
Inflammation in human cerebral aneurysms: pathogenesis, diagnostic imaging, genetics, and therapeutics


Abstract
Intracranial aneurysms are a life-threatening cerebrovascular pathology with a probability of spontaneous rupture. Current intervention techniques carry inherent risk. Recent investigation has reinforced inflammation’s role in the pathophysiological process of cerebral aneurysms. These data suggest alternative diagnostic and noninvasive therapeutic strategies. Furthermore, novel characteristics of the underlying disease have been elucidated through distinct bioinformatic and gene expression profile analyses. This article will emphasize the most recent investigation, highlighting findings of clinical significance and etiological relevance.
Keywords
Introduction
Intracranial aneurysms (IAs) are a common multifactorial cerebrovascular disease. While having a 3.2% prevalence rate in the general population and 1.1% annual rupture risk, aneurysms remain lethal upon rupture in 40% of cases.[1-3] Risk for aneurysmal subarachnoid hemorrhage (SAH) is higher in hypertensive patients, smokers, heavy drinkers, and females.[2,4] The current interventional techniques, surgical clippings and endovascular occlusions, remain invasive despite advancements in technology. Although SAH risk factors are known, a more complete understanding of the complex pathophysiology underlying IA formation, progression, and rupture is needed. The majority of evidence from intensive investigation has implicated a mounting inflammatory response during the aneurysm pathogenesis.[5,6] This data ultimately provides promising targets for in vivo molecular imaging and noninvasive IA therapeutics. This article will discuss inflammation as it pertains to IA pathogenesis, with a focus on the most recent investigation. Furthermore, it will offer a review of recent genetic and bioinformatic analyses, highlighting findings of pathological significance and methodological diversity. The final sections will further illuminate both innovations in diagnostic imaging of aneurysmal inflammation and experimentally efficacious noninvasive attempts at IA prevention and regression.
Pathogenesis
Hemodynamic insult is considered to be one of the first steps in activating the cerebral vessel wall’s inflammatory response.[7] There is a continuous balance between hemodynamic stress and the integrity of the vessel wall.[5] Upon the hemodynamic insult, this balance is perturbed, leading to vessel wall weakening. Dilation results, as extracellular matrix is degraded by increased levels of matrix metalloproteinases (MMP) compounded by concomitant apoptotic death of vascular smooth muscle cells (VSMCs).[7] Initially, the vessel wall is highly organized. Integral disturbances lead to less organization within the aneurysm wall and fewer distinct layers.[5] Simultaneously, MMP activation has been found to facilitate flow-induced internal elastic lamina (IEL) fragmentation.[8] Similar IEL fragmentation is a feature commonly observed in atherosclerotic lesions.[9]
Vascular smooth muscles cells (VSMCs), mainly found in the media layer, are recognized as major producers of matrix in the vessel wall. Upon endothelial injury, intimal thickening occurs as VSMCs migrate into the intima and proliferate.[7,10] Phenotypic transformation is seen in these migrated VSMCs as environmental change induces a switch from a contractile phenotype to a synthetic pro-inflammatory matrix remodeling phenotype in these cells.[11] Nakajima et al.[12] showed phenotypically modulated VSMCs have lower expression of contractile smooth muscle myosin heavy chain isoforms when immunohistochemically stained. Furthermore, the group observed decreases in contractile protein staining intensity in ruptured IA, suggesting VSMC phenotypic modulation is related to a rupture mechanism.
