
The expanding spectrum of pediatric anti-glutamic acid decarboxylase antibody mediated CNS disease - a chance association?

Abstract
Central nervous system autoimmunity in the pediatric age group represents an evolving constellation of various syndromes distinct from the adult age group. One of the rarely described pathogenic auto-antibodies (ab) is the one directed against glutamic acid decarboxylase (GAD). While its pathogenic role is controversial, literature concerning adult patients abounds with heterogeneous presentations with epilepsy often as part of limbic encephalitis or chronic temporal lobe epilepsy and cerebellar ataxia accompanying endocrinopathies or paraneoplastic disorders. Diagnosis is often delayed until late adulthood. The authors report hitherto under-reported syndromes in the pediatric age group. The first case was a 3-year-old boy with sub-acute myoclonus-ataxia following a flu-like illness akin to para-infectious cerebellitis. The second case was a 7-year-old girl with long-standing chronic extratemporal partial epilepsy and electrical status epilepticus in sleep (ESES) with right hemiparesis and developmental delay. Investigations revealed two-four fold elevations in titres of GAD-65-ab. The absence of systemic diseases like diabetes and the dramatic clinical response to steroids as well as intravenous immunoglobulin in both the cases argued for GAD-ab mediated neuronal injury rather than a chance association. The concern exists regarding other potentially co-existent auto-ab to gamma-amino butyric acid and glycine receptors, and demonstration of intrathecal synthesis of GAD-ab would be ideal. This entity should be contemplated in children presenting with acute/sub-acute onset episodic or progressive ataxia or refractory cryptogenic focal epilepsy syndromes, epileptic encephalopathy such as ESES and worsening neurological deficits. These children ought to be maintained on regular follow-up for monitoring evolution of other autoimmune disorders in adult life.
Keywords
Introduction
The spectrum of autoimmune encephalitis is ever expanding, with presentations outside the distinctively symptomatic groups being recognized every day. The array of intraneuronal and cell surface antibodies are fairly well elucidated with the former associated with paraneoplastic etiology and poor response to immunosuppressive agents.[1] While the clinical spectrum of non-paraneoplastic encephalitis, like anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis and voltage-gated potassium channel (VGKC) encephalitis, has been well described,[1] the presentations of anti-glutamic acid decarboxylase antibody (GAD-ab) positive encephalitis are unclear. GAD-ab is directed to an intracellular enzyme and therefore considered to be the unlikely pathogenic moiety in itself. However, epilepsy and cerebellar ataxia represent the 2 most common neurological syndromes described in adults with this antibody.[2]In vitro, GAD-ab from patients with neurological syndromes induce a suppression of gamma amino-butyric acid (GABA) release.[3] The diagnostic value of low titres of GAD-ab in a patient with a neurological syndrome is unknown, as opposed to high titres as was seen in recently described case series in patients with autoimmune endocrinopathies, like type 1 diabetes mellitus (DM1) or central nervous system (CNS) autoimmunity, such as limbic encephalitis.[2,4]
The largest reported cohort of 9 adult patients with GAD-ab mediated limbic encephalitis was notable for a poor response to treatment in comparison with VGKC antibody positive encephalitis, however, only a minority had been given the benefit of immunomodulation with immuoglobulin.[4] Currently, anti-GAD-ab disorders of the CNS in the pediatric age-group are rarely reported. Here we report a case series in the pediatric age group with variable clinical presentations, course and treatment response with the 2 patients demonstrating definite elevations in anti-GAD 65 antibody titres, adding to the evolving clinical conundrum of CNS autoimmunity in childhood.
