep medications[117].
A limited study showed that PD-associated impulse control disorders, as well as depression and anxiety, may be reduced with CBT[121]. With over 40% of patients who take oral dopamine agonists experience impulse control disorders, CBT could be a new method to treat serious associated adverse effects of the necessary medications taken[44]. In addition, a new, telephone-based CBT treatment has promising results, outperforming typical treatments for depression, anxiety, and QoL measurements. CBT is limited by workforce, physical, and geographic barriers; telephone-based treatment bypasses these difficulties and holds the potential to still offer personalized neuropsychiatric care. However, there is no controlled research for this effect. While remote delivery of PD treatment is growing, insurance limits may apply in some states for telemental health services, including telephone-based CBT[122].
Conclusion
L-DOPA remains the most accepted form of treatment for PD, as it is used as a dopamine replacement for this neurodegenerative disease. While other dopamine agonists are successful at controlling symptoms of PD early on in the onset of the disease, L-DOPA is the most effective pharmaceutical at helping to improve QoL, especially once symptoms become more unmanageable with other anti-parkinsonian medications. There is no known cure for PD, but alternative drug, surgical and behavioral therapies exist for the treatment of PD, and new therapies are being developed to help mitigate the side effects and symptoms of this progressive disease. Physical, occupational, and speech therapies provide non-drug alternatives that can be used in adjunct with medications, or separately for those who prefer more natural approaches. They can help treat individual symptoms as they arise. There is still a need to further explore other treatments, and more studies can delve into the under-researched therapies for PD, but the future of PD treatment is promising for patient-specific care that is more effective and with minimal side effects.
Declarations
Authors’ contributionsPerformed data acquisition and data analysis as well as helping with the writing in the text: Lee TK, Yankee EL
Availability of data and materialNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Rizek P, Kumar N, Jog MS. An update on the diagnosis and treatment of Parkinson disease. CMAJ 2016;188:1157-65.
2. Jameson JL, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J. Harrison’s principles of internal medicine. 12th ed. New York: McGraw-Hill Companies; 1991.
3. Georgiou A, Demetriou CA, Christou YP, et al. Genetic and environmental factors contributing to Parkinson’s disease: a case-control study in the cypriot population. Front Neurol 2019;10:1047.
4. Mehra S, Sahay S, Maji SK. α-Synuclein misfolding and aggregation: implications in Parkinson’s disease pathogenesis. Biochim Biophys Acta Proteins Proteom 2019;1867:890-908.
5. Martino R, Candundo H, Lieshout PV, Shin S, Crispo JAG, Barakat-Haddad C. Onset and progression factors in Parkinson’s disease: a systematic review. Neurotoxicology 2017;61:132-41.
6. Calne S, Schoenberg B, Martin W, Uitti RJ, Spencer P, Calne DB. Familial Parkinson’s disease: possible role of environmental factors. Can J Neurol Sci 1987;14:303-5.
8. Carlsson A, Fornstedt B. Possible mechanisms underlying the special vulnerability of dopaminergic neurons. Acta Neurol Scand Suppl 1991;136:16-8.
9. Bekris LM, Mata IF, Zabetian CP. The genetics of Parkinson disease. J Geriatr Psychiatry Neurol 2010;23:228-42.
10. Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 2008;79:368-76.
11. Suchowersky O, Reich S, Perlmutter J, Zesiewicz T, Gronseth G, Weiner WJ; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: diagnosis and prognosis of new onset Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006;66:968-75.
12. Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol 2009;8:464-74.
13. Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol 2006;5:235-45.
14. Fullard ME, Morley JF, Duda JE. Olfactory Dysfunction as an Early Biomarker in Parkinson’s Disease. Neurosci Bull 2017;33:515-25.
15. Mahlknecht P, Seppi K, Poewe W. The Concept of Prodromal Parkinson’s Disease. J Parkinsons Dis 2015;5:681-97.
16. Seppi K, Ray Chaudhuri K, Coelho M, et al; the collaborators of the Parkinson’s Disease Update on Non-Motor Symptoms Study Group on behalf of the Movement Disorders Society Evidence-Based Medicine Committee. Update on treatments for nonmotor symptoms of Parkinson’s disease-an evidence-based medicine review. Mov Disord 2019;34:180-98.