Endothelial cells are also affected by the hemodynamic insult. As blood flows, mechanical stimulus has notable affects on the cells of the vascular system.[13] Shear stress influences arterial physiology, in particular, the cellular function of the vessel wall.[13] Consequently, IAs develop in vessel regions exposed to high hemodynamic stress such as arterial bifurcations and sharp angles.[7] Endothelial cells under laminar flow become aligned with the flow but endothelial cells under turbulent flow become cuboidal due to F-actin reorganization.[14] Experimentally, endothelial cells respond to hemodynamic stress with the up-regulation of the inflammatory mediator, prostaglandin E receptor 2 (EP2), during the formation of cerebral aneurysms.[15] When shear stress was applied to primary endothelial cells, EP2 was found to be up-regulated.[15] The stimulation of EP2 in primary endothelial cells also led to the activation of the transcription factor NF-κB, a well-studied transcriptional mediator of inflammation in IA.[15]
As cerebral vessel walls undergo change during aneurysm development, the formation of new vessels, angiogenesis, also contributes to aneurysmal progression.[16] Angiogenesis indirectly advances the inflammatory process of aneurysm progression by aiding in the delivery of inflammatory cells to vessel walls. Hoh et al.[16] recently showed that stromal cell-derived factor-1 (SDF-1) is expressed in murine carotid IAs, murine circle of Willis IAs, and human cerebral aneurysms. The study found increased levels of progenitor cells expressing the SDF-1 receptor, CXCR4, in mice with carotid and cerebral aneurysms. Study findings also showed SDF-1 to promote endothelial cell migration, endothelial cell tube formation (angiogenesis), and macrophage migration.[16] Inhibition of SDF-1 in the murine model significantly decreased the amount of endothelial cells and capillaries in the aneurysm wall, and the overall rate of aneurysm formation was reduced.[16]
Mediators of inflammation
Human and animal studies have both shown that inflammatory cells and mediators are involved in IA pathogenesis [Figure 1]. A number of these inflammatory cells and mediators are highlighted in this section, with a special focus on the most recent investigation. Mast cells infiltration into the aneurysm vessel wall has been illustrated throughout the pathophysiological chronology of IA development.[17] Ishibashi et al.[17] analyzed mast cells levels at different time intervals following aneurysm induction in a rat model and discovered that the number of mast cells in the IA walls significantly increases over time. To further understand mast cells’ role in the aneurysm pathogenesis, mast cell degranulation inhibitors, tranilast and emedastine, were administered into an aneurysm-induced rat model. The inhibitors had an impact on aneurysm progression by decreasing aneurysm size, as well as the extent of media thinning in the induced IAs. Mast cells’ contribution to the inflammatory process was simultaneously studied through the analysis of mast cell degranulation’s effect on cultured arterial smooth muscle cells of rat IAs. The group found that mast cell degranulation promoted MMP-2, MMP-9, and inducible nitric oxide synthase (iNOS) expression.[17] Based on previous findings that iNOS deficiency leads to a decrease in apoptotic smooth muscle cell death, Ishibashi et al.[17] and Sadamasa et al.[18] suggested that the inhibition of mast cell degranulation impacts the process of media thinning through its induction of iNOS. The authors also concluded that the decrease in media thinning results from mast cells’ modulation of MPP-2 and MMP-9 expression.[17,19] Overall, these data suggest that the degranulation of mast cells plays a role in IA formation.