Case report
Case 1
A 3-year-old boy, product of a non-consanguineous parentage with normal birth and development presented to us with subacute onset incoordination of upper and lower limbs along with scanning dysarthria. A flu-like prodrome was noted nearly 20 days prior to onset. Around 10 days into illness parents had also noted sudden jerky movement of extremities. His examination was notable for pancerebellar involvement with multifocal stimulus sensitive myoclonus. There was no evidence of any behavioural or cognitive decline, limb weakness, seizures, opsoclonus or any extrapyramidal involvement with no history of drug/toxin exposure. Blood biochemistry and serology were normal. Based on the possibility of a post-infective or a paraneoplastic immune mediated myoclonic ataxia syndrome, magnetic resonance imaging (MRI), cerebro-spinal fluid (CSF) study including lactate levels, electroencephalogram (EEG) and somato-sensory evoked potentials were ordered and were normal. A search for a neoplastic focus with ultrasonography abdomen and chest X-ray was negative. Twenty-four hours urine vallinly mandelic acid and metanephrine tests were conducted to exclude occult neuroblastoma and urine aminoacid estimation to exclude alkaptonuria was also normal. He was empirically started on a course of intravenous (IV) methylprednisolone after excluding any active infection. Over the period of the next 2 weeks, he had resolution of all his symptoms and the steroids were tapered off over 6 weeks. However, he presented to us 6 months later with recurrence of the same complaints with much more severe symptoms along with irritability, hyperactivity and temper tantrums. Considering his recurrent course and apparent steroid responsiveness, a further search was done with repeat MRI, including MRI abdomen, CSF oligoclonal bands and autoimmune panel of antibodies including NMDA, VGKC, and GAD 65 as well as anti-aquaporin antibodies. His antibody panel revealed an elevated GAD 65-abtitre of 10.7 IU/mL (0.0-5.0 IU/mL) by enzyme-linked immunosorbent assay (ELISA) and the rest of the investigations were negative including blood tandem mass spectroscopy. Considering the severity of symptoms he was started on a simultaneous course of intravenous methylprednisolone (20 mg/kg for 5 days) and immunoglobulin (400 mg/kg for 5 days) with which all his symptoms completed remitted in 1 week. He was maintained on oral steroids with plans for a longer duration of maintenance and slow taper. After nearly 24 months of follow-up he is symptom free with preserved motor and cognitive abilities and is presently off steroids.
Case 2
A 7-year-old girl, product of a non-consanguineous parentage with normal birth and development history, presented with habitual seizures since 2 years of age without any initial precipitating event. Her seizure semiology was suggestive of frontal lobe epilepsy with more than 95% events being nocturnal events occurring out of sleep and characterized by head and eye deviation to the right side with right upper limb abduction, clonic jerks and right facial jerks. Since the onset of seizures parents noted delay in subsequent development as well as a shift of handedness from right to left. She presented to us nearly 4 years into her illness by which time she had frequent clustering of right hemiclonic seizures, which were drug refractory and in addition to serially prolonged episodes of Todd’s paresis. Her examination revealed a developmental age of 3.5 years with right pyramidal signs. There were no epilepsia partialis continua. The initial MRI taken 1 year into the illness revealed non-specific volume loss over the left posterior cortex. A 12-h video EEG recording [Figure 1B-D] detected 6 complex partial seizure of left hemspheric semiology, five of left frontocentral ictal onset and one of left posterior head region onset. The interictal data showed frequent left frontocentral, left posterior temporal and occipital interracial