
Encephalitis associated with autoantibodies binding to γ-aminobutyric acid-A, γ-aminobutyric acid-B and glycine receptors: immunopathogenic mechanisms and clinical characteristics

Abstract
Recent, discoveries of neural antibodies have facilitated the diagnosis of immune-mediated, immunotherapy-responsive neurologic disorders. Antibodies that target inhibitory central nervous system receptors, such as γ-aminobutyric acid-B, γ-aminobutyric acid-A, and glycine receptors, disrupt inhibitory regulatory synaptic functions, and lead to neuronal hyperexcitability. The myriad of neurologic, manifestations associated with these antibodies includes seizures, encephalopathy, muscle rigidity and stiffness. This article provides a review of the immunopathogenic mechanisms and the clinical and therapeutic implications of autoimmune encephalitis associated with these antibodies that target inhibitory receptors.
Keywords
Introduction
Recent, discoveries of neural antibodies that bind to antigenic targets in the brain have led to a paradigm shift in the clinical approach to patients presenting with encephalopathy,[1-4] cognitive change,[5] and refractory seizures.[4,6] With a wider availability of neural antibody testing, a significant proportion of the patients, who were previously diagnosed with encephalitis of undetermined etiology have been shown to have neurologic symptoms caused by an underlying autoimmune disorder and some of these patients respond favorably to immunosuppressive treatments.[7] Neural antibodies that, target channels or receptors on the neuronal cell surface can interfere with the function of these proteins, leading to altered neuronal excitability, and a myriad of neurologic syndromes that mirror genetic and pharmacologically induced disorders of the target receptors.[8] In this review, we describe the immunopathogenic mechanisms of autoimmune encephalitis associated with antibodies targeting the inhibitory synaptic receptors γ-aminobutyric acid-B (GABAB), γ-aminobutyric acid-A (GABAA), and glycine receptors (GlyRs), together with their clinical and therapeutic implications.
Anti-GABAB receptor and anti-GABAA receptor encephalitis
GABA, the main inhibitory neurotransmitter in the brain, binds to metabotropic and ionotropic receptors to regulate neuronal activity. To date, GABAB receptor (GABABR) and GABAA receptor (GABAAR) have been identified as antigenic targets of autoimmunity [Table 1].
Key demographic, clinical, and AI/oncologic associations of the GABABR, GABAAR, and GlyR-α1 antibodies
| GABABR | GABAAR | GlyR-α1 | |
|---|---|---|---|
| Clinical features | Limbic encephalitis (memory impairment, hallucination, confusion, behavior changes) with early and prominent seizures; Less commonly: cerebellar ataxia, opsoclonus-myoclonus, brainstem encephalitis, chorea, myelopathy, peripheral neuropathy, and myopathy | High serum antibodies concentration: rapidly progressive encephalopathy, refractory seizures, and status epilepticus; Low serum antibodies concentration: seizures, SMS, opsoclonus-myoclonus, behavioral change, psychosis, confusion, chorea, ataxia, hallucinations, and hemiparesis | Axial/limb spasms, rigidity, and myoclonus (SMS) + brainstem signs (PERM) Optic neuropathy, seizures, cognitive impairment, autonomic disturbance, respiratory failure, and transverse myelitis also reported in isolation or with SMS or PERM |
| Onset | Mostly subacute/acute | Acute/subacute | Subacute > acute > chronic |
| Age group | Wide range, children, and adults | Wide range, children, and adults | Wide range, children, and adults |
| Gender, male:female | 1.3:1 | 2:1 | 1:1 |
| Inflammatory CSF* | 73% | 40% | 50%, but OCB frequently negative |
| MRI | 65% abnormal (medial temporal > extratemporal changes) | High serum antibodies concentration: 100% multifocal temporal and extratemporal T2/FLAIR hyperintensities | < 30% T2/FLAIR abnormalities in temporal lobes, SC abnormalities rarely reported (short, multifocal, and LETM) |
| EMG | - | - | 60% abnormal (continuous motor activity, stimulus induced motor activity) |
| Other coexisting neural antibodies | 56% (VGCC, AGNA, GAD-65, VGKC-complex, NMDAR, ANNA-1, -2 and -3,CRMP-5 IgG, amphiphysin, BRSK2) | 70% (AChR, NMDAR, GABAB, GAD-65, VGKC-complex) | Rare (GAD-65, MOG, NMDAR, aquaporin-4, VGKC-complex) |
| Associated tumors | 62% (SCLC most common, also neuroendocrine lung, malignant melanoma, esophageal, malignant melanoma, thymus anaplastic carcinoid) | 15% (invasive thymoma, Hodgkin lymphoma) | Approximately 10% to date (breast cancer, lymphoma (both Hodgkin and NHL), leukemia, lung cancer, melanoma) |
| Fatalities (%) | 36 | 20 | < 10 |
Anti-GABABR encephalitis
The GABABR is a metabotropic G-protein-coupled receptor on presynaptic, postsynaptic, and extrasynaptic membranes, expressed in both the central and peripheral nervous systems, particularly the hippocampus, thalamus, and cerebellum.[9] This receptor is a heterodimer comprising two subunits: GABAB1 and GABAB2. Both subunits need to be co-expressed in order to form a functional receptor.[9,10] The extracellular domain of the GABAB1 subunit binds to GABA while the GABAB2 subunit couples the receptor with the effector G protein.[11] Antibodies to GABABR bind to the B1 subunit of the GABABR,[4] the component that is required for GABA binding and receptor function.