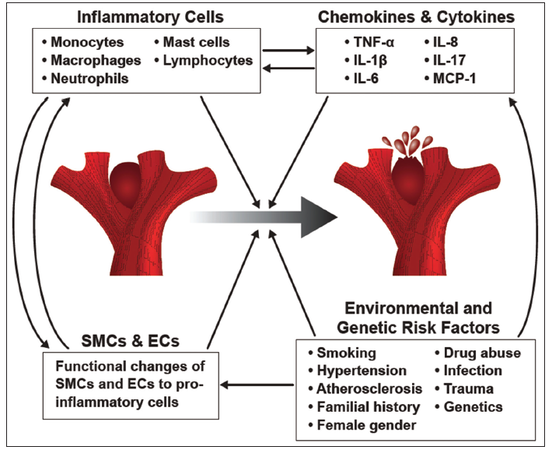
Figure 1. Summary of the factors contributing to cerebral aneurysm pathogenesis. Inflammatory cells, cytokines, chemokines, changes to the vascular smooth muscle cells and endothelial cells, and environmental as well as genetic risk factors all play a role in the development of cerebral aneurysms and their progression to rupture
T cells and macrophage infiltration have found to be associated with human cerebral aneurysm rupture.[20] Kanematsu et al.[21] studied leukocyte infiltration into the aneurysm wall of an aneurysm-induced mouse model and found macrophages to be one of the dominant leukocytes present. Cerebral aneurysm incidence was found to be significantly attenuated in mice with lower macrophage levels as well as in mice with inhibited monocyte chemotactic protein-1 (MCP-1), a macrophage chemoattractant, when compared with wildtype mice.[21] Certain proteinases secreted by macrophages have been found to be involved in the vascular remodeling of aneurysm formation. Aoki et al.[19] demonstrated that in aneurysm-induced rats, the expression of MMP-2 and MMP-9, was present in the arterial wall undergoing the beginning stages of aneurysm formation. This expression was found to increase alongside the progression of the studied cerebral aneurysms. Macrophages were determined to be the main secretes of the MMPs in the study.[19] Further connection has been found between macrophage presence and aneurysm rupture. Hasan et al.[22] showed that in human cerebral aneurysms, M1 and M2 macrophages were found to be in equal proportions; however, M1 macrophages presented in higher levels than M2 macrophages in ruptured human cerebral aneurysms. This means that there were more macrophages promoting inflammation, M1 macrophages, than macrophages working to repair the vessel tissue and decrease inflammation, M2 macrophages. This imbalance was also correlated with an increase in mast cell presence in ruptured aneurysms.[22]
The role of the inflammatory mediators, chemokines, has been studied in aneurysm formation. Chemokines promote chemotaxis in particular, the migration of inflammatory cells during the inflammatory response. In recent years, Chalouhi et al.[23] demonstrated that plasma concentrations of RANTES, MIG, IP-10, eotaxin, interleukin 8 (IL-8), IL-17, and the monocyte chemoattractant protein-1 were significantly increased in the lumen of unruptured human cerebral aneurysms when compared to femoral arterial plasma of the same 16 patients. In addition, the study found increased plasma concentrations of MCP-1 in unruptured aneurysms.[23] This reaffirms the work of Aoki et al.[24] who observed increased MCP-1 expression in rat arterial walls. These data indicate that inflammatory cells are being actively recruited to the aneurysm wall as a result of high chemokine levels, further contributing to IA formation and eventual rupture.[23]
Aoki et al.[25] confirmed in rodent models that tumor necrosis factor-α (TNF-α) levels are higher in cerebral aneurysms, particularly in the endothelial cells of these lesions, during their formation and progression when compared to the TNF-α levels of control cerebral arteries. Aoki et al.[25] further investigated the TNF-α-TNFR1 signaling pathway and found that IA formation was suppressed in rodents deficient in TNFR1. The authors concluded that TNFR1 deficiency blocks NF-κB activation and MCP-1 expression, ultimately inhibiting macrophage infiltration. Intervening with this signaling pathway may serve as a future therapeutic strategy.[25] TNF-α has been previously identified as a contributor to the phenotypic modulation of VSMCs in vivo following hemodynamic stress.[26] The transcription factor, NF-κB, has also been found to be essential in the activation and recruitment of macrophages as it influences the expression of a number of pro-inflammatory genes.[5,6] Aoki et al.[27] demonstrated the activation of NF-κB and expression of downstream genes; these changes seen in the vessel wall of the early stages of aneurysm development in a rat model. The study specifically showed that mice deficient in NF-κB p50 subunit had a lower incidence of aneurysm formation and macrophage infiltration into the aneurysm wall. Furthermore, Aoki et al.[28] showed that the up-regulation of interleukin-1β (IL-1β) and activation of NF-κB significantly reduced collagen biosynthesis, a process linked to IA progression and rupture.