17. Triarhou LC. Dopamine and Parkinson’s Disease. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013. Available from: https://www.ncbi.nlm.nih.gov/books/NBK6271/ [Last accessed on 30 Aug 20].
18. Nieoullon A, Kerkerian-Le Goff L. Cellular interactions in the striatum involving neuronal systems using “classical” neurotransmitters: possible functional implications. Mov Disord 1992;7:311-25.
19. Robertson R, Clarke C, Boyce S, Sambrook M, Crossman A. The role of striatopallidal neurones utilizing gamma-aminobutyric acid in the pathophysiology of MPTP-induced parkinsonism in the primate: evidence from [3H] flunitrazepam autoradiography. Brain Research 1990;531:95-104.
20. Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson’s disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007;27:494-506.
21. Sahay S, Ghosh D, Singh PK, Maji SK. Alteration of structure and aggregation of α-synuclein by familial Parkinson’s disease associated mutations. Curr Protein Pept Sci 2017;18:656-76.
22. Bohnen NI, Albin RL. The cholinergic system and Parkinson disease. Behav Brain Res 2011;221:564-73.
23. Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rüb U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J Neurol 2002;249 Suppl 3:III/1-5.
25. Scatton B, Javoy-agid F, Rouquier L, Dubois B, Agid Y. Reduction of cortical dopamine, noradrenaline, serotonin and their metabolites in Parkinson’s disease. Brain Res 1983;275:321-8.
26. Halliday G, Blumbergs P, Cotton R, Blessing W, Geffen L. Loss of brainstem serotonin- and substance P-containing neurons in Parkinson’s disease. Brain Res 1990;510:104-7.
27. Cash R, Ruberg M, Raisman R, Agid Y. Adrenergic receptors in Parkinson’s disease. Brain Res 1984;322:269-75.
28. Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol 2012;11:986-98.
29. Cullufi P, Tabaku M, Beetz C, et al. Comprehensive clinical, biochemical and genetic screening reveals four distinct GBA genotypes as underlying variable manifestation of Gaucher disease in a single family. Mol Genet Metab Rep 2019;21:100532.
30. Tayebi N, Stubblefield BK, Park JK, et al. Reciprocal and nonreciprocal recombination at the glucocerebrosidase gene region: implications for complexity in Gaucher disease. Am J Hum Genet 2003;72:519-34.
31. Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 2004;81:70-3.
32. Lang AE, Gill S, Patel NK, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol 2006;59:459-66.
33. Nutt JG, Burchiel KJ, Comella CL, et al; ICV GDNF Study Group. Implanted intracerebroventricular. Glial cell line-derived neurotrophic factor. Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 2003;60:69-73.
34. Boado RJ, Pardridge WM. Comparison of blood-brain barrier transport of glial-derived neurotrophic factor (GDNF) and an IgG-GDNF fusion protein in the rhesus monkey. Drug Metab Dispos 2009;37:2299-304.
35. Mohl B, Berman BD, Shelton E, Tanabe J. Levodopa response differs in Parkinson’s motor subtypes: A task-based effective connectivity study. J Comp Neurol 2017;525:2192-201.
36. Anwar A, Saleem S, Akhtar A, Ashraf S, Ahmed MF. Juvenile Parkinson disease. Cureus 2019;11:e5409.
37. Gomez Arevalo G, Jorge R, Garcia S, Scipioni O, Gershanik O. Clinical and pharmacological differences in early- versus late-onset Parkinson’s disease. Mov Disord 1997;12:277-84.
38. Espay AJ, Lang AE. Common myths in the use of levodopa in Parkinson disease: when clinical trials misinform clinical practice. JAMA Neurol 2017;74:633-4.
39. Spiegel J, Hellwig D, Samnick S, et al. Striatal FP-CIT uptake differs in the subtypes of early Parkinson’s disease. J Neural Transm (Vienna) 2007;114:331-5.