epileptiform discharges with intrahemispheric and secondary bilateral synchrony, with sleep records showing electrical status epilepticus in sleep (ESES) [Figure 1A]. As she was on a combination of carbamazepine, phenobarbitone and levetiracetam, a possibility of sodium-channel blocker mediated worsening with ESES was considered and carbamazepine was gradually withdrawn and replaced by valproic acid. One month later she presented with increased nocturnal seizures, and she was commenced on lamotrigine which was subsequently withdrawn due to drug allergy. After another month, she developed simple partial and complex partial status epilepticus with worsening right hemiparesis. She was treated with a fosphenytoin-midazolam infusion along with continued polytherapy in view of seizure clusters and a 5-day pulse of IV methylprednisolone was administered, considering the possibility of left hemispheric focal encephalitis. The repeat MRI showed diffuse bilateral generalised parenchymal atrophy with asymmetric dilatation of ventricles (left more than right) with subtle asymmetric loss of grey-white differentiation over the left posterior quadrant [Figure 2]. Her CSF evaluation including immunoglobulin G (IgG) index at that time was normal. Her serum autoimmune panel comprising of blood and CSF: NMDAR antibody, VGKC antibody, anti-thyroid antibodies and anti-GAD antibody, which revealed elevated GAD 65-ab titre of 21 IU/mL by ELISA (0.0-5.0 IU/mL). Following discharge, she developed a phenytoin allergy. She was withdrawn off oral steroids over 2 months with only infrequent brief nocturnal seizures and near complete recovery of hemiparesis. Three months later she was re-admitted with seizure clustering and right hemiparesis. Following the initiation of IV steroids, she developed complex partial status epilepticus. She required ventilation with midazolam anesthesia and intravenous lacosamide, following which she recovered over 1 week. She was administered IV immunoglobulin 2 g/kg over 5 days with which her hemiparesis also recovered. She was maintained on cyclical immunoglobulin 1 g/kg 6-8 weekly for 3 cycles along with a tapering schedule of oral steroids. Presently 12 months into follow-up she experiences brief nocturnal simple partial seizures, her right hemiparesis has recovered by more than 80% and schooling has resumed. She is maintained on a regular schedule of valproic acid, lacosamide, clobazam and levetiracetam. Her repeat GAD 65-ab titre remains elevated (11 IU/mL).
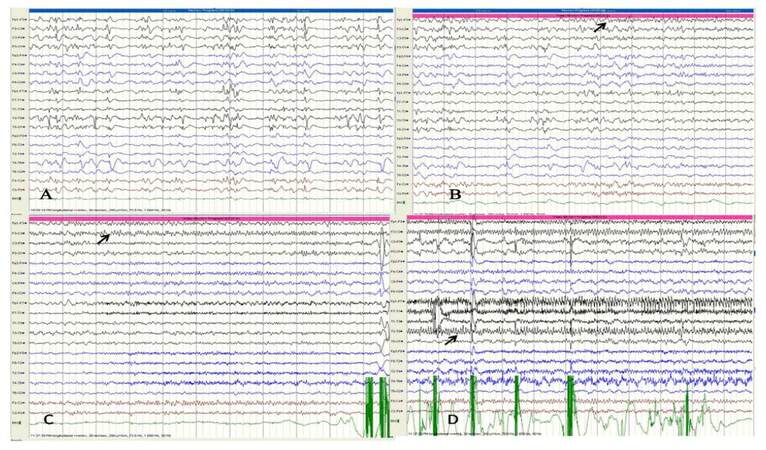
Figure 1. (A) Interictal activity in Case 2 during the first video EEG, consistent with an atypical left hemispheric dominant electrical status epilepticus in sleep; (B) ictal onset (arrow) in the form of low voltage fast activity over the left fronto-centro-parietal regions during hemiclonic seizure; (C) propogation of ictal activity over the left centro-parietal region (arrow); and (D) ictal activity during the established hemiclonic phase (arrow) demonstrating involvement of the left hemisphere with spread to the left posterior cortex. EEG: electroencephalogram
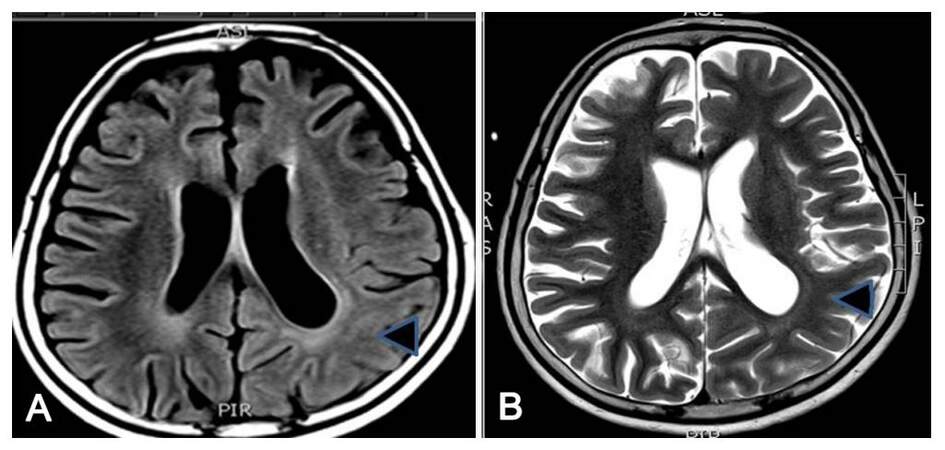
Figure 2. (A) FLAIR axial image; (B) T2-Weighted axial image. FLAIR and T2W axial MRI sequences in case 2 demonstrating global atrophy with loss of grey white matter differentiation over the left posterior quadrant. This was done 1 month after the first episode of complex partial status epilepticus. FLAIR: fluid attenuated inversion recovery; T2W: T2-weighted; MRI: magnetic resonance imaging
Discussion