GABABR exert inhibitory regulatory effects on synaptic transmission by inhibiting presynaptic voltage-gated calcium channel-mediated neurotransmitter release and by activating postsynaptic potassium channels, resulting in hyperpolarization of neuronal membranes, and inhibition of adenylate cyclase.[11,12] GABABR dysfunction is implicated in a variety of neurological disorders such as epilepsy, in which genetic mutations may play a role.[13,14] Pharmacological disruption of the GABABR leads to seizures, cognitive deficits, and behavioral changes,[10,15,16] all of which may be seen in autoimmune anti-GABABR limbic encephalitis.
Antibodies to GABABR were first described in 15 patients with limbic encephalitis, in whom subacute early onset of seizures was a distinctive feature.[4] Seizures were predominantly of temporal lobe onset with secondary generalization, and 3 of the 15 patients developed status epilepticus. Memory impairment, confusion, hallucinations, and behavioral changes consistent with limbic involvement were frequently seen. Electroencephalography (EEG) changes included epileptiform discharges, electrographic ictal activity, and/or temporal lobe slowing.
Magnetic resonance imaging (MRI) brain imaging typically demonstrates unilateral or bilateral increased T2/fluid attenuated inversion recovery signal changes in the medial temporal region, consistent with limbic encephalitis.[4,17] Extratemporal changes in the grey and white matter, cerebellum, basal ganglia, and brainstem have also been reported.[4,17-19] Cerebrospinal fluid (CSF) examination may yield lymphocytic pleocytosis, elevated protein, and oligoclonal bands.[4] As with other types of limbic encephalitis, the EEG, MRI brain, and/or CSF exam may be normal, and should not preclude the diagnosis or presumptive treatment when the clinical presentation is suspicious.
Limbic encephalitis, the most common neurologic manifestation of anti-GABABR encephalitis,[4,20,21] typically occurs in the setting of very high GABABR antibody titers.[4,20] A widening phenotypic spectrum of anti-GABABR disorders including cerebellar ataxia,[17,20,22,23] opsoclonus-myoclonus,[17,18] and brainstem encephalitis[19] is now appreciated. Extralimbic presentations may be explained by the high expression of GABABR outside of the hippocampus, including the cerebellum.[9] Other rarer neurological features of anti-GABABR encephalitis include chorea, myelopathy, peripheral neuropathy, and myopathy, particularly in patients with lower antibody titers.[4,20] It is uncertain, whether all of these extralimbic neurologic manifestations can be attributed solely to the GABABR antibodies or whether the co-existence of other neural autoantibodies may contribute to the increasingly diverse neurological features being reported. Known neural antibody accompaniments to the GABABR antibodies include the 65 kDa isoform of glutamate decarboxylase (GAD-65), voltage-gated calcium channels (N-type and P/Q type), voltage-gated potassium-complex (VGKC-complex), and neuronal nuclear and cytoplasmic antibodies [such as antineuronal nuclear autoantibody (ANNA-1), ANNA-3, collapsing response mediator protein-5 IgG, and anti-glial nuclear autoantibody/SOX-1 antibodies].[4,20-22]
In comparison to other cell surface neural antibody associated encephalitis, anti-GABABR encephalitis is probably uncommon. In a clinical service laboratory, only 7 of 3,989 (0.2%) patients with suspected autoimmune encephalopathy were found to have the GABABR antibody.[20] There is no obvious gender predisposition for this neurologic disorder. A paraneoplastic etiology is diagnosed in approximately half of the patients with GABABR antibodies.[4,17,20,21] The neurologic disorder usually precedes the diagnosis of malignancy, and the most frequently encountered tumor is small cell lung cancer.[4,20] Tumors are more likely to be detected in older patients.[4] Although, lung tumors from patients with GABABR encephalitis have not been studied for GABABR expression, samples of archived small cell lung cancers from patients without encephalitis were found to react with both guinea pig and human GABAB IgG. This suggests that the GABABR could be expressed by small cell lung cancer and could potentially trigger an autoimmune reaction.[4] Other oncologic associations of anti-GABABR encephalitis include neuroendocrine lung tumor, multiple myeloma, esophageal carcinoma, malignant melanoma, and carcinoid of the thymus.[4,19,20,22,23]
Neurological improvement has been reported in up to 90% of patients with anti-GABABR encephalitis who received immunotherapy and appropriate cancer treatment (if the tumor was detected).[4,17,20,21] As the cases reported so far were retrospectively ascertained, there was heterogeneity in the immunotherapies used. A variety of immunotherapie have been used successfully, including various combinations of first-line agents, corticosteroids, intravenous immunoglobulin (IVIg), and/or plasma exchange (PLEX).[4,20] Patients who do not respond to these treatments warrant second line and maintenance treatments, such as rituximab, cyclophosphamide, mycophenolate mofetil, and azathioprine.[17,20,23,24] Neurological improvement may be incomplete or not sustained. Despite optimal immunosuppressive treatment, patients with GABABR antibodies can deteriorate due to tumor progression, chemotherapy-related complications, and/or treatment-resistant relapses.[4,17,22-24] Further studies to elucidate the optimal treatment regimens are needed. The presence of an underlying small cell lung cancer and the co-existence of other paraneoplastic neural antibodies targeting intracellular (neuronal nuclear and cytoplasmic) antigens have been suggested as poor prognostic indicators.[17,20]
