
Neurological diseases associated with autoantibodies targeting the voltage-gated potassium channel complex: immunobiology and clinical characteristics
 0
0
Abstract
Voltage-gated potassium channels (VGKCs) represent a group of tetrameric signaling proteins with several functions, including modulation of neuronal excitability and neurotransmitter release. Moreover, VGKCs give a key contribution to the generation of the action potential. VGKCs are complexed with other neuronal proteins, and it is now widely known that serum autoantibodies directed against VGKCs are actually directed against the potassium channel subunits only in a minority of patients. By contrast, these autoantibodies more commonly target three proteins that are complexed with alpha-dendrotoxin-labeled potassium channels in brain extracts. These three proteins are contactin-associated protein-2 (Caspr-2), leucine-rich, glioma inactivated 1 (LGI-1)-protein and the protein Tag-1/contactin-2. Neoplasms are detected only in a minority of seropositive patients for VGKC complex-IgG and do not significantly associate with Caspr-2 or LGI-1. Among all the cancers described in association with VGKC complex-IgG, lung carcinoma, thymoma, and hematologic malignancies are the most commonly detected. We will review all the major neurological conditions associated with VGKC complex-IgG. These include Isaacs’ syndrome, Morvan syndrome, limbic encephalitis, facio-brachial dystonic seizures, chorea and other movement disorders, epilepsy, psychosis, gastrointestinal neuromuscular diseases, a subacute encephalopathy that mimics Creutzfeldt-Jakob prion disease both clinically and radiologically and autoimmune chronic pain. The vast majority of these conditions are reversible by immunotherapy, and it is becoming increasingly recognized that early diagnosis and detection of VGKC complex-IgG is critical in order to rapidly start the treatment. As a result, VGKC complex-IgG are now part of the investigation of patients with unexplained subacute onset of epilepsy, memory or cognitive problems, or peripheral nerve hyperexcitability syndromes.
Keywords
Introduction
Voltage-gated potassium channels (VGKCs) represent a group of tetrameric signaling proteins with several functions, including modulation of neuronal excitability and neurotransmitter release.[1] Moreover, VGKCs contribute to the generation of the action potential. Neurological autoimmune and paraneoplastic syndromes involve only a small number of VGKCs, notably the “Shaker” type Kv1 channels (Kv1.1, Kv1.2, Kv1.6), sensitive to alpha-dendrotoxin.[2] VGKCs are complexed with other neuronal proteins, and it is now widely known that serum autoantibodies directed against VGKCs are actually directed against the potassium channel subunits only in a minority of patients. In contrast, these autoantibodies more commonly target three proteins that are complexed with alpha-dendrotoxin-labeled potassium channels in brain extracts.[3] These three proteins are contactin-associated protein-2 (Caspr-2), which is localized at the juxtaparanodes in myelinated axons and associates with Transient axonal glycoprotein 1,postsynaptic density-95/discs large/zonula occludens-1, and the ankyrin-spectrin protein;[4] leucine-rich, glioma inactivated 1 (LGI-1) protein that is most strongly expressed in the hippocampus; and the protein Tag-1/contactin-2, associated with Caspr-2.
Bien et al.[5] demonstrated that T-cell cytotoxicity is not a major contributor for the pathogenesis of the neurological syndromes associated with VGKCs, whereas antibody and complement-mediated neuronal cell damage are prevalent.
Neoplasms are detected only in a minority of seropositive patients for VGKC complex-IgG (16% in the experience of Mayo Clinic)[2,4] and do not significantly associate with Caspr-2 or LGI-1. Among the tumors that are believed to be associated with VGKC complex-IgG, lung carcinoma, thymoma and hematologic malignancies are the most commonly described.[2]
We will review all the major neurological conditions associated with VGKC complex-IgG. These include Isaacs’ syndrome,[6] Morvan syndrome (MoS),[7] limbic encephalitis (LE),[8,9] facio-brachial dystonic seizures (FBDS),[10,11] chorea and other movement disorders,[12] epilepsy,[13] psychosis,[14] gastrointestinal neuromuscular diseases,[15,16] a subacute encephalopathy that mimics Creutzfeldt-Jakob prion disease both clinically and radiologically[2] and autoimmune chronic pain.[17]
Peripheral nerve hyperexcitability
Motor nerves
Isaacs’ syndrome (neuromyotonia) Immune-mediated neuromyotonia, also known as Isaacs’ syndrome, is the most severe phenotype of peripheral nerve hyperexcitability. It is characterized by spontaneous and continuous muscular activity resulting from repetitive motor unit action potentials of peripheral origin. The syndrome was described for the first time by Isaacs in 1961.[18] Isaacs also established the peripheral nerve origin of the discharges by documenting the persistence of abnormal electromyographic activity after proximal nerve block. The main clinical features of the neuromyotonia are muscle twitching at rest (visible myokymia), cramps and muscle stiffness, impaired muscle relaxation after voluntary contraction (pseudomyotonia), along with hyperhidrosis. Patients may also suffer from weakness. Other symptoms include myokymia of the limb, trunk,[19] face[20] and tongue[21] muscles. In some patients, hypertrophy of muscles can occur due to continuous muscle activity.[22] The main electromyographic hallmark of the neuromyotonia is the presence of spontaneous, continuous doublet, triplet or multiplet single motor unit discharges, firing at a high intraburst frequency (30-300 Hz).[23] In addition, fibrillation potentials and fasciculations are often present, the former indicating the discharge of a single muscle fibers. About 40% of patients with acquired neuromyotonia have detectable anti-VGKC antibodies[24] and the percentage increases to 80% if there is an associated thymoma. Interestingly, the dysfunction of peripheral nerve VGKCs can be also due to genetic cause, that is, episodic ataxia type I. In fact, episodic ataxia type I is caused by a mutation of the potassium channel gene KCNA1 on chromosome 12.[25]