40. Pascual A, Hidalgo-Figueroa M, Gómez-Díaz R, López-Barneo J. GDNF and protection of adult central catecholaminergic neurons. J Mol Endocrinol 2011;46:R83-92.
41. AlDakheel A, Kalia LV, Lang AE. Pathogenesis-targeted, disease-modifying therapies in Parkinson disease. Neurotherapeutics 2014;11:6-23.
42. Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA 2014;311:1670-83.
43. Koller WC, Rueda MG. Mechanism of action of dopaminergic agents in Parkinson’s disease. Neurology 1998;50:S11-4. discussion S44-8
44. Armstrong MJ, Okun MS. Diagnosis and treatment of Parkinson disease: a review. JAMA 2020;323:548-60.
46. Ossig C, Reichmann H. Treatment of Parkinson’s disease in the advanced stage. J Neural Transm (Vienna) 2013;120:523-9.
47. Zibetti M, Merola A, Artusi CA, et al. Levodopa/carbidopa intestinal gel infusion in advanced Parkinson’s disease: a 7-year experience. Eur J Neurol 2014;21:312-8.
48. Zibetti M, Rizzone M, Merola A, et al. Sleep improvement with levodopa/carbidopa intestinal gel infusion in Parkinson disease. Acta Neurol Scand 2013;127:e28-32.
49. Honig H, Antonini A, Martinez-Martin P, et al. Intrajejunal levodopa infusion in Parkinson’s disease: a pilot multicenter study of effects on nonmotor symptoms and quality of life. Mov Disord 2009;24:1468-74.
50. Pessoa RR, Moro A, Munhoz RP, Teive HAG, Lees AJ. Apomorphine in the treatment of Parkinson’s disease: a review. Arq Neuropsiquiatr 2018;76:840-8.
51. Dellapina E, Gerdelat-Mas A, Ory-Magne F, et al. Apomorphine effect on pain threshold in Parkinson’s disease: a clinical and positron emission tomography study. Mov Disord 2011;26:153-7.
52. Vanle B, Olcott W, Jimenez J, Bashmi L, Danovitch I, IsHak WW. NMDA antagonists for treating the non-motor symptoms in Parkinson’s disease. Transl Psychiatry 2018;8:117.
53. Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology 1998;50:1323-6.
54. Dashtipour K, Tafreshi AR, Pahwa R, Lyons KE. Extended-release Amantadine for Levodopa-induced Dyskinesia. Expert Rev Neurother 2019;19:293-9.
55. Brocks DR. Anticholinergic drugs used in Parkinson’s disease: an overlooked class of drugs from a pharmacokinetic perspective. J Pharm Pharm Sci 1999;2:39-46.
56. Zahoor I, Shafi A, Haq E. Pharmacological treatment of Parkinson’s disease. In: Stoker TB, Greenland JC, editors. Parkinson’s disease: pathogenesis and clinical aspects. Brisbane (AU): Codon Publications; 2018. pp. 129-44.
57. Contin M, Riva R, Albani F, Baruzzi A. Pharmacokinetic optimisation in the treatment of Parkinson’s disease. Clin Pharmacokinet 1996;30:463-81.
58. Chung KA, Lobb BM, Nutt JG, Horak FB. Effects of a central cholinesterase inhibitor on reducing falls in Parkinson disease. Neurology 2010;75:1263-9.
59. Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993;260:1130-2.
60. Pascual A, Hidalgo-Figueroa M, Piruat JI, Pintado CO, Gómez-Díaz R, López-Barneo J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat Neurosci 2008;11:755-61.
61. Arenas E, Trupp M, Åkerud P, Ibáñez CF. GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo. Neuron 1995;15:1465-73.
62. Gill SS, Patel NK, Hotton GR, et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med 2003;9:589-95.
64. Hadaczek P, Wu G, Sharma N, et al. GDNF signaling implemented by GM1 ganglioside; failure in Parkinson’s disease and GM1-deficient murine model. Exp Neurol 2015;263:177-89.