A genetic approach
Progress in understanding IA pathogenesis has been slowed by its multifactorial nature. Heritable genetic variance has long been recognized as an IA risk factor. Indeed familial genetic predisposition leads to increases in IA prevalence.[29,30] The establishment of gene-prevalence and pathway-prevalence correlations may illuminate novel facets of IA pathophysiology worthy of noninvasive therapeutic intervention. This section will discuss variety recent genetic methods and their significance with respect to the inflammatory pathophysiology of IA.
The aforementioned considerations motivated the use of genome-wide association study (GWAS) to identify common genetic variants in patients who harbor IAs. Recent GWAS have identified several novel IA susceptibility loci, many unsurprisingly associated with structural genes.[31,32] Comprehensive meta-analysis of all genetic association studies (including GWASs) of sporadic IA found 19 single nucleotide polymorphism associations across the expansive international discovery cohort.[33] The strongest three of these associations were robust to sensitivity analyses for statistical heterogeneity and ethnicity.[33] Critically, this meta-analysis confirmed that the previously studied proinflammatory cytokine IL-6 G572C single nucleotide polymorphism was also statistically associated with sporadic IA.[6] However, initial sensitivity analysis revealed statistical heterogeneity among sample studies. Specifically, when one Chinese study was excluded, the association no longer held.[33] These variable results suggest more work is needed to precisely define this genetic association and role of IL-6 in IA biology.
In the genomic era, many candidate gene association studies and GWAS have identified common genetic variants that predispose individuals to IA. These methods have recently been bolstered by pathway and network-based analysis (PANOGA) of GWAS data, which ultimately highlights uncommon genetic variants in common pathologically relevant biological pathways. These approaches make use of protein-protein interaction networks. For example, such PANOGA analysis of GWAS data recently implicated pathways conserved across aneurysmal population cohorts, including (named by KEGG term) T-cell receptor signaling and chemokine signaling.[34] Such methods reinforce inflammations importance in sporadic IA formation and will play an important role in elucidating the complex genetic etiology of IA formation and rupture.
As noted above, common genetic variance only explains a fraction of the heritability in cases of complex disease. Furthermore, low-frequency predisposing genetic abnormalities are typically found in isolated populations with small founder populations, subsequent bottleneck affects, and genetic drift. One such population, with a higher rate of IA incidence, is the thoroughly characterized genetic isolate of Finland.[35] A recent study revealed four novel low-frequency risk loci utilizing an alternative genetic approach.[36] The study focused on the Finnish population, enriched the percentage of familial IA patients in the discovery sample, and increased genome-wide coverage through imputing genotyped variants against the 1000 genomes project reference panel (v3, March 2012 release).[36] The first novel variant rs74972714 at 2q23.3 is located 40 Kb downstream of LYPD6.[36] LYPD6 can inhibit transcriptional activity of the AP-1 transcription factor complex, a key activated mediator of inflammation in endothelial cells subject to atherogenic blood flow conditions.[37,38] The variant rs113816216 at 5q31.3 is located in the intron of FSTL4, a poorly characterized gene.[36] However, the FSTL4 paralog FSTL1 encodes a protein that induces innate immunity by acting as a toll-like receptor-4 (TLR-4) agonist.[39] The variant rs150927513 at 7p22.1 is located in the intron of Radil.[36] Radil has been implicated in the control of neutrophil adhesion and chemotaxis.[40] Finally, two IA associated common variants, rs12472355 and rs919433 at 2q33.1 were located 30 Kb upstream and intronic of ANKRD44, respectively.[36] ANKRD44 is a likely subunit of protein phosphotase-6 whose functions include inhibition of NF-κB activation.[41,42] Ultimately, this study suggests varied investigative methodologies may yield novel associations in diseases with complex inheritance patterns and illustrates the use of population isolates as genetic tools. Collectively the genetic analyses presented in this section further highlight inflammation’s role in IA biology.