The acquired, immune-mediated form of the neuromyotonia has been described in association with many autoimmune diseases, such as myasthenia gravis, Addison disease, vitiligo, Hashimoto thyroiditis, pernicious anemia, celiac disease, and rheumatoid arthritis.[26] It is well established that neuromyotonia may also be paraneoplastic. In such cases, the pathophysiology is likely due to cross-reactivity of tumor antigens and VGKCs.[26] Most cases of paraneoplastic neuromyotonia are related to small cell lung carcinoma[27,28] and thymoma,[29,30] but it has also been reported an association with Hodgkin lymphoma,[31] bladder[32] and ovarian carcinoma.[33] Membrane-stabilizing drugs, namely the anticonvulsants carbamazepine, phenytoin, sodium valproic acid, lamotrigine are used for symptomatic relief in patients with Isaacs’ syndrome as they usually provide significant improvement of stiffness, muscle spasms, and pain. Their mechanism of action helps reducing neuronal repetitive firing through interaction with VGKCs. If the response is not sufficient, oral corticosteroid may be prescribed, and nonsteroid immunosuppressive drugs such as azathioprine and methotrexate may also be considered as treatment.[34] Plasma exchange often produces clinical improvement lasting about 6 weeks with a significant fall in VGKC antibody titers.[34] Intravenous immunoglobulins are also indicated for severe neuromyotonia, providing short-term relief. In the paraneoplastic form of the neuromyotonia, treatment of malignancy is warranted.[34]
Sensory nerves
Chronic pain Klein et al.[17] found that the 50% of VGKC-complex antibody positive patients experience pain in isolation (28%) or with accompanying neurologic manifestations (72%), not attributable to an alternative cause. VGKC-complex antibodies related pain is subacute in onset, chronic in course, neuropathic, nociceptive, regional, or diffuse. It is significantly associated with Caspr-2 antibody positivity, but not with LGI-1 antibody,[17] and occurs in isolation or with recognized neurologic manifestations of VGKC-complex autoimmunity.[11,35,36] It is characterized by prominent morbidity and in some cases may require to be treated by narcotics. It has been hypothesized that pain related to VGKC antibodies is due to the hyperexcitability of nociceptive pathways, although such involvement is difficult to be demonstrated, and patients’ symptoms are often disproportionate to objectively measured neuropathic dysfunction. It has been demonstrated that VGKCs act synergistically with the potassium/sodium hyperpolarization-activated cyclic nucleotide-gated ion channel 2 (HCN2), that is an inward rectifying channel acting as a regulator of nociceptive pain.[37] VGKC and HCN2 act synergistically to maintain nociceptive afferent sensory neural thresholds,[38,39] and it is reasonable to hypothesize that VGKC-complex antibodies may interfere with their functional activity.
Membrane-stabilizing antiepileptic drugs may have some benefit in patients with VGKC-complex autoimmune chronic pain. Interestingly, 81% of patients described by Klein et al.[17] experienced improvement in their pain by immunotherapy, allowing narcotics to be discontinued in some cases.
More recently, Rosch et al.[40] reported two cases of ganglioside antibody-negative pediatric Guillain-Barrè syndrome associated with Caspr-2 antibodies. Both patients experienced a full recovery. Thus, Caspr-2 might be a possible autoimmune target in Guillain-Barrè syndrome. Certainly, further studies are needed in order to fully understand the relevance of Caspr-2 as an autoantigen in the pathophysiology of Guillain-Barrè syndrome.
Autonomic nerves
Gastrointestinal neuromuscular disorders described in association with VGPC complex-IgG Gastrointestinal neuromuscular diseases are characterized by symptoms of intestinal neuromuscular dysfunction.[41] These disorders may be attributable to congenital or, more frequently, acquired conditions. An autoimmune pathophysiology has been proposed to explain acquired gastrointestinal neuromuscular diseases. Interestingly, inflammatory neuropathy is common in patients affected by gastrointestinal neuromuscular diseases and autoantibodies directed against neuronal antigens are present in some patients. VGKC complex-IgG have been detected in the sera of patients with primary and paraneoplastic slow-transit constipation,[15,16] primary achalasia,[42] postinfective irritable bowel syndrome associated with inflammatory enteric neuropathy,[43] chronic intestinal pseudo obstruction,[44] and esophageal and colonic dysfunction secondary to infection by the protozoan parasite trypanosoma cruzi (Chagas’ disease).[45] The presence of VGKC-Ab (or other antineuronal antibodies) in the early phases of gastrointestinal neuromuscular diseases still remains contentious. However, if present, early detection, followed by proper immunotherapy could be important in order to prevent the progressive deterioration of gut function.
Central nervous system manifestations
LE
LE is generally characterized by a subacute and progressive onset of episodic memory deficits, disorientation, and recurrent seizures. Additional features are hallucinations, sleep-cycle disturbances, agitation, and delusions. There is histological evidence of mesial temporal lobe inflammation. LE can be associated with several antibodies including anti-Hu,[2] anti-CV2/CRMP5,[46] anti-Ri,[47,48] anti-Ma2,[49,50] and anti-amphiphysin.[51,52] Antibodies targeting neuronal cell surface antigens, such as ion channels and ligand-gated ion channels have been recently identified. VGKC complex-IgG are a good example of the second category of antibodies. VGKC antibodies were first reported in 2001 in two patients affected by reversible LE[53] and then in two series in 2004.[8,9] VGKC-LE has been described in association with antibodies against LGI-1 in 80-90% of patients[54] or Caspr-2 in 5-10%.[55] Very few patients have contactin-2 antibodies, and some patients have no specific target.[56] VGKC-LE is frequently diagnosed in the absence of associated tumors.[57,58] In a recent study, only the 21.4% of the patients showed malignancies.[59] Hyponatremia is a characteristic feature of VGKC-LE. It is present in about 60% of patients, and it is initially resistant to treatment, but it usually resolves as the VGKC complex-IgG titers decline.[9] The serum hyponatremia usually follows a syndrome of inappropriate antidiuretic hormone (SIADH) secretion pattern. Intermittent and episodic hypothermia, along with neuropathic pain, both responsive to immunotherapy, have been reported in patients affected by VGKC-LE.[60] A severe sleep disorder, characterized by insomnia, deep diurnal drowsiness and complete disappearance of rapid eye movement sleep has also been associated with VGKC-LE.[61] Autonomic dysfunctions have been extensively described in VGKC-LE patients. Vincent et al.[9] reported sweating and hypersecretion due to an effect of the antibodies on the postganglionic sympathetic neurons. More recently, episodic bradycardia has been recognized as a prodrome of LGI-1-LE[62] and in all the patients reported, led to pacemaker implantation. Interestingly, in other cell-surface antibody-associated neurological disorders (i.e. N-methyl-D-aspartate receptor antibody encephalitis) bradycardia has been rarely reported.