65. Tatarewicz SM, Wei X, Gupta S, Masterman D, Swanson SJ, Moxness MS. Development of a maturing T-cell-mediated immune response in patients with idiopathic Parkinson’s disease receiving r-metHuGDNF via continuous intraputaminal infusion. J Clin Immunol 2007;27:620-7.
66. Chiricozzi E, Lunghi G, Di Biase E, Fazzari M, Sonnino S, Mauri L. GM1 ganglioside is a key factor in maintaining the mammalian neuronal functions avoiding neurodegeneration. Int J Mol Sci 2020;21:868.
67. Hawthorne GH, Bernuci MP, Bortolanza M, Tumas V, Issy AC, Del-Bel E. Nanomedicine to overcome current Parkinson’s treatment liabilities: a systematic review. Neurotox Res 2016;30:715-29.
68. Re F, Gregori M, Masserini M. Nanotechnology for neurodegenerative disorders. Maturitas 2012;73:45-51.
69. Kulkarni AD, Vanjari YH, Sancheti KH, Belgamwar VS, Surana SJ, Pardeshi CV. Nanotechnology-mediated nose to brain drug delivery for Parkinson’s disease: a mini review. J Drug Target 2015;23:775-88.
70. Tsai MJ, Huang YB, Wu PC, et al. Oral apomorphine delivery from solid lipid nanoparticles with different monostearate emulsifiers: pharmacokinetic and behavioral evaluations. J Pharm Sci 2011;100:547-57.
72. Harris JP, Burrell JC, Struzyna LA, et al. Emerging regenerative medicine and tissue engineering strategies for Parkinson’s disease. NPJ Parkinsons Dis 2020;6:4.
73. Dostrovsky JO, Hutchison WD, Lozano AM. The globus pallidus, deep brain stimulation, and Parkinson’s disease. Neuroscientist 2002;8:284-90.
74. Beuter A, Lefaucheur JP, Modolo J. Closed-loop cortical neuromodulation in Parkinson’s disease: an alternative to deep brain stimulation? Clin Neurophysiol 2014;125:874-85.
75. Laitinen LV, Bergenheim AT, Hariz MI. Leksell’s posteroventral pallidotomy in the treatment of Parkinson’s disease. J Neurosurg 1992;76:53-61.
76. Obeso JA, Rodríguez MC, Gorospe A, Guridi J, Alvarez L, Macias R. Surgical treatment of Parkinson’s disease. Baillieres Clin Neurol 1997;6:125-145.
77. Gross RE. What happened to posteroventral pallidotomy for Parkinson’s disease and dystonia? Neurotherapeutics 2008;5:281-93.
78. Habets JGV, Heijmans M, Kuijf ML, Janssen MLF, Temel Y, Kubben PL. An update on adaptive deep brain stimulation in Parkinson’s disease. Mov Disord 2018;33:1834-43.
79. Lee DJ, Dallapiazza RF, De Vloo P, Lozano AM. Current surgical treatments for Parkinson’s disease and potential therapeutic targets. Neural Regen Res 2018;13:1342-5.
80. Follett KA, Weaver FM, Stern M, et al; CSP 468 Study Group. Pallidal versus subthalamic deep-brain stimulation for Parkinson’s disease. N Engl J Med 2010;362:2077-91.
81. Stefani A, Lozano AM, Peppe A, et al. Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson’s disease. Brain 2007;130:1596-607.
82. Thevathasan W, Coyne TJ, Hyam JA, et al. Pedunculopontine nucleus stimulation improves gait freezing in Parkinson disease. Neurosurgery 2011;69:1248-53. discussion 1254
83. Kalampokini S, Lyros E, Lochner P, Fassbender K, Unger MM. Effects of subthalamic nucleus deep brain stimulation on facial emotion recognition in Parkinson’s disease: a critical literature review. Behav Neurol 2020;2020:4329297.
84. Parastarfeizabadi M, Kouzani AZ. Advances in closed-loop deep brain stimulation devices. J Neuroeng Rehabil 2017;14:79.
85. Yasuhara T, Kameda M, Sasaki T, Tajiri N, Date I. Cell Therapy for Parkinson’s disease. Cell Transplant 2017;26:1551-9.