Inflammatory gene expression profiling
Many studies have attempted to quantify characteristic gene expression patterns in IA. These potentially powerful and informative studies (as reviewed extensively by Chalouhi et al.[5]) are limited by inherent differences in cohort size, quantification techniques, status of sample aneurysms examined (onset vs. rupture), and control tissue used. As such, the results have been unsurprisingly heterogeneous. This section will discuss recent novel approaches, including a discussion of peripheral blood cell transcriptome analysis in patients with aneurysmal SAH.
To tackle the problem of biological heterogeneity among sample aneurysms, Nakaoka et al.[43] determined the gene expression levels in 8 ruptured cerebral arteries (within 24 h post SAH), 5 unruptured cerebral aneurysms, and 10 control aneurysms (superficial temporal arteries) utilizing agilent microarrays. Critically, utilizing hierarchical clustering and nonnegative matrix factorization, the samples were classified into homogeneous subgroups showing similar gene expression patterns. Focusing on ruptured IA in an early onset age (average age 46.6) sub group, and unruptured IA allowed the investigators to note expressional differences. They observed increased levels of markers for both macrophage infiltration (CD163) and oxidative stress (myeloperoxidase). Significant increases in S100/ calgranulin expression, which acts through TLR-2 to recruit macrophages/monocytes and neutrophils, were similarly reported as selectively overexpressed in early ruptured IA. TLR-6 that forms heterodimers with TLR-2, was also significantly overexpressed, ultimately suggesting this pro-inflammatory signaling pathway is involved in IA wall inflammation and rupture.[43] Finally, Krüppel-like transcription factors (KLF) KLF-2, KLF-12, and KLF-15 were all down-regulated in the early ruptured IA subgroup. KLF-2 is known to possess anti-inflammatory functionality.[44] This study was the first to show distinct gene expression differences between early and late ruptured IA. In addition, these data further implicate macrophage-mediated inflammation in IA rupture.
Differential gene expression in peripheral blood has been observed in response to both brain arteriovenous malformations and abdominal aortic aneurysms.[45,46] The first peripheral blood transcriptome analysis in patients with SAH from ruptured IA was recently carried out by Pera et al.[47] The group found that the gene expression profile of venous blood obtained prior to neurosurgical intervention differed significantly from control patients who did not harbor IA. The Illumina HumanHT-12 v4 microarrays revealed T-lymphocyte subpopulation specific transcripts were down-regulated, whereas transcripts related to neutrophils and monocytes were up-regulated. Based on these data, the authors developed a L/MN index, defined as the mean folds of standardized peripheral blood expression levels of lymphocyte related genes against expression of neutrophil and monocyte related genes. This ratio was statistically associated with clinical prognosis; the L/MN value was lower in patients who died during hospitalization compared with RA patients who survived. When blood gene expression profiles are compared with transcriptomic analyses of IA tissue, inflammation is the common denominator.[47] However, these inflammatory changes likely reflect a systemic response to ruptured IA bleeding. These data further suggest novel molecular biomarkers, which may prove beneficial for optimum management of aneurysmal SAH patients.