FBDS represent a typical manifestation that may precede the development of LGI-1 LE.[11] The clinical features of FBDS will be extensively discussed elsewhere in this review. They may be characterized by facial twitching, hand and leg posturing. The antiepileptic drugs do not usually reduce seizure activity. In contrast, early initiation of plasma exchange and immunosuppression help to avoid the development of full-blown LE.
Routine cerebrospinal fluid (CSF) analysis may reveal a mild lymphocytosis in some patients and protein, and glucose levels may be modestly raised or within normal limits. Polymerase chain reaction is obviously negative for herpes simplex virus and other neurotropic viruses while oligoclonal bands may be present, but rarely unmatched with serum bands.[9]
In a recent magnetic resonance imaging (MRI) study on patients affected by VGKC-LE[59] initial MRI findings included unilateral or bilateral amygdala and/or hippocampal enlargement and T2 hyperintensity in 78.6% of patients at some time point during the disease course. Restricted diffusion, mild ill-defined contrast enhancement, and extratemporal findings were also reported. Interestingly, more than a quarter of the patients with initially negative MRI or only unilateral abnormalities then progressed to bilateral involvement, supporting the hypothesis of radiologic progression of the disease. It is still a matter of debate whether these changes reflect persistent inflammation or alternatively they are secondary to recurrent seizures. In fact, patients with VGKC-LE have a very high frequency of epileptic seizures,[58] and this has been hypothesized to be related to the development of mesial temporal sclerosis, often seen in follow-up VGKC-LE patients. Patients with VGKC-LE and high signal in the medial temporal lobes typically develop hippocampal atrophy as the high signal declines.[63,64] It is not clear yet whether the cases of otherwise “cryptogenic” mesial temporal sclerosis are at least partly related to a remote effect of VGKC autoimmunity.
FBDS
FBDS are very brief highly distinctive seizures associated with VGKC-complex antibodies, almost always in the LGI-1 subtype. They carry a high chance of developing VGKC-LE, and their recognition should prompt consideration of immunotherapies in order to prevent the onset of LE. The FBDS was first described in 2008 by Irani et al.[10] and then better characterized in 2011.[11] As compared to the initial descriptions, it is now evident that the age of onset of FBDS is broad, varying from 28 to 92 years,[65,66] possibly with a small male prevalence.[11,65] The daily frequency of FBDS is high, ranging from 6 to 360 attacks per day at their peak.[11] Emotions and movements are common triggers of FBDS,[11,65] and a sensory aura or auditory hallucinations may precede them.[65] Every FBDS is characterized by a dystonic posturing of the arm, both proximally and distally, and may involve also the ipsilateral face and less commonly, the trunk and the ipsilateral leg. It is worth noting that events involving the leg alone have been rarely observed.[65] Furthermore, synchronous bilateral dystonia and rapidly alternating events have been reported.[65] Either side can be involved, but FBDS are usually unilateral on any occasion.[11] If FBDS can be classified as tonic seizures, or as a movement disorder, namely a form of dystonia, is still a matter of debate[67] and data to support the former and the latter hypothesis are summarized in Table 1. FBDS are often accompanied by ictal automatisms and may be, followed by fear, agitation and speech arrest.[65] The duration of FBDS was reported to last < 3 s in the early description,[11] however it is now clear that they may last also between 10 and 30 s.[65] Serum sodium levels are often reduced in FBDS patients presenting also with cognitive impairment but are rarely low during the period with facio-brachial dystonic seizures alone.[11,65] If the patient experiences FBDS alone, with no cognitive impairment, routine MRI is unremarkable in the vast majority of cases.[11,65] However, routine MRI showed a high signal change in the putamen in a patient described by Irani et al.[65] in 2013 and a gadolinium-enhancing lesion involving the caudate and globus pallidus in a patient that we described in 2013.[68] When cognitive impairment is present, both unilateral and bilateral medial temporal lobe and also caudate signal changes have been described.[11,65] Interestingly, the basal ganglia signal changes are controlateral to the facio-brachial dystonic seizures in all the patient described so far.[65,68] Electroencephalography shows ictal epileptiform activity in a minority of patients with a frontotemporal, frontal, or temporal focus.[11] The LE following FBDS is clinically undistinguishable from the other VGKC-LE with amnesia, confusion, hallucinations, sleep disturbances and other nondystonic seizure types.[11] The initial treatment with antiepileptic drugs is ineffective in the majority of the patients, while the initiation of immunotherapy with corticosteroids, intravenous immunoglobulin, and plasma exchange has proven to be useful in order to reduce the frequency of FBDS.[11,65] The cessation of FBDS after the immunotherapy is associated with a significant reduction in serum VGKC complex/LGI-1 antibody titers.[65] Antiepileptic drugs are also responsible for adverse cutaneous reactions in a high proportion of patients and the involvement of both eyes and mouth has been reported in one patient,[65] while the documented psychiatric side effects of steroids (including paranoia and hypomania) generally improve by tapering steroids. In patients with persisting VGKC-complex/LGI1-antibodies, relapses of FBDS have been described during the steroid tapering period, and all the relapses showed an absolute response, after increasing the corticosteroid dose.
Data to support the hypothesis of FBDS as tonic seizures, or as a movement disorder
| Movement disorders | Epileptic seizures |
|---|---|
| Loss of consciousness not always noted | The majority of patients experiences loss of awareness, although not during every attack |
| Electroencephalography: epileptic activity in a minority of patients (24%) | Electroencephalography: focal slowing or epileptiform changes in 24% of cases with FBDS is a significant proportion (very brief duration of the attacks arising from spatially limited or deep foci) |
| Functional neuroimaging: altered glucose metabolism in different cerebral regions, including basal ganglia | Brief duration and highly stereotyped attacks |
| Poor response to antiepileptic drugs | LGI-1 antibodies associated with typical medial temporal lobe seizures in the context of limbic encephalitis, often refractory to anticonvulsants |
| Chorea and other movement disorders associated with VGKC | The frequent ictal presence of automatisms, and fear, agitation and speech arrest after the motor event |
The recognition of FBDS is critical because the response to anti-epileptic drugs is unsatisfactory while response to immunotherapy is excellent, and early initiation of immunosuppressant therapy offers the chance to modify the course of this neurological disorder avoiding the development of full-blown LE.