87. Axelsen TM, Woldbye DPD. Gene Therapy for Parkinson’s disease, an update. J Parkinsons Dis 2018;8:195-215.
88. Hitti FL, Yang AI, Gonzalez-Alegre P, Baltuch GH. Human gene therapy approaches for the treatment of Parkinson’s disease: an overview of current and completed clinical trials. Parkinsonism Relat Disord 2019;66:16-24.
89. Gonzalez-Barrios JA, Lindahl M, Bannon MJ, et al. Neurotensin polyplex as an efficient carrier for delivering the human GDNF gene into nigral dopamine neurons of hemiparkinsonian rats. Mol Ther 2006;14:857-65.
90. Huang R, Han L, Li J, et al. Neuroprotection in a 6-hydroxydopamine-lesioned Parkinson model using lactoferrin-modified nanoparticles. J Gene Med 2009;11:754-63.
92. Bankiewicz KS, Eberling JL, Kohutnicka M, et al. Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp Neurol 2000;164:2-14.
93. Borrione P, Tranchita E, Sansone P, Parisi A. Effects of physical activity in Parkinson’s disease: a new tool for rehabilitation. World J Methodol 2014;4:133-43.
94. Radder DLM, Sturkenboom IH, van Nimwegen M, Keus SH, Bloem BR, de Vries NM. Physical therapy and occupational therapy in Parkinson’s disease. Int J Neurosci 2017;127:930-43.
95. Mahler LA, Ramig LO, Fox C. Evidence-based treatment of voice and speech disorders in Parkinson disease. Curr Opin Otolaryngol Head Neck Surg 2015;23:209-15.
96. Dixon L, Duncan D, Johnson P, et al. Occupational therapy for patients with Parkinson’s disease. Cochrane Database Syst Rev 2007:CD002813.
97. Fisher BE, Wu AD, Salem GJ, et al. The effect of exercise training in improving motor performance and corticomotor excitability in people with early Parkinson’s disease. Arch Phys Med Rehabil 2008;89:1221-9.
98. Shu HF, Yang T, Yu SX, et al. Aerobic exercise for Parkinson’s disease: a systematic review and meta-analysis of randomized controlled trials. PLoS One 2014;9:e100503.
99. Tambosco L, Percebois-Macadré L, Rapin A, Nicomette-Bardel J, Boyer FC. Effort training in Parkinson’s disease: a systematic review. Ann Phys Rehabil Med 2014;57:79-104.
100. Mehrholz J, Friis R, Kugler J, Twork S, Storch A, Pohl M. Treadmill training for patients with Parkinson’s disease. Cochrane Database Syst Rev 2010:CD007830.
101. Herman T, Giladi N, Hausdorff JM. Treadmill training for the treatment of gait disturbances in people with Parkinson’s disease: a mini-review. J Neural Transm (Vienna) 2009;116:307-18.
102. Frenkel-Toledo S, Giladi N, Peretz C, Herman T, Gruendlinger L, Hausdorff JM. Treadmill walking as an external pacemaker to improve gait rhythm and stability in Parkinson’s disease. Mov Disord 2005;20:1109-14.
103. Maki BE. Gait changes in older adults: predictors of falls or indicators of fear. J Am Geriatr Soc 1997;45:313-20.
104. Arfa-Fatollahkhani P, Safar Cherati A, Habibi SAH, Shahidi GA, Sohrabi A, Zamani B. Effects of treadmill training on the balance, functional capacity and quality of life in Parkinson’s disease: a randomized clinical trial. J Complement Integr Med 2019;17:/j/jcim.
105. Herman T, Giladi N, Gruendlinger L, Hausdorff JM. Six weeks of intensive treadmill training improves gait and quality of life in patients with Parkinson’s disease: a pilot study. Arch Phys Med Rehabil 2007;88:1154-8.
106. Protas EJ, Mitchell K, Williams A, Qureshy H, Caroline K, Lai EC. Gait and step training to reduce falls in Parkinson’s disease. NeuroRehabilitation 2005;20:183-90.