Our case series demonstrates the heterogeneity of clinical presentations of presumed GAD mediated autoimmunity in the pediatric age-group. Despite the inability to ascertain intrathecal synthesis of GAD-ab, treatment was directed by clinical suspicion of the same and subsequent responses. Sero-prevalence of GAD-ab raises controversies on its role in CNS autoimmunity. GAD is the rate limiting enzymein the formation of inhibitory neurotransmitter GABA. The enzyme has 2 isoforms-GAD 65 and GAD 67, which differ substantially in their amino terminal, but function in synergy to maintain a physiological GABA level, with the former more active during stress and the latter assuming more of a housekeeping function.[5] GAD 65 is an intracellular protein, but it has been suggested that it could be exposed on the cell surface during exocytosis from GABA-ergic neurons, allowing a pathogenic ab-antigen interaction to occur. GAD 65-specific autoantibodies are also seen in some patients with other neurologic diseases, such as myoclonus, stiff person syndrome, pure cerebellar ataxia.[2,5] High GAD-ab levels, usually more than 100-fold higher that those found in DM1, are present in up to 80% of patients with stiff person syndrome (SPS), a subgroup of patients with late onset isolated cerebellar ataxia, epilepsy or brain stem dysfunction and are usually associated with type 1 diabetes or poly-endocrine autoimmunity, both of which were not seen in our patients.[6]
The causative immunological mechanisms remain speculative at best, and the clinical spectrum of GAD-ab associated disease may depend on the specific immunological response that is elicited. While it is believed that GAD-ab are unlikely to be pathogenic, in vitro studies suggest that IgG from patients can mediate effects on cerebellar neurons.[3,7] One explanation could be the intracellular uptake of these antibodies with subsequent inhibition of GABA synthesis, as has been demonstrated with ampiphysin antibodies associated with paraneoplastic SPS.[8] A more plausible explanation is that other unknown antibodies in sera positive for GAD-ab are the pathogenic moiety; the possible co-existence of antibodies for other membrane antigens, i.e. GABA-B receptor and glycine receptor requires further evaluation.[9,10] Furthermore, GAD-ab titers are significantly higher in the sera of adult patients with CNS involvement, some of whom also have evidence of intrathecal synthesis.[2] Due to lack of availability we could not confirm the antibody status using cell-based assays. There is enough evidence in literature that the epitopes of classical intracellular or onconeuronal antigens (Hu, Yo, Ri, CRMP5, Ma2, amphiphysin) are resistant to protein denaturation, and hence detectable by immunoblot or ELISA, as well as immunohistochemistry using mammalian brain, most commonly rat or mouse.[11] This is contrasted to antibodies to cell surface or synaptic protein ssuch as those against NMDA and VGKC wherein the reactivity is usually lost when the antigen is denatured so that these antibodies cannot be detected by standard immunoblot or ELISA. Detection of these antibodies requires either an immunohistochemistry protocol adapted to cell surface antigens, the use of cultures of live neurons, or cell-based assays in which recombinant antigens are expressed in mammalian cells. It is evident that the specificity and sensitivity of these assays vary among laboratories even when the same techniques are used. Because the reading of the tests is done by visual assessment, the interpretation of low serum titers can be misleading, and some sera produce non-specific background reactivity that may be interpreted as a positive result, although this rarely occurs when CSF is used. Another study however demonstrated absence of serum cross reactivity to NMDA and VGKC antibody in patients with suspected anti GAD mediated epilepsy with titres ranging between 6 to > 200,000 IU/mL with only 7 out of 15 subjects demonstrating high titres > 1,000 IU/mL.[12] This variability in serum titres is also demonstrated in Table 1. This indicates that low titres, as also demonstrated in another case series, need not be neglected in a clinically relevant scenario.[13] However, clinical and serological follow-up are likely to ascertain the significance of these mildly elevated titres in pediatric patients reported here.