Chorea and other movement disorders associated with VGPC complex-IgG
Epilepsy
In recent years, a number of studies have reported antibodies targeting neuronal cell surface antigens in patients with epilepsy, including antibodies against the VGKC-complex and the N-methyl-D-aspartate glutamate receptor.[70-72] The role of such antibodies in the pathogenesis of epilepsy has not been fully established,[73] but they have been found more likely to occur in patients with focal epilepsy of undetermined cause rather than in patients with structural/metabolic focal epilepsy.[71] In particular, VGKC-complex antibodies are of interest in epilepsy, as they target VGKC that play an important role in regulating neuronal excitability. Lilleker et al.[73] found that in patients with unexplained adult onset epilepsy, VGKC-complex antibodies were positive in the 8% of cases and the 4.2% of the patients had titers higher than 400 pM. Patients with VGKC-complex antibodies titers higher than 400 pM had focal seizures and in 4 out the 6 described cases the seizure focus was diagnosed in the temporal lobe. These patients reported rising abdominal sensation, olfactory hallucination, piloerection, dejà vu, oromasticatory automatism and depersonalization. Two patients had generalized tonic-clonic seizures in addition to focal seizures, and one patient had FBDS. No patient had the clinical syndrome of LE, and no patient was diagnosed with cancer. The sera of all 6 patients were tested for LGI-1 and Caspr-2 antibodies. The serum of the patient who presented with FBDS tested positive for LGI-1 antibodies; one patient was positive for Caspr-2 antibodies; all the other patients were negative for both LGI-1 and Caspr-2 antibodies. Brain MRI was normal in all patients except in one patient showing increased signal in the left hippocampus and amygdala that was attributed to recent seizures. Interestingly, all the patients improved with immunotherapy, although not all of them have been rendered seizure-free. Furthermore, Iorio et al.[72] found that patients with neural antibodies not responding to antiepileptic drugs may benefit from immunotherapy. They studied 81 patients (39 patients with epilepsy and other neurological symptoms and/or autoimmune diseases responsive to antiepileptic drugs and 42 patients with AED-resistant epilepsy). Neural autoantibodies were detected in 22% of patients, mostly from the antiepileptic drug-resistant epilepsy group. Three patients with antiepileptic drug-resistant epilepsy had anti-LGI-1 antibodies. Twelve patients received immunotherapy and 9 (75%) achieved a > 50% reduction in seizure frequency. Interestingly, LGI-1 is also linked to seizures of genetic etiology. Mutations in the LGI-1 gene are responsible for autosomal dominant temporal lobe epilepsy with auditory features (buzzing and tinnitus).[74-76] Indeed, the identification of autoantibodies, such as those targeting VGKC-complex, has changed paradigms in the diagnosis and management of epilepsy and has expanded the phenotypic spectrum of autoimmune disorders. In future, the discovery of new autoantibodies may also further expand the range of the autoimmune epilepsies.
Psychiatric manifestations
Several psychiatric manifestations have been described in patients with VGKC-complex antibodies.[12,14] These are often affective-predominant and include confusion, memory impairment, personality changes, depression, agitation, hallucinations, and anxiety. A clinical improvement has been reported in the majority of the patients who received immunotherapy.[12] Neuropsychiatric presentations are significantly more common in patients with higher autoantibody values, and clinical improvements are more likely in patients treated early. Further studies are needed in order to clarify the exact prevalence of VGKC-complex antibodies in patients from the general population with psychiatric manifestations.
Subacute encephalopathy that mimics Creutzfeldt-Jakob prion disease associated with VGKC complex-IgG
Rossi et al.[77] described three patients that were referred with possible prion disease. Their clinical picture was in keeping with autoimmune encephalitis, and they had very high VGKC-complex/LGI-1 antibodies. Otherwise, low titers of neuronal antibodies occurs rarely in suspected patients with sporadic Creutzfeldt-Jakob disease (sCJD) and when present should be interpreted with caution. Atypical features in sCJD, such as FBDS, hyponatremia[36] and autonomic dysfunction, may suggest an autoimmune disorder.[77] A high titer of VGKC-complex Ab (LGI-1 negative) was also identified in a 61-year-old Caucasian man with a novel prion protein (PRNP) gene mutation and Gerstmann-Straüssler-Scheinker disease, but despite 1 year of aggressive immunosuppressive treatment the patient died.[78]
Interestingly, nonspecific markers of neuronal degeneration in CSF such as 14.3.3 and S100B proteins may test positive in patients with VGKC complex-IgG encephalitis, thus being not completely reliable for the definite diagnosis of sCJD.[77]
The demographic, clinical and neuroradiological characteristics of the patients positive for VGKC complex-IgG (except for FBDS, dysautonomia, and hyponatremia) are not distinguishable from CJD[36,77] and most patients fulfilled the World Health Organization diagnostic criteria for sCJD.[36,79] Thus, it is critical to consider autoimmune encephalitis in the differential diagnosis of sCJD in order to promptly test for the relevant antibodies.