Anti-GABAAR encephalitis
The GABAAR is a ligand-gated ion channel located at synaptic and extrasynaptic sites that functions to mediate fast inhibitory synaptic transmission.[25,26] Activation of the GABAAR triggers opening of intrinsic chloride channels, thereby eliciting an inhibitory postsynaptic potential.[27] Disruption of GABAAR results in increased neuronal excitability and seizures.[27] Mutations in the α1 and β3 subunits of the GABAAR gene have been implicated in epilepsy syndromes.[27-29] Benzodiazepine and barbiturate, medications used for the treatment of seizures and status epilepticus, enhance GABAergic inhibition to exert an anticonvulsant effect.[30]
GABAARs are pentamers comprising combinations of five subunits that form chloride ion channels. Different combinations of subunits result in functional heterogeneity. Synaptic GABAARs, which contain the α (α1-3), β, and γ subunits, are responsible for phasic inhibition. By contrast, extrasynaptic and perisynaptic GABAARs, which are responsible for tonic inhibition, comprise α4 or α6 subunits combined with β and δ subunits.[31] The GABAAR antibody binds to the α1,β3, or both subunits of the synaptic GABAAR.[32,33] GABAAR antibodies reduce the density of the GABAAR at synaptic sites when applied to rat hippocampal neurons, suggesting that antibody binding leads to the relocation of GABAAR from the synaptic membrane.[32,33] This phenomenon is similar to the loss of synaptic GABAAR and resultant neuronal hyperexcitability observed in epilepsy and status epilepticus.[27] The combined reinforcing effects of antibody-mediated synaptic GABAAR relocation, together with the status epilepticus-induced loss of GABAAR, could support a postulated model to explain the severity of seizures in patients with anti-GABAAR encephalitis.[33]
Recently, 18 patients with autoimmune encephalitis and prominent seizures were described with GABAAR antibodies, 6 of whom had very high antibody titers.[33] Patients with high titers in both serum and CSF developed a particularly rapid, severe progressive encephalopathy with refractory seizures and/or status epilepticus, for which intensive care admission for pharmacologically-induced coma was required. Other reported clinicalmanifestations in GABAAR antibody seropositive patients are opsoclonus-myoclonus, affective problems, hallucinations, mutism, aphasia, memory impairment, hemiparesis, chorea, cerebellar ataxia, and Stiff-man syndrome (SMS).[32,33] GABABR, GAD-65, N-methyl-D-aspartate receptor (NMDAR), leucine-rich, glioma-inactivated 1 and contactin-associated protein-like 2 antibodies frequently co-exist in these patients.[32,33] A propensity for other neurological autoimmune conditions such as myasthenia gravis has also been noted.[32,33]
GABAAR antibodies are reported in both children and adults (age 2-74 years), but larger cohorts need to be characterized.[33] A low frequency of tumors in seropositive patients has been reported. In the initially published study of 18 patients, only one patient was found to have cancer (Hodgkin lymphoma).[33] Two recent additionally reported cases had invasive thymoma.[32]
The electroencephalograms of patients with anti-GABAAR encephalitis may demonstrate generalized slowing suggestive of encephalopathy, multifocal ictal and interictal discharges, or status epilepticus.[33] CSF findings range from normal to lymphocytic pleocytosis.[33] Distinctive to GABAAR antibodies, the majority of patients, especially those with high antibody titers, had extensive temporal and extratemporal MRI brain abnormalities[33] which could be a consequence of autoimmune inflammation in the brain or prolonged ictal activity. The extensive radiologic changes contrast with those of patients with limbic encephalitis associated with other neuronal synaptic and cell surface antibodies, such as NMDAR and VGKC-complex antibodies, in which MRI abnormalities are often confined to the mesial temporal regions.
Despite the severity of their presentation, 80% of the patients reported with anti-GABAAR encephalitis demonstrate partial or complete recovery with a combination of immunotherapy, antiepileptic drugs, and supportive treatment.[32,33] In severe cases, multiple immunotherapies may be required. Treatment options are the same as with GABABR and GlyR antibody mediated disorders. In addition to immunotherapy, early recognition and treatment of epilepsy, as well as supportive treatment (including ventilation support) are pivotal.