Presentations with CNS manifestations in non-neoplastic GAD positive patients (bold font indicative of patients with paediatric GAD-ab mediated diseases)
| Author | No. of subjects (age at diagnosis) | Presentation | Serum Ab titre Mean (SD) | MRI | CSF Ab | Treatment | Outcome |
|---|---|---|---|---|---|---|---|
| Honnorat et al.[6] | 14 (40-70 years) | Cerebellar ataxia | 37,300 (30,460) U/mL | Cerebellar atrophy/ normal | Intra-thecal synthesis in 6/9 | NA | Variable |
| McKnight et al.[16] | 5 (3-36 years) | Chronic Drug resistant epilepsy | > 1,000 U/mL in 3; > 10 U/mL in 2 | Normal in all | NA | NA | Chronic epilepsy |
| Mata et al.[15] | 2 (20, 47 years) | Memory decline, seizures | 72-87.5 U/mL | Temporal lobe HI | 46-54.1 U/mL | Steroids, IVIG, PLEX | Partial benefit |
| Saiz et al.[2] (largest series) | 50 (13-79 years) | Variable (predominant ataxia, SPS, drug resistant epilepsy) | > 2,000 U/mL | Temporal lobe HI, variable | High IgG index | Variable | Variable |
| Ozkan et al.[17] | 2 (9 months, 6 years) | Acute ataxia, status epilepticus with involuntary movements | 1.48-1.79 U/mL (ref < 1) | Normal | 2.16 U/mL | Steroids, IVIG | Improved |
| Malter et al.[16] | 9 (17-66 years) | Cognitive decline, seizure (presentation as limbic encephalitis) | 1,798-12,030 U/mL | Medial temporal hyperintensity, PET hypometabolism | 29-235 U/mL | Steroids, IVIG | None seizure free |
| Present series | 2 (3, 7 years) | Chronic extratemporal partial epilepsy with ESES, subacute myoclonus ataxia | 10.7-21 U/mL (ref 0-5.0 U/mL) | Grey matter loss with subcortical HI | NA | Steroids, IVIG | Improved |
Table 1 reflects the rarity of pediatric GAD-ab mediated CNS autoimmunity. In a large series, the spectrum of GAD-ab positive spectrum of diseases was associated with a variety of neurological and non-neurological entities.[2] The mean age was between 50-60 years in this series. In the adult population a female gender predilection with predominant presentation as limbic encephalitis is noted. In contrast, presentations of both our patients were unique. The first child presented with a subacute ataxia-myoclonus syndrome with good response to steroids that was previously undescribed in adult series. This constellation has been previously noted in anti-NR1 receptor NMDA-ab mediated encephalitis in addition to the well described paraneoplastic opsoclonus-myoclonus syndrome.[14] The other child had refractory focal epilepsy with lateralizing neurological deficits and a prolonged course of 4 years resembling a left hemispheric focal encephalitis versus a large malformation of cortical development. The latter’s MRI features were also more in favour of focal encephalitis in the absence of discrete cortical pathology with the development of progressive grey matter volume loss, which may also be attributed to refractory seizures and the effect of anti-epileptic drugs. However, the clinical scenario was distinct from the well-described limbic encephalitis. In most adult series, GAD-ab were requested at the time of diagnosis of type 1 diabetes more than a decade or two after the onset of the epilepsy. As evident in Table 1, patients in series No. 2 had drug-resistant temporallobe epilepsy associated with hippocampal sclerosis, with 1 patient diagnosed to have celiac disease.[15] Patients with epilepsy reported in the largest series have ranged from chronic epilepsy with hippocampal sclerosis or co-existent heterotopias and one patient was diagnosed to harbour GAD-ab many years after the diagnosis of epilepsy following development of oscillopsia with nystagmus and subsequent detection of CSF oligoclonal IgG bands and intrathecal synthesis of GAD-ab.