Morvan’s syndrome
Morvan’s “fibrillary chorea” or MoS was first described by the French physician Augustin Marie Morvan[80] in 1890 in a patient who exhibited myokymia combined with excessive sweating and disordered sleep. It is a rare entity characterized by peripheral and central nervous system (CNS) involvement, specifically, neuromyotonia, hallucinations, delirium, insomnia, and autonomic disturbance [Table 2].[80,81] Peripheral nerve involvement is mainly characterized by neuromyotonia, but neuropathic pain in the feet and/or legs and back, areflexia and a stocking-type sensory loss may also be present.[81] In some cases, insomnia may be severe, amounting to not less than complete lack of sleep (agrypnia) for weeks or months in a row.[82] Common encephalopathic manifestations are spatial and temporal disorientation, confusion, amnesia, hallucinations and agitation. Epileptic seizures, including generalized tonic-clonic seizures and partial seizures consistent with FBDS, are present in about one-third of the cases.[11] Compulsive behaviors, stereotypies, and reduplicative paramnesias can be part of the CNS involvement.[83] Autonomic disturbance includes hyperhidrosis, pruritus, drooling, severe constipation, urinary incontinence, excessive lacrimation, and cardiac arrhythmias.[84] Autonomic system dysfunction has been described in the 93% of MoS patients, being hyperhidrosis and cardiovascular instability the most common manifestations.[81] Weight loss, skin lesions or itching, and hyponatremia due to the SIADH secretion are also possible symptoms. MoS usually presents with a slow, insidious onset over months to years[85] and is almost exclusively seen in males. In about the 90% of cases, it spontaneously goes into remission, while in the remaining 10% of cases leads to death.[86]
Symptoms and signs of Morvan's syndrome
| Peripheral nervous system | Central nervous system | Autonomic system | Systemic features |
|---|---|---|---|
| Neuromyotonia | Insomnia | Hyperhidrosis | Weight loss |
| Neuropathic pain | Disorientation/confusion | Tachycardia | Skin lesions |
| Areflexia | Amnesia | Blood pressure abnormalities | Hyponatremia |
| Stocking-type sensory loss | Hallucinations | Drooling | |
| Agitation | Constipation | ||
| Delusions | Urinary incontinence | ||
| Seizures | Excessive lacrimation |
Patients with MoS may have an associated underlying tumor, including thymoma, that is the most common, lung cancer,[87] sigmoid cancer,[88] testicular cancer and lymphoma, thus indicating the paraneoplastic nature of the disease.[89] On the other hand, patients without an associated tumor have been also been studied, and they generally experience a good clinic response to immunotherapy.[81] Interestingly, it has been described the occurrence of MoS after scrotal tap and injection of sclerosing agent for the treatment of hydrocele in 5 males.[90] Some MoS cases associated with thymomas and myasthenia gravis have also been reported.[91] VGKC-complex antibody serum levels are increased in the 90% of the patients [Figure 1] and although these are directed against LGI-1, Caspr-2, or commonly both, Caspr-2 antibodies predominate and are always associated with thymoma. Fewer patients have been reported also with contactin-2 antibodies.[81] It is intriguing that low levels of Caspr-2 mRNA have been detected in the human prostate: although Caspr-2 is predominantly expressed in the nervous system, the male reproductive system may be a source of the antigen and MoS onset after scrotal drainage[90] may be a crucial factor to break the immune tolerance. Moreover, thymectomy and thymoma chemotherapy may act as disease triggers, thus suggesting that thymic tumors may also harbor the antigenic targets, in particular, Caspr-2. CSF analysis in MoS usually shows normal protein, glucose, white cell count, and IgG index. Oligoclonal bands may be detected. Marked changes in circadian serum levels of neurohormones have been described, with serum levels of melatonin and prolactin substantially lower than normal and without a circadian rhythm of release.[87] Plasma levels of norepinephrine were found to be high throughout the 24 h period, without the physiological nocturnal decrease.[83,87] Increased serum levels of cortisol were also observed.[87] A negative MRI is a characteristic finding in MoS,[81,88] but frontal T2 hyperintensity in one patient and bilateral hippocampal T2 high signal in another patient have also been reported[81] and in these cases the diagnosis of “LE associated with neuromyotonia” should be more appropriate. MoS treatment is based on immunotherapies, including plasma exchange, intravenous immunoglobulin, corticosteroids, azathioprine, cyclosporine, and cyclophosphamide. In the paraneoplastic forms of MoS, the management of the underlying malignancy is mandatory.
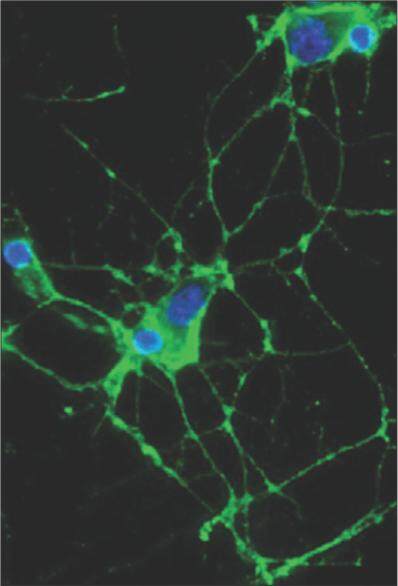
Figure 1. Indirect immunofluorescence showing IgG in the serum of a patient with Morvan's syndrome binding to the surface of a live rat hippocampal neurons
Conclusion
The clinical spectrum of the neurological disorders associated with VGKC complex-IgG is rapidly expanding, and new associated conditions have been described in the last years. The vast majority of these disorders are reversible by immunotherapy, and it is becoming increasingly recognized that early diagnosis and detection of VGKC complex-IgG is critical in order to rapidly start the treatment. As a result, VGKC complex-IgG are now part of the investigation of patients with unexplained subacute onset of epilepsy, memory or cognitive problems, or peripheral nerve hyperexcitability syndromes. It is still not fully understood how VGKC complex-IgG could cause such a range of different clinical presentations. The accelerated development that the research on antibody-mediated syndrome has had in the last period has been exciting and has made possible to diagnose and to treat clinical syndromes that would have otherwise been poorly defined. Certainly, the development and validation of experimental models of VGKC autoimmunity will represent the next critical challenge in order to clearly elucidate how the antibodies get into the CNS and understand if also antibody-binding, internalization and loss of the target surface antigens, along with complement activation, are involved in the physiopathology. In fact, there is a need to extend our understanding of the pathophysiological mechanisms of these syndromes in order to improve their diagnosis, and ultimately, to develop more effective targeted therapies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stühmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev 2005;57:473-508.
3. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, Peles E, Buckley C, Lang B, Vincent A. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734-48.
4. Klein CJ, Lennon VA, Aston PA, McKeon A, O'Toole O, Quek A, Pittock SJ. Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol 2013;70:229-34.
5. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, Jellinger KA, Reuss DE, Ribalta T, Schlegel J, Sutton I, Lassmann H, Bauer J. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 2012;135:1622-38.
6. Hart IK, Waters C, Vincent A, Newland C, Beeson D, Pongs O, Morris C, Newsom-Davis J. Autoantibodies detected to expressed K+channels are implicated in neuromyotonia. Ann Neurol 1997;41:238-46.
7. Barber PA, Anderson NE, Vincent A. Morvan's syndrome associated with voltage-gated K+channel antibodies. Neurology 2000;54:771-2.
8. Thieben MJ, Lennon VA, Boeve BF, Aksamit AJ, Keegan M, Vernino S. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology 2004;62:1177-82.
9. Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N, Clover L, Parkinson A, Bien CG, Omer S, Lang B, Rossor MN, Palace J. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004;127:701-12.
10. Irani SR, Buckley C, Vincent A, Cockerell OC, Rudge P, Johnson MR, Smith S. Immunotherapy-responsive seizure-like episodes with potassium channel antibodies. Neurology 2008;71:1647-8.
11. Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR, Schott JM, Armstrong RJ, A SZ, Bleasel A, Somerville ER, Smith SM, Vincent A. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892-900.
12. Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008;70:1883-90.
13. Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006;71:135-41.
14. Somers KJ, Lennon VA, Rundell JR, Pittock SJ, Drubach DA, Trenerry MR, Lachance DH, Klein CJ, Aston PA, McKeon A. Psychiatric manifestations of voltage gated potassium-channel complex autoimmunity. J Neuropsychiatry Clin Neurosci 2011;23:425-33.
15. Knowles CH, Lang B, Clover L, Scott SM, Gotti C, Vincent A, Martin JE. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol 2002;37:166-70.
16. Dhamija R, Tan KM, Pittock SJ, Foxx-Orenstein A, Benarroch E, Lennon VA. Serologic profiles aiding the diagnosis of autoimmune gastrointestinal dysmotility. Clin Gastroenterol Hepatol 2008;6:988-92.
17. Klein CJ, Lennon VA, Aston PA, McKeon A, Pittock SJ. Chronic pain as a manifestation of potassium channel-complex autoimmunity. Neurology 2012;79:1136-44.
18. Isaacs H. A syndrome of continuous muscle-fibre activity. J Neurol Neurosurg Psychiatry 1961;24:319-25.
19. Greenhouse AH, Bicknell JM, Pesch RN, Seelinger DF. Myotonia, myokymia, hyperhidrois and wasting of muscle. Neurology 1967;17:263-8.
20. Irani PF, Purohit AV, Wadia NH. The syndrome of continuous muscle fiber activity. Evidence to suggest proximal neurogenic causation. Acta Neurol Scand 1977;55:273-88.
21. Black JT, Garcia-Mullin R, Good E, Brown S. Muscle rigidity in a newborn due to continuous peripheral nerve hyperactivity. Arch Neurol 1972;27:413-25.
22. Vasilescu C, Alexianu M, Dan A. Muscle hypertrophy and a syndrome of continuous motor unit activity in prednisone-responsive Guillain-Barré polyneuropathy. J Neurol 1984;231:276-9.
24. Shillito P, Molenaar PC, Vincent A, Leys K, Zheng W, van den Berg RJ, Plomp JJ, van Kempen GT, Chauplannaz G, Wintzen AR, et al. Acquired neuromyotonia: evidence for autoantibodies directed against K+channels of peripheral nerves. Ann Neurol 1995;38:714-22.
25. Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 1994;8:136-40.
26. Rana SS, Ramanathan RS, Small G, Adamovich B. Paraneoplastic Isaacs' syndrome: a case series and review of the literature. J Clin Neuromuscul Dis 2012;13:228-33.
27. Walsh JC. Neuromyotonia: an unusual presentation of intrathoracic malignancy. J Neurol Neurosurg Psychiatry 1976;39:1086-91.
28. Partanen VS, Soininen H, Saksa M, Riekkinen P. Electromyographic and nerve conduction findings in a patient with neuromyotonia, normocalcemic tetany and small-cell lung cancer. Acta Neurol Scand 1980;61:216-26.
29. García-Merino A, Cabello A, Mora JS, Lia-o H. Continuous muscle fiber activity, peripheral neuropathy, and thymoma. Ann Neurol 1991;29:215-8.
30. Perini M, Ghezzi A, Basso PF, Montanini R. Association of neuromyotonia with peripheral neuropathy, myasthenia gravis and thymoma: a case report. Ital J Neurol Sci 1994;15:307-10.
31. Caress JB, Abend WK, Preston DC, Logigian EL. A case of Hodgkin's lymphoma producing neuromyotonia. Neurology 1997;49:258-9.
32. Forte F, Pretegiani E, Battisti C, Sicurelli F, Federico A. Neuromyotonia as paraneoplastic manifestation of bladder carcinoma. J Neurol Sci 2009;280:111-2.
33. Issa SS, Herskovitz S, Lipton RB. Acquired neuromyotonia as a paraneoplastic manifestation of ovarian cancer. Neurology 2011;76:100-1.
34. Skeie GO, Apostolski S, Evoli A, Gilhus NE, Hart IK, Harms L, Hilton-Jones D, Melms A, Verschuuren J, Horge HW. Guidelines for the treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol 2006;13:691-9.
35. McKnight K, Jiang Y, Hart Y, Cavey A, Wroe S, Blank M, Shoenfeld Y, Vincent A, Palace J, Lang B. Serum antibodies in epilepsy and seizure-associated disorders. Neurology 2005;65:1730-6.
36. Geschwind MD, Tan KM, Lennon VA, Barajas RF Jr, Haman A, Klein CJ, Josephson SA, Pittock SJ. Voltage-gated potassium channel autoimmunity mimicking creutzfeldt-jakob disease. Arch Neurol 2008;65:1341-6.
37. Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 2011;333:1462-6.
38. Takeda M, Tsuboi Y, Kitagawa J, Nakagawa K, Iwata K, Matsumoto S. Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol Pain 2011;7:5.
39. Oca-a M, Cendán CM, Cobos EJ, Entrena JM, Baeyens JM. Potassium channels and pain: present realities and future opportunities. Eur J Pharmacol 2004;500:203-19.
40. Rosch RE, Bamford A, Hacohen Y, Wraige E, Vincent A, Mewasingh L, Lim M. Guillain-Barre syndrome associated with Caspr2 antibodies: two paediatric cases. J Peripher Nerv Syst 2014;19:246-9.
41. Knowles CH, De Giorgio R, Kapur RP, Bruder E, Farrugia G, Geboes K, Lindberg G, Martin JE, Meier-Ruge WA, Milla PJ, Smith VV, Vandervinden JM, Veress B, Wedel T. The London Classification of gastrointestinal neuromuscular pathology: report on behalf of the Gastro 2009 International Working Group. Gut 2010;59:882-7.