GlyR antibody encephalitis
Glycine, a key neurotransmitter for fast postsynaptic inhibitory neurons in the CNS, has a complex functional pathway that involves pre- and post-synaptic GlyR interacting with other neurotransmitters (GABA and glutamate), NMDAR, and postsynaptic anchoring proteins like gephyrin. Antibodies directed at any of these targets may affect the glycinergic system, resulting in neurological dysfunction.[34] Whether due to strychnine (a GlyR antagonist) poisoning, genetic mutations of the GlyR gene (hereditary hyperekplexia), or immune-mediated encephalitis, GlyR dysfunction may be associated with severe muscle spasms, stiffness, agitation, seizures, myoclonus, autonomic instability, and/or respiratory failure.[35,36]
GlyRs, pentamers of α1-α4 and β-subunit proteins, are ligand-gated chloride ion channels, widely distributed in the CNS. They are predominantly expressed in the olfactory bulb, retina, hippocampus, brainstem (auditory, visual, vestibular, and sensory nuclei), cerebellum, and spinal cord.[37,38] Glycine binding mediates opening of the GlyR chloride channel, resulting in hyperpolarization of the membrane potential and reduced neuronal excitability. The GlyR antibody targets the α1 subunit of the postsynaptic GlyR and is associated with hyperexcitable neurologic disorders [Table 1].[39]Gephyrin allows multiple GlyR to cluster together on the synaptic membrane. Antibodies to gephyrin, an anchoring protein in the postsynaptic GlyR, have been described only in a single case to date.[40]
Classic neurologic manifestations associated with GlyR antibodies are progressive encephalomyelitis with rigidity and myoclonus (PERM) and SMS.[39,41,42] PERM and SMS were first described as separate clinical entities, but today these 2 conditions are considered to belong to a continuum of CNS hyperexcitability disorders. Patients with PERM and SMS share common features of rigidity, painful spasms, autonomic disturbances, hyperekplexia, and myoclonus. The widespread distribution of hyperexcitability and brainstem involvement classically distinguishes PERM from SMS and associated psychiatric symptoms such as anxiety are more commonly observed in SMS patients.[43-46] The autoimmune nature of these conditions, and specifically the involvement of GlyR antibodies in some cases was not appreciated until recently.[41,42] SMS was initially associated with antibodies to GAD-65 (60-70% of cases),[47,48] gephyrin (1 case),[40] and amphyiphysin (< 5% cases, in the setting of both small cell lung and breast cancers).[48,49] In 2008, Hutchinson et al.[41] reported the first case of PERM with GlyR antibodies. It remains unresolved whether all of these antibodies are truly pathogenic.[50,51] McKeon et al.[42] reported that 10 of 81 (12%) patients with SMS spectrum disorders were positive for GlyR antibodies. Interestingly, GlyR seropositivity was associated with better responsiveness to immunotherapy regardless of GAD-65 status, suggesting a pathogenic role. There are no passive transfer animal models of GlyR antibodies to date.[39,42]
The spectrum of GlyR antibody encephalitis manifestations is now widening beyond classic PERM and SMS. Various combinations of psychiatric disturbances, cognitive dysfunction, seizures (focal/generalized epilepsy and new-onset status epilepticus), and movement disorders, autonomic instability with central hypoventilation, pseudobulbar and/or-oculomotor dysfunction, steroid responsive optic neuropathy, and transverse myelopathy have now been described with GlyR antibodies.[39,52-54]
The onset of symptoms in GlyR antibody neurologic syndromes is typically acute to subacute. In the largest case series of 52 GlyR antibody positive patients with a variety of presentations, there appears to be no sex predominance and all age groups are vulnerable.[39] Patients frequently have a history of other autoimmune disorders.[39] As with other autoimmune encephalopathies, GlyR antibodies may co-exist with other antibodies, such as NMDAR, GAD-65, VGKC-complex, myelin oligodendrocyte glycoprotein, and aquaporin-4 antibodies although this is rare.[39,42,53,55,56] Tumors are identified in less than 20% of cases (thymoma, lymphoma, breast cancer, small cell lung carcinoma and leukemia).[39,42,57,58]
GlyR antibodies may be detected in both serum and/or the CSF.[39,59] CSF evaluation is possibly more sensitive than serum, therefore testing both is recommended.[42] CSF lymphocytic pleocytosis or raised protein may be seen, and oligoclonal bands were negative in 50-70% of 2 case series recently published.[39,56] Imaging is typically normal. Rarely, MRI temporal lobe T2-weighted abnormality with subsequent hippocampal volume loss is detected, particularly in cases associated with significant seizure activity. EEG may be normal, or show features of focal or generalized ictal activity.[59]