[2] Although the frequency of high GAD-ab levels in patients with epilepsy is low and ranges from 0% to 4%, GAD-ab-positive patients are more likely to have chronic drug-resistant epilepsy.[16,17] Rasmussen’s encephalitis-like presentation has also been reported in a 6.5-year-old male with type I DM who presented with epilepsia partialis continua for days with detectable GAD-ab.[18] The presentation of Case 2 as ESES, refractory partial epilepsy and focal deficits with GAD-ab is previously undescribed in the literature, although a report of onco-neural antibody mediated ESES in a child with neuroblastoma and opsoclonus-myoclonus syndrome exists.[19] While the response of GAD-ab mediated epilepsies has been shown to be far from satisfactory in terms of seizure outcomes following immune-modulation or surgery, especially in temporal lobe epilepsies,[20] our experience has demonstrated significant improvement with immunomodulation during sub-acute worsening of encephalopathy, seizures, focal deficits in Case 2 with chronic extratemporal partial epilepsy. Though GAD-ab may just reflect the presence or later risk of concomitant DM1 and other endocrine autoimmune disorders, longitudinal follow-up in our patients is likely to be provide more insight. The antibody titres were not extremely elevated as in reference cases in Table 1, however, the possibility of an alternative diagnosis has been reliably excluded by investigations. Despite the inability to demonstrate intrethecal synthesis, the dramatic response to immunomodulation demonstrated in both patients highlights the significance of evaluating for these antibodies in children with chronic refractory partial epilepsy with epileptic encephalopathy of uncertain etiology and acute-subacute myoclonus-ataxia syndromes.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Patient consent
Obtained.
Ethics approval
Ethics approval was obtained from Institute Review Board for case reports.
REFERENCES
1. Vincent A, Bien C, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011;10:759-72.
2. Saiz A, Blanco Y, Sabater L, González F, Bataller L, Casamitjana R, Ramió-Torrentà L, Graus F. Spectrum of neurological syndromes associatedwith glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain 2008;131:2553-63.
3. Manto MU, Laute MA, Aguera M, Rogemond V, Pandolfo M, Honnorat J. Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases. Ann Neurol 2007;61:544-51.
4. Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol 2010;67:470-8.
6. Honnorat J, Saiz A, Giometto B, Vincent A, Brieva L, de Andres C, Maestre J, Fabien N, Vighetto A, Casamitjana R, Thivolet C, Tavolato B, Antoine J, Trouillas P, Graus F. Cerebellar ataxia with anti-glutamic acid decarboxylase antibodies: study of 14 patients. Arch Neurol 2001;58:225-30.
7. Takenoshita H, Shizuka-Ikeda M, Mitoma H, Song S, Harigaya Y, Igeta Y, Yaguchi M, Ishida K, Shoji M, Tanaka M, Mizusawa H, Okamoto K. Presynapticinhibition of cerebellar GABAergic transmission by glutamatedecarboxylase autoantibodies in progressive cerebellar ataxia. J Neurol Neurosurg Psychiatry 2001;70:386-9.
8. Geis C, Weishaupt A, Hallermann S, Grünewald B, Wessig C, Wultsch T, Reif A, Byts N, Beck M, Jablonka S, Boettger MK, Üçeyler N, Fouquet W, Gerlach M, Meinck HM, Sirén AL, Sigrist SJ, Toyka KV, Heckmann M, Sommer C. Stiff person syndrome-associated autoantibodies to amphiphysin mediate reduced GABAergic inhibition. Brain 2010;133:3166-80.
9. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, Friedman D, Skeen MB, Grisold W, Kimura A, Ohta K, Iizuka T, Guzman M, Graus F, Moss SJ, Balice-Gordon R, Dalmau J. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010;9:67-76.