42. Kraichely RE, Farrugia G, Pittock SJ, Castell DO, Lennon VA. Neural autoantibody profile of primary achalasia. Dig Dis Sci 2010;55:307-11.
43. Törnblom H, Lang B, Clover L, Knowles CH, Vincent A, Lindberg G. Autoantibodies in patients with gut motility disorders and enteric neuropathy. Scand J Gastroenterol 2007;42:1289-93.
44. Viallard JF, Vincent A, Moreau JF, Parrens M, Pellegrin JL, Ellie E. Thymoma-associated neuromyotonia with antibodies against voltage-gated potassium channels presenting as chronic intestinal pseudo-obstruction. Eur Neurol 2005;53:60-3.
45. Hubball AW, Lang B, Souza MA, Curran OD, Martin JE, Knowles CH. Voltage-gated potassium channel (K (v) 1) autoantibodies in patients with chagasic gut dysmotility and distribution of K (v) 1 channels in human enteric neuromusculature (autoantibodies in GI dysmotility). Neurogastroenterol Motil 2012;24:719-28, e344.
46. Kellinghaus C, Kraus J, Blaes F, Nabavi DG, Schabitz WR. CRMP-5-autoantibodies in testicular cancer associated with limbic encephalitis and choreiform dyskinesias. Eur Neurol 2007;57:241-3.
47. Harloff A, Hummel S, Kleinschmidt M, Rauer S. Anti-Ri antibodies and limbic encephalitis in a patient with carcinoid tumour of the lung. J Neurol 2005;252:1404-5.
48. Mitchell AN, Bakhos CT, Zimmerman EA. Anti-Ri-associated paraneoplastic brainstem cerebellar syndrome with coexisting limbic encephalitis in a patient with mixed large cell neuroendocrine lung carcinoma. J Clin Neurosci 2015;22:421-3.
49. Al-Thubaiti I, Al-Hayek K, Binfalah M. Anti-Ma-associated encephalitis due to dysgerminoma in a woman with Swyer syndrome. Neurology 2013;80:1439-40.
50. Scheid R, Voltz R, Guthke T, Bauer J, Sammler D, von Cramon DY. Neuropsychiatric findings in anti-Ma2-positive paraneoplastic limbic encephalitis. Neurology 2003;61:1159-61.
51. Moon J, Lee ST, Shin JW, Byun JI, Lim JA, Shin YW, Kim TJ, Lee KJ, Park KI, Jung KH, Jung KY, Lee SK, Chu K. Non-stiff anti-amphiphysin syndrome: clinical manifestations and outcome after immunotherapy. J Neuroimmunol 2014;274:209-14.
52. Dorresteijn LD, Kappelle AC, Renier WO, Gijtenbeek JM. Anti-amphiphysin associated limbic encephalitis: a paraneoplastic presentation of small-cell lung carcinoma. J Neurol 2002;249:1307-8.
53. Buckley C, Oger J, Clover L, Tüzün E, Carpenter K, Jackson M, Vincent A. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001;50:73-8.
54. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, Cowell JK, Dalmau J. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776-85.
55. Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014;76:168-84.
56. Malter MP, Frisch C, Schoene-Bake JC, Helmstaedter C, Wandinger KP, Stoecker W, Urbach H, Surges R, Elger CE, Vincent AV, Bien CG. Outcome of limbic encephalitis with VGKC-complex antibodies: relation to antigenic specificity. J Neurol 2014;261:1695-705.
57. Graus F, Saiz A, Lai M, Bruna J, López F, Sabater L, Blanco Y, Rey MJ, Ribalta T, Dalmau J. Neuronal surface antigen antibodies in limbic encephalitis: clinical-immunologic associations. Neurology 2008;71:930-6.
58. Quek AM, Britton JW, McKeon A, So E, Lennon VA, Shin C, Klein C, Watson RE, Jr, Kotsenas AL, Lagerlund TD, Cascino GD, Worrell GA, Wirrell EC, Nickels KC, Aksamit AJ, Noe KH, Pittock SJ. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol 2012;69:582-93.
59. Kotsenas AL, Watson RE, Pittock SJ, Britton JW, Hoye SL, Quek AM, Shin C, Klein CJ. MRI findings in autoimmune voltage-gated potassium channel complex encephalitis with seizures: one potential etiology for mesial temporal sclerosis. AJNR Am J Neuroradiol 2014;35:84-9.
60. Jacob S, Irani SR, Rajabally YA, Grubneac A, Walters RJ, Yazaki M, Clover L, Vincent A. Hypothermia in VGKC antibody-associated limbic encephalitis. J Neurol Neurosurg Psychiatry 2008;79:202-4.
61. Montiel P, Sellal F, Clerc C, Richard P, Bataillard M. Limbic encephalitis with severe sleep disorder associated with voltage-gated potassium channels (VGKCs) antibodies. Rev Neurol (Paris) 2008;164:181-4.
62. Naasan G, Irani SR, Bettcher BM, Geschwind MD, Gelfand JM. Episodic bradycardia as neurocardiac prodrome to voltage-gated potassium channel complex/leucine-rich, glioma inactivated 1 antibody encephalitis. JAMA Neurol 2014;71:1300-4.
63. Urbach H, Soeder BM, Jeub M, Klockgether T, Meyer B, Bien CG. Serial MRI of limbic encephalitis. Neuroradiology 2006;48:380-6.
64. Bien CG, Elger CE. Limbic encephalitis: a cause of temporal lobe epilepsy with onset in adult life. Epilepsy Behav 2007;10:529-38.
65. Irani SR, Stagg CJ, Schott JM, Rosenthal CR, Schneider SA, Pettingill P, Pettingill R, Waters P, Thomas A, Voets NL, Cardoso MJ, Cash DM, Manning EN, Lang B, Smith SJ, Vincent A, Johnson MR. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013;136:3151-62.
66. Fidzinski P, Jarius S, Gaebler C, Boegner F, Nohr R, Ruprecht K. Faciobrachial dystonic seizures and antibodies to Lgi1 in a 92-year-old patient: a case report. J Neurol Sci 2014;347:404-5.
67. Striano P. Faciobrachial dystonic attacks: seizures or movement disorder? Ann Neurol 2011;70:179-80.
68. Plantone D, Renna R, Grossi D, Plantone F, Iorio R. Teaching NeuroImages: basal ganglia involvement in facio-brachial dystonic seizures associated with LGI1 antibodies. Neurology 2013;80:e183-4.