A combination of immunotherapies (corticosteroids, IVIg, PLEX, cyclophosphamide), pharmacological therapies targeting symptoms of motor hyperexcitability and pain (clonazepam, diazepam, baclofen, gabapentin), and anticonvulsants (levetiracetam) are required to control clinical symptoms.[39,42] Eighty percent of patients with GlyR antibodies showed a substantial response to immunotherapy.[39,42] Two cases were reported that responded dramatically to thymectomy in addition to other immunotherapy.[39,60] In the largest case series to date, 6 out of 52 (12%) patients continued to have sporadic relapses whilst on treatment.[39]
Conclusion
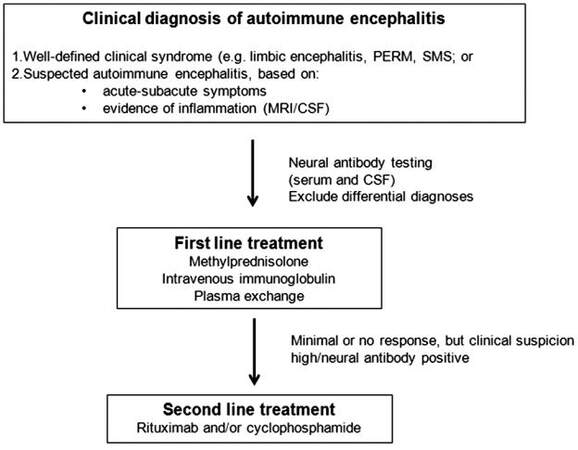
An increased awareness of the autoimmune mechanisms underlying cases of noninfective encephalitis and/or refractory seizures has led to increased recognition, earlier treatment, and improved outcomes in a subgroup of patients previously considered untreatable. Antibodies targeting the inhibitory receptors GABAB, GABAA, and glycine are three more recently appreciated, but important antibodies to consider in refractory seizure disorders and encephalitis of unclear etiology. A high index of suspicion and an awareness of the expanding clinical spectrum of these antibody-mediated disorders should prompt early neural antibody testing in patients with typical constellations of neurological symptoms, in particular refractory seizure disorders and encephalitis of unclear etiology. Once identified early, these conditions may be responsive to immunotherapy. There are sparse data to recommend one immunotherapeutic regime over another. Large cohort studies of patients with anti-NMDAR encephalitis suggest that first line therapy should comprise corticosteroids, IVIg, and/or PLEX, followed by second line immunotherapy (cyclophosphamide, rituximab, or both) in patients who fail to respond to initial treatment.[61] A practical approach, guided by the literature on autoimmune encephalitis with antibodies against neuronal surface antigens, is suggested in Figure 1. Immunotherapy needs to be complemented by supportive, symptomatic medical therapy. There is a consensus that early treatment confers better outcomes. Age and antibody appropriate tumor screening should be performed in all cases and may be aided by testing for other co-existing neural antibodies.[62]
The neurologic hyperexcitability effects of antibody binding to GABAA, GABAB, and GlyRs (and potentially other receptors in the future) reflect the important functions mediated by these inhibitory neuronal synaptic receptors. More research is needed in order to better understand this novel category of immune-mediated encephalitis. Further studies could focus on immunopathogenic mechanisms of these antibodies in causing disease, as these may be potential targets for directed treatment. To date, the numbers of patients reported with these antibodies remain small, with most cases retrospectively identified. With increasing access to testing for neural antibodies, the clinical spectrum of these autoimmune encephalitides may continue to expand. Systematic studies of prospectively identified, newly diagnosed cases should help to provide data on the long-term course of the disease, prognostic factors, and optimal immunotherapeutic regimes.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A, Baehring JM, Shimazaki H, Koide R, King D, Mason W, Sansing LH, Dichter MA, Rosenfeld MR, Lynch DR. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61:25-36.
2. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, Mata S, Kremens D, Vitaliani R, Geschwind MD, Bataller L, Kalb RG, Davis R, Graus F, Lynch DR, Balice-Gordon R, Dalmau J. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009;65:424-34.
3. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, Cowell JK, Dalmau J. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776-85.
4. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, Friedman D, Skeen MB, Grisold W, Kimura A, Ohta K, Iizuka T, Guzman M, Graus F, Moss SJ, Balice-Gordon R, Dalmau J. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010;9:67-76.
5. Flanagan EP, McKeon A, Lennon VA, Boeve BF, Trenerry MR, Tan KM, Drubach DA, Josephs KA, Britton JW, Mandrekar JN, Lowe V, Parisi JE, Pittock SJ. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010;85:881-97.
6. Quek AM, Britton JW, McKeon A, So E, Lennon VA, Shin C, Klein C, Watson RE Jr, Kotsenas AL, Lagerlund TD, Cascino GD, Worrell GA, Wirrell EC, Nickels KC, Aksamit AJ, Noe KH, Pittock SJ. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol 2012;69:582-93.
7. Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis 2012;54:899-904.
8. Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011;77:179-89.
9. Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Molecular structure and physiological functions of GABA(B) receptors. Physiol Rev 2004;84:835-67.