10. Hutchinson M, Waters P, McHugh J, Gorman G, O'Riordan S, Connolly S, Hager H, Yu P, Becker CM, Vincent A. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology 2008;71:1291-92.
11. Rosenfeld MR, Titulaer MJ, and Dalmau J. Paraneoplastic syndromes and autoimmune encephalitis: five new things. Neurol Clin Pract 2012;2:215-23.
12. Liimatainen S, Peltola J, Hietaharju A, Sabater L, Lang B. Lack of antibodies to NMDAR or VGKC-complex in GAD and cardiolipin antibody-positive refractory epilepsy. Epilepsy Res 2014;108:592-6.
13. Ozkan M, Aksoy A, Çenesiz F, Atay NE, Yüksel D. The association of anti-glutamic acid decarboxylase antibodies with different neurological findings in childhood. Epilepsy Behav 2012;25:464-7.
14. Player B, Harmelink M, Bordini B, Weisgerber M, Girolami M, Croix M. Pediatric opsoclonus-myoclonus-ataxia syndrome associated with anti-N-methyl-D-aspartate receptor encephalitis. Pediatr Neurol 2015;53:456-8.
15. Matà S, Muscas GC, Naldi I, Rosati E, Paladini S, Cruciatti B, Bisulli F, Paganini M, Mazzi G, Sorbi S, Tinuper P. Non-paraneoplastic limbic encephalitis associated with anti-glutamic acid decarboxylase antibodies. J Neuroimmunol 2008;199:155-9.
16. McKnight K, Jiang Y, Hart Y, Cavey A, Wroe S, Blank M, Shoenfeld Y, Vincent A, Palace J, Lang B. Serum antibodies in epilepsy and seizure-associated disorders. Neurology 2005;65:1730-6.
17. Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol 2010;67:470-8.
18. Olson JA, Olson DM, Sandborg C, Alexander S, Buckingham B. Type 1 diabetes mellitus and epilepsia partialis continua in a 6-year-old boy with elevated anti-GAD65 antibodies. Pediatrics 2002;109:E50.
19. Hu LY, Shi XY, Feng C, Wang JW, Yang G, Lammers SH, Yang XF, Ebrahimi-Fakhari D, Zou LP. An 8-year old boy with continuous spikes and waves during slow sleep presenting with positive onconeuronal antibodies. Eur J Paediatr Neurol 2015;19:257-61.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Menon D, Menon RN, Kumar H, Radhakrishnan A, Kannoth S, Nair M, Thomas S. The expanding spectrum of pediatric anti-glutamic acid decarboxylase antibody mediated CNS disease - a chance association?. Neurosciences 2016;3:219-24. http://dx.doi.org/10.20517/2347-8659.2016.02
AMA Style
Menon D, Menon RN, Kumar H, Radhakrishnan A, Kannoth S, Nair M, Thomas S. The expanding spectrum of pediatric anti-glutamic acid decarboxylase antibody mediated CNS disease - a chance association?. Neuroimmunology and Neuroinflammation. 2016; 3: 219-24. http://dx.doi.org/10.20517/2347-8659.2016.02
Chicago/Turabian Style
Menon, Deepak, Ramshekhar N. Menon, Hardeep Kumar, Ashalatha Radhakrishnan, Sudheeran Kannoth, Muralidharan Nair, Sanjeev Thomas. 2016. "The expanding spectrum of pediatric anti-glutamic acid decarboxylase antibody mediated CNS disease - a chance association?" Neuroimmunology and Neuroinflammation. 3: 219-24. http://dx.doi.org/10.20517/2347-8659.2016.02
ACS Style
Menon, D.; Menon RN.; Kumar H.; Radhakrishnan A.; Kannoth S.; Nair M.; Thomas S. The expanding spectrum of pediatric anti-glutamic acid decarboxylase antibody mediated CNS disease - a chance association?. Neurosciences. 2016, 3, 219-24. http://dx.doi.org/10.20517/2347-8659.2016.02
About This Article
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 13
likes
Like This Article 13
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.