69. Tofaris GK, Irani SR, Cheeran BJ, Baker IW, Cader ZM, Vincent A. Immunotherapy-responsive chorea as the presenting feature of LGI1-antibody encephalitis. Neurology 2012;79:195-6.
70. Niehusmann P, Dalmau J, Rudlowski C, Vincent A, Elger CE, Rossi JE, Bien CG. Diagnostic value of N-methyl-D-aspartate receptor antibodies in women with new-onset epilepsy. Arch Neurol 2009;66:458-64.
71. Brenner T, Sills GJ, Hart Y, Howell S, Waters P, Brodie MJ, Vincent A, Lang B. Prevalence of neurologic autoantibodies in cohorts of patients with new and established epilepsy. Epilepsia 2013;54:1028-35.
72. Iorio R, Assenza G, Tombini M, Colicchio G, Della Marca G, Benvenga A, Damato V, Rossini PM, Vollono C, Plantone D, Marti A, Batocchi AP, Evoli A. The detection of neural autoantibodies in patients with antiepileptic-drug-resistant epilepsy predicts response to immunotherapy. Eur J Neurol 2015;22:70-8.
73. Lilleker JB, Jones MS, Mohanraj R. VGKC complex antibodies in epilepsy: diagnostic yield and therapeutic implications. Seizure 2013;22:776-9.
74. Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, Sáenz A, Poza JJ, Galán J, Gesk S, Sarafidou T, Mautner VF, Binelli S, Staub E, Hinzmann B, French L, Prud'homme JF, Passarelli D, Scannapieco P, Tassinari CA, Avanzini G, Marti-Masso JF, Kluwe L, Deloukas P, Moschonas NK, Michelucci R, Siebert R, Nobile C, Perez-Tur J, Lopez de Munain A. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet 2002;11:1119-28.
75. Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Martinelli Boneschi F, Choi C, Morozov P, Das K, Teplitskaya E, Yu A, Cayanis E, Penchaszadeh G, Kottmann AH, Pedley TA, Hauser WA, Ottman R, Gilliam TC. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 2002;30:335-41.
76. Ottman R, Winawer MR, Kalachikov S, Barker-Cummings C, Gilliam TC, Pedley TA, Hauser WA. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology 2004;62:1120-6.
77. Rossi M, Mead S, Collinge J, Rudge P, Vincent A. Neuronal antibodies in patients with suspected or confirmed sporadic Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2014;pii:jnnp-2014-308695.
78. Jones M, Odunsi S, du Plessis D, Vincent A, Bishop M, Head MW, Ironside JW, Gow D. Gerstmann-Straüssler-Scheinker disease: novel PRNP mutation and VGKC-complex antibodies. Neurology 2014;82:2107-11.
79. World Health Organization. Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: report of a WHO consultation. In: Emerging and Other Communicable Diseases, Surveillance and Control. Geneva, Switzerland: World Health Organization; 1998. pp. 1-26.
80. Morvan A. De la chorèe fibrillaire. Gazz Hebd Mèd Chir 1890;27:173-200. (In French)
81. Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C, Zuliani L, Watanabe O, Lang B, Buckley C, Vincent A. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol 2012;72:241-55.
83. Spinazzi M, Argentiero V, Zuliani L, Palmieri A, Tavolato B, Vincent A. Immunotherapy-reversed compulsive, monoaminergic, circadian rhythm disorder in Morvan syndrome. Neurology 2008;71:2008-10.
84. Löscher WN, Wanschitz J, Reiners K, Quasthoff S. Morvan's syndrome: clinical, laboratory, and in vitro electrophysiological studies. Muscle Nerve 2004;30:157-63.
85. Cottrell DA, Blackmore KJ, Fawcett PR, Birchall D, Vincent A, Barnard S, Walls TJ. Sub-acute presentation of Morvan's syndrome after thymectomy. J Neurol Neurosurg Psychiatry 2004;75:1504-5.
86. Plazzi G, Montagna P, Meletti S, Lugaresi E. Polysomnographic study of sleeplessness and oneiricisms in the alcohol withdrawal syndrome. Sleep Med 2002;3:279-82.
87. Liguori R, Vincent A, Clover L, Avoni P, Plazzi G, Cortelli P, Baruzzi A, Carey T, Gambetti P, Lugaresi E, Montagna P. Morvan's syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain 2001;124:2417-26.
88. Abou-Zeid E, Boursoulian LJ, Metzer WS, Gundogdu B. Morvan syndrome: a case report and review of the literature. J Clin Neuromuscul Dis 2012;13:214-27.
89. Misawa T, Mizusawa H. Anti-VGKC antibody-associated limbic encephalitis/Morvan syndrome. Brain Nerve 2010;62:339-45.
90. Sharma S, Sharma P. Morvan syndrome: after scrotal sac drainage and chemical instillation in hydrocele. Neurol India 2013;61:300-2.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Plantone D, Renna R, Koudriavtseva T. Neurological diseases associated with autoantibodies targeting the voltage-gated potassium channel complex: immunobiology and clinical characteristics. Neurosciences 2016;3:69-78. http://dx.doi.org/10.4103/2347-8659.169883
AMA Style
Plantone D, Renna R, Koudriavtseva T. Neurological diseases associated with autoantibodies targeting the voltage-gated potassium channel complex: immunobiology and clinical characteristics. Neuroimmunology and Neuroinflammation. 2016; 3: 69-78. http://dx.doi.org/10.4103/2347-8659.169883
Chicago/Turabian Style
Plantone, Domenico, Rosaria Renna, Tatiana Koudriavtseva. 2016. "Neurological diseases associated with autoantibodies targeting the voltage-gated potassium channel complex: immunobiology and clinical characteristics" Neuroimmunology and Neuroinflammation. 3: 69-78. http://dx.doi.org/10.4103/2347-8659.169883
ACS Style
Plantone, D.; Renna R.; Koudriavtseva T. Neurological diseases associated with autoantibodies targeting the voltage-gated potassium channel complex: immunobiology and clinical characteristics. Neurosciences. 2016, 3, 69-78. http://dx.doi.org/10.4103/2347-8659.169883
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 19 clicks
Cite This Article 19 clicks

 Like This Article 43
likes
Like This Article 43
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.