10. Gassmann M, Shaban H, Vigot R, Sansig G, Haller C, Barbieri S, Humeau Y, Schuler V, Muller M, Kinzel B, Klebs K, Schmutz M, Froestl W, Heid J, Kelly PH, Gentry C, Jaton AL, Van der Putten H, Mombereau C, Lecourtier L, Mosbacher J, Cryan JF, Fritschy JM, Luthi A, Kaupmann K, Bettler B. Redistribution of GABAB (1) protein and atypical GABAB responses in GABAB(2)-deficient mice. J Neurosci 2004;24:6086-97.
11. Benarroch EE. GABAB receptors: structure, functions, and clinical implications. Neurology 2012;78:578-84.
12. Bowery NG, Bettler B, Froestl W, Gallagher JP, Marshall F, Raiteri M, Bonner TI, Enna SJ. International Union of Pharmacology. XXXIII. Mammalian gamma-aminobutyric acid(B) receptors: structure and function. Pharmacol Rev 2002;54:247-64.
13. Gambardella A, Manna I, Labate A, Chifari R, La Russa A, Serra P, Cittadella R, Bonavita S, Andreoli V, LePiane E, Sasanelli F, Di Costanzo A, Zappia M, Tedeschi G, Aguglia U, Quattrone A. GABA(B) receptor 1 polymorphism (G1465A) is associated with temporal lobe epilepsy. Neurology 2003;60:560-3.
14. Wang X, Sun W, Zhu X, Li L, Wu X, Lin H, Zhu S, Liu A, Du T, Liu Y, Niu N, Wang Y, Liu Y. Association between the gamma-aminobutyric acid type B receptor 1 and 2 gene polymorphisms and mesial temporal lobe epilepsy in a Han Chinese population. Epilepsy Res 2008;81:198-203.
15. Enna SJ, Bowery NG. GABA (B) receptor alterations as indicators of physiological and pharmacological function. Biochem Pharmacol 2004;68:1541-8.
16. Mombereau C, Kaupmann K, Froestl W, Sansig G, van der Putten H, Cryan JF. Genetic and pharmacological evidence of a role for GABA(B) receptors in the modulation of anxiety- and antidepressant-like behavior. Neuropsychopharmacology 2004;29:1050-62.
17. Hoftberger R, Titulaer MJ, Sabater L, Dome B, Rozsas A, Hegedus B, Hoda MA, Laszlo V, Ankersmit HJ, Harms L, Boyero S, de Felipe A, Saiz A, Dalmau J, Graus F. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology 2013;81:1500-6.
18. Kruer MC, Hoeftberger R, Lim KY, Coryell JC, Svoboda MD, Woltjer RL, Dalmau J. Aggressive course in encephalitis with opsoclonus, ataxia, chorea, and seizures: the first pediatric case of gamma-aminobutyric acid type B receptor autoimmunity. JAMA Neurol 2014;71:620-3.
19. Mundiyanapurath S, Jarius S, Probst C, Stocker W, Wildemann B, Bosel J. GABA-B-receptor antibodies in paraneoplastic brainstem encephalitis. J Neuroimmunol 2013;259:88-91.
20. Jeffery OJ, Lennon VA, Pittock SJ, Gregory JK, Britton JW, McKeon A. GABAB receptor autoantibody frequency in service serologic evaluation. Neurology 2013;81:882-7.
21. Kim TJ, Lee ST, Shin JW, Moon J, Lim JA, Byun JI, Shin YW, Lee KJ, Jung KH, Kim YS, Park KI, Chu K, Lee SK. Clinical manifestations and outcomes of the treatment of patients with GABAB encephalitis. J Neuroimmunol 2014;270:45-50.
22. Boronat A, Sabater L, Saiz A, Dalmau J, Graus F. GABA(B) receptor antibodies in limbic encephalitis and anti-GAD-associated neurologic disorders. Neurology 2011;76:795-800.
23. Jarius S, Steinmeyer F, Knobel A, Streitberger K, Hotter B, Horn S, Heuer H, Schreiber SJ, Wilhelm T, Trefzer U, Wildemann B, Ruprecht K. GABAB receptor antibodies in paraneoplastic cerebellar ataxia. J Neuroimmunol 2013;256:94-6.
24. DeFelipe-Mimbrera A, Masjuan J, Corral I, Villar LM, Graus F, Garcia-Barragan N. Opsoclonus-myoclonus syndrome and limbic encephalitis associated with GABAB receptor antibodies in CSF. J Neuroimmunol 2014;272:91-3.
25. Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 2005;6:215-29.