107. Morris ME, Iansek R, Matyas TA, Summers JJ. Stride length regulation in Parkinson’s disease. Normalization strategies and underlying mechanisms. Brain 1996;119:551-68.
108. Aguiar LPC, Da Rocha PA, Morris M. Therapeutic dancing for Parkinson’s disease. Int J Gerontol 2016;10:64-70.
109. Hackney ME, Earhart GM. Effects of dance on gait and balance in Parkinson’s disease: a comparison of partnered and nonpartnered dance movement. Neurorehabil Neural Repair 2010;24:384-92.
110. Cheung C, Bhimani R, Wyman JF, et al. Effects of yoga on oxidative stress, motor function, and non-motor symptoms in Parkinson’s disease: a pilot randomized controlled trial. Pilot Feasibility Stud 2018;4:162.
111. Sturkenboom IHWM, Graff MJL, Hendriks JCM, et al. Efficacy of occupational therapy for patients with Parkinson’s disease: a randomised controlled trial. Lancet Neurol 2014;13:557-66.
112. Ehgoetz Martens KA, Pieruccini-Faria F, Almeida QJ. Could sensory mechanisms be a core factor that underlies freezing of gait in Parkinson’s disease? PLoS One 2013;8:e62602.
113. Ehgoetz Martens KA, Ellard CG, Almeida QJ. Does anxiety cause freezing of gait in Parkinson’s disease? PLoS One 2014;9:e106561.
114. Dashtipour K, Tafreshi A, Lee J, Crawley B. Speech disorders in Parkinson’s disease: pathophysiology, medical management and surgical approaches. Neurodegener Dis Manag 2018;8:337-48.
115. Fox CM, Ramig LO, Ciucci MR, Sapir S, McFarland DH, Farley BG. The science and practice of LSVT/LOUD: neural plasticity-principled approach to treating individuals with Parkinson disease and other neurological disorders. Semin Speech Lang 2006;27:283-99.
116. Herd CP, Tomlinson CL, Deane KH, et al. Comparison of speech and language therapy techniques for speech problems in Parkinson’s disease. Cochrane Database Syst Rev 2012;2012:CD002814.
117. Mitchell MD, Gehrman P, Perlis M, Umscheid CA. Comparative effectiveness of cognitive behavioral therapy for insomnia: a systematic review. BMC Fam Pract 2012;13:40.
118. Videnovic A. Management of sleep disorders in Parkinson’s disease and multiple system atrophy. Mov Disord 2017;32:659-68.
119. Morin CM, Bélanger L, LeBlanc M, et al. The natural history of insomnia: a population-based 3-year longitudinal study. Arch Intern Med 2009;169:447-53.
120. Holbrook AM, Crowther R, Lotter A, Cheng C, King D. Meta-analysis of benzodiazepine use in the treatment of insomnia. CMAJ 2000;162:225-33.
121. Okai D, Askey-Jones S, Samuel M, et al. Trial of CBT for impulse control behaviors affecting Parkinson patients and their caregivers. Neurology 2013;80:792-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lee TK, Yankee EL. A review on Parkinson's disease treatment. Neurosciences 2021;8:222. http://dx.doi.org/10.20517/2347-8659.2020.58
AMA Style
Lee TK, Yankee EL. A review on Parkinson's disease treatment. Neuroimmunology and Neuroinflammation. 2021; 8: 222. http://dx.doi.org/10.20517/2347-8659.2020.58
Chicago/Turabian Style
Lee, Tori K., Eva L. Yankee. 2021. "A review on Parkinson's disease treatment" Neuroimmunology and Neuroinflammation. 8: 222. http://dx.doi.org/10.20517/2347-8659.2020.58
ACS Style
Lee, TK.; Yankee EL. A review on Parkinson's disease treatment. Neurosciences. 2021, 8, 222. http://dx.doi.org/10.20517/2347-8659.2020.58
About This Article
Copyright

Data & Comments
Data



 Cite This Article 550 clicks
Cite This Article 550 clicks

 Like This Article 10
likes
Like This Article 10
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.