26. Tretter V, Moss SJ. GABA(A) receptor dynamics and constructing GABAergic synapses. Front Mol Neurosci 2008;1:7.
27. Benarroch EE. GABAA receptor heterogeneity, function, and implications for epilepsy. Neurology 2007;68:612-4.
28. Macdonald RL, Kang JQ, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol 2010;588:1861-9.
29. Tanaka M, Olsen RW, Medina MT, Schwartz E, Alonso ME, Duron RM, Castro-Ortega R, Martinez-Juarez IE, Pascual-Castroviejo I, Machado-Salas J, Silva R, Bailey JN, Bai D, Ochoa A, Jara-Prado A, Pineda G, Macdonald RL, Delgado-Escueta AV. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet 2008;82:1249-61.
30. Macdonald RL, Kelly KM. Antiepileptic drug mechanisms of action. Epilepsia 1995;36 Suppl 2:S2-12.
31. Gonzalez MI. The possible role of GABAA receptors and gephyrin in epileptogenesis. Front Cell Neurosci 2013;7:113.
32. Ohkawa T, Satake S, Yokoi N, Miyazaki Y, Ohshita T, Sobue G, Takashima H, Watanabe O, Fukata Y, Fukata M. Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis. J Neurosci 2014;34:8151-63.
33. Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R, McCracken L, Martinez-Hernandez E, Mason WP, Kruer MC, Ritacco DG, Grisold W, Meaney BF, Alcala C, Sillevis-Smitt P, Titulaer MJ, Balice-Gordon R, Graus F, Dalmau J. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014;13:276-86.
34. Benarroch EE. Glycine and its synaptic interactions: functional and clinical implications. Neurology 2011;77:677-83.
35. Martinez-Martinez P, Molenaar PC, Losen M, de Baets MH. Glycine receptor antibodies in PERM: a new channelopathy. Brain 2014;137:2115-6.
36. Chung SK, Vanbellinghen JF, Mullins JG, Robinson A, Hantke J, Hammond CL, Gilbert DF, Freilinger M, Ryan M, Kruer MC, Masri A, Gurses C, Ferrie C, Harvey K, Shiang R, Christodoulou J, Andermann F, Andermann E, Thomas RH, Harvey RJ, Lynch JW, Rees MI. Pathophysiological mechanisms of dominant and recessive GLRA1 mutations in hyperekplexia. J Neurosci 2010;30:9612-20.
38. Waldvogel HJ, Baer K, Eady E, Allen KL, Gilbert RT, Mohler H, Rees MI, Nicholson LF, Faull RL. Differential localization of gamma-aminobutyric acid type A and glycine receptor subunits and gephyrin in the human pons, medulla oblongata and uppermost cervical segment of the spinal cord: an immunohistochemical study. J Comp Neurol 2010;518:305-28.
39. Carvajal-Gonzalez A, Leite MI, Waters P, Woodhall M, Coutinho E, Balint B, Lang B, Pettingill P, Carr A, Sheerin UM, Press R, Lunn MP, Lim M, Maddison P, Meinck HM, Vandenberghe W, Vincent A. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain 2014;137:2178-92.
40. Butler MH, Hayashi A, Ohkoshi N, Villmann C, Becker CM, Feng G, De Camilli P, Solimena M. Autoimmunity to gephyrin in Stiff-man syndrome. Neuron 2000;26:307-12.
41. Hutchinson M, Waters P, McHugh J, Gorman G, O'Riordan S, Connolly S, Hager H, Yu P, Becker CM, Vincent A. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology 2008;71:1291-2.
42. McKeon A, Martinez-Hernandez E, Lancaster E, Matsumoto JY, Harvey RJ, McEvoy KM, Pittock SJ, Lennon VA, Dalmau J. Glycine receptor autoimmune spectrum with Stiff-man syndrome phenotype. JAMA Neurol 2013;70:44-50.
43. Moersch FP, Woltman HW. Progressive fluctuating muscular rigidity and spasm ("stiff-man" syndrome); report of a case and some observations in 13 other cases. Proc Staff Meet Mayo Clin 1956;31:421-7.
44. Molina JA, Porta J, Garcia-Morales I, Bermejo PF, Jimenez-Jimenez FJ. Treatment with intravenous prednisone and immunoglobin in a case of progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry 2000;68:395-6.
45. Whiteley AM, Swash M, Urich H. Progressive encephalomyelitis with rigidity. Brain 1976;99:27-42.
46. Campbell AM, Garland H. Subacute myoclonic spinal neuronitis. J Neurol Neurosurg Psychiatry 1956;19:268-74.
47. Solimena M, Folli F, Denis-Donini S, Comi GC, Pozza G, De Camilli P, Vicari AM. Autoantibodies to glutamic acid decarboxylase in a patient with Stiff-man syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med 1988;318:1012-20.
48. McKeon A, Robinson MT, McEvoy KM, Matsumoto JY, Lennon VA, Ahlskog JE, Pittock SJ. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol 2012;69:230-8.
49. Pittock SJ, Lucchinetti CF, Parisi JE, Benarroch EE, Mokri B, Stephan CL, Kim KK, Kilimann MW, Lennon VA. Amphiphysin autoimmunity: paraneoplastic accompaniments. Ann Neurol 2005;58:96-107.
50. Baizabal-Carvallo JF, Jankovic J. Stiff-person syndrome: insights into a complex autoimmune disorder. J Neurol Neurosurg Psychiatry 2015;86:840-8.
51. Pleasure D. GlyRalpha1, GAD65, amphiphysin, and gephyrin autoantibodies: leading or supporting roles in stiff-person disorders? JAMA Neurol 2013;70:16-7.
52. Woodhall M, Coban A, Waters P, Ekizoglu E, Kurtuncu M, Shugaiv E, Turkoglu R, Akman-Demir G, Eraksoy M, Vincent A, Tuzun E. Glycine receptor and myelin oligodendrocyte glycoprotein antibodies in Turkish patients with neuromyelitis optica. J Neurol Sci 2013;335:221-3.
53. Martinez-Hernandez E, Sepulveda M, Rostasy K, Hoftberger R, Graus F, Harvey RJ, Saiz A, Dalmau J. Antibodies to aquaporin 4, myelin-oligodendrocyte glycoprotein, and the glycine receptor alpha1 subunit in patients with isolated optic neuritis. JAMA Neurol 2015;72:187-93.
54. Ekizoglu E, Tuzun E, Woodhall M, Lang B, Jacobson L, Icoz S, Bebek N, Gurses C, Gokyigit A, Waters P, Vincent A, Baykan B. Investigation of neuronal autoantibodies in two different focal epilepsy syndromes. Epilepsia 2014;55:414-22.
55. Turner MR, Irani SR, Leite MI, Nithi K, Vincent A, Ansorge O. Progressive encephalomyelitis with rigidity and myoclonus: glycine and NMDA receptor antibodies. Neurology 2011;77:439-43.
56. Stern WM, Howard R, Chalmers RM, Woodhall MR, Waters P, Vincent A, Wickremaratchi MM. Glycine receptor antibody mediated Progressive Encephalomyelitis with Rigidity and Myoclonus (PERM): a rare but treatable neurological syndrome. Pract Neurol 2014;14:123-7.
57. Derksen A, Stettner M, Stocker W, Seitz RJ. Antiglycine receptor-related stiff limb syndrome in a patient with chronic lymphocytic leukaemia. BMJ Case Rep 2013; doi: 10.1136/bcr-2013-008667.
58. Kyskan R, Chapman K, Mattman A, Sin D. Antiglycine receptor antibody and encephalomyelitis with rigidity and myoclonus (PERM) related to small cell lung cancer. BMJ Case Rep 2013; doi: 10.1136/bcr-2013-010027.
59. Zuliani L, Ferlazzo E, Andrigo C, Casano A, Cianci V, Zoccarato M, Leite MI, Waters P, Woodhall M, Della Mora E, Morra M, Giometto B, Aguglia U, Vincent A. Glycine receptor antibodies in 2 cases of new, adult-onset epilepsy. Neurol Neuroimmunol Neuroinflamm 2014;1:e16.
60. Clerinx K, Breban T, Schrooten M, Leite MI, Vincent A, Verschakelen J, Tousseyn T, Vandenberghe W. Progressive encephalomyelitis with rigidity and myoclonus: resolution after thymectomy. Neurology 2011;76:303-4.
61. Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, Honig LS, Benseler SM, Kawachi I, Martinez-Hernandez E, Aguilar E, Gresa-Arribas N, Ryan-Florance N, Torrents A, Saiz A, Rosenfeld MR, Balice-Gordon R, Graus F, Dalmau J. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157-65.
62. Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012;83:638-45.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Quek AML, O’Toole O. Encephalitis associated with autoantibodies binding to γ-aminobutyric acid-A, γ-aminobutyric acid-B and glycine receptors: immunopathogenic mechanisms and clinical characteristics. Neurosciences 2016;3:86-92. http://dx.doi.org/10.4103/2347-8659.170633
AMA Style
Quek AML, O’Toole O. Encephalitis associated with autoantibodies binding to γ-aminobutyric acid-A, γ-aminobutyric acid-B and glycine receptors: immunopathogenic mechanisms and clinical characteristics. Neuroimmunology and Neuroinflammation. 2016; 3: 86-92. http://dx.doi.org/10.4103/2347-8659.170633
Chicago/Turabian Style
Quek, Amy May Lin, Orna O’Toole. 2016. "Encephalitis associated with autoantibodies binding to γ-aminobutyric acid-A, γ-aminobutyric acid-B and glycine receptors: immunopathogenic mechanisms and clinical characteristics" Neuroimmunology and Neuroinflammation. 3: 86-92. http://dx.doi.org/10.4103/2347-8659.170633
ACS Style
Quek, AML.; O’Toole O. Encephalitis associated with autoantibodies binding to γ-aminobutyric acid-A, γ-aminobutyric acid-B and glycine receptors: immunopathogenic mechanisms and clinical characteristics. Neurosciences. 2016, 3, 86-92. http://dx.doi.org/10.4103/2347-8659.170633
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 29
likes
Like This Article 29
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.