Direct in vivo imaging of inflammation
The recent development of novel in vivo imaging techniques directly targeting specific immune cell subsets and inflammatory enzymatic biomarkers allows clinicians to quantitatively access the inflammatory status of pathologically relevant tissue.[48] Given the role of macrophages in IA pathogenesis, several studies conducted by Hasan et al.[49-52] investigated the possibility of in vivo aneurysm wall macrophage quantification through infusion of a carbohydrate coated superparamagnetic iron oxide nanoparticle, ferumoxytol (AMAG Pharmaceuticals, Lexington, Massachusetts, USA). Ferumoxytol is cleared by macrophages in either the arterial lumen or subendothelium of IAs, thus allowing for delayed visualization of macrophage activity as a surrogate biomarker for inflammatory status of a particular lesion.[53] The optimal imaging chronology for this novel ferumoxytol enhanced MRI was found to be 5 mg/ kg ferumoxytol with imaging at 72 h postinjection.[49] A critical observation was made in a subsequent follow-up study. The authors found that the time of ferumoxytol uptake may be indicative of aneurysmal stability and rupture propensity. Those exhibiting early uptake (24 h postinjection) ruptured within 6 months.[51] Furthermore, immunostaining of surgically resected aneurysm dome tissue revealed increased expression of COX-2, mPGES, and the number of M1 variety macrophages in aneurysms with early uptake. It should be noted that this suggests imaging beyond 24 h may prove unnecessary with respect to rupture risk stratification; however, 72 h remains the optimal timing for imaging macrophages in the walls of IA’s. This time interval may still be used to access the efficacy of anti-inflammatory therapeutics.[51] Additionally, previous studies that provided evidence of inflammatory changes in ruptured aneurysms had fallen short of providing evidence of its involvement prior to rupture, rather than as a response to rupture.[5] These data, therefore, provide the first direct evidence that inflammation is a causal factor in IA rupture progression. Finally, the group was able to use this novel noninvasive technique to monitor the therapeutic effects of aspirin as it curtailed the inflammatory progression of cerebral aneurysms in human patients.[54] In this study, the findings of ferumoxytol enhanced MRI correlated well with a subsequent immunohistochemical assessment.[54] Taken together, these studies suggest that ferumoxytol enhanced MRI may indicate active inflammation in aneurysm walls and allow neurosurgeons to optimize decisions regarding intervention or observation of cerebral aneurysms.
Recent histological evidence suggests that the myeloperoxidase, a secretable oxidoreductase of azurophilic granules of polymorphonuclear cells (primarily neutrophil granulocytes), may potentially be used as a tissue-specific biomarker of inflammation and cerebral aneurysm instability.[55] Previously, Deleo et al.[56] developed a method to access MPO enzymatic activity as an inflammatory biomarker in a rabbit model of IA. The group used clinical field strength MRI and an MPO specific paramagnetic substrate, di-5-hydroxytryptamide of gadopentetate dimeglumine, as an MR contrast agent. Following endovascular injection of Escherichia coli lipopolysaccharide, which resulted in inflammatory cell infiltration into the aneurysm wall and increased active MPO expression, the investigators found the MR enhancement ratios were consistent with the inflammatory changes.[56] Ultimately, these studies suggest enzymatically specific MR imaging may help identify aneurysms with a significant rupture propensity.
These studies further highlight inflammation’s role in the progression and rupture of cerebral aneurysms. In vivo targeted molecular imaging may ultimately provide the needed noninvasive metric required for optimal management of IA. Given the strong association of inflammation and macrophage infiltration with IA rupture, IA experts have agreed on the importance of these findings and suggested that larger scale studies are needed.[53]
Therapeutics targeting inflammation
Studies focused on developing noninvasive IA therapeutics reaffirm inflammation’s pathophysiological role in IA formation and progression. Hasan et al.[57] investigated the anti-inflammatory effect of acetylsalicylic acid (aspirin) on the progression of aneurysm to rupture. A secondary analysis of the ISUIA study revealed that the aspirin decreased patients’ risk of aneurysm rupture by 60%. Furthermore, the group found ruptured aneurysms had higher levels of cyclooxygenase-2 (COX-2) and microsomal prostaglandin E2 synthase 1 (mPGES-1) expression.[58] An exploration of acetylsalicylic acid’s effect on inflammatory mediators through ferumoxytol enhanced MRI and immunostaining found aspirin-treated patients to have both decreased macrophage infiltration and COX-2 and mPGES-1 expression.[52] These pro-inflammatory enzymes were found to be overexpressed in ruptured IA tissue. Taken together, these findings suggest that low doses of aspirin (81 mg daily for 3 months) may effectively attenuate inflammation in IA, preventing acute SAH.[57]
Angiotensin 1-7 has also been explored as a potential therapeutic option as Ang 1-7 is an antagonist to Ang 2. Ang 2 has been shown to increase the expression of various pro-inflammatory cytokines as well as promote blood vessel extracellular matrix remodeling.[59] Peña Silva et al.[60] explored the therapeutic effect of Ang 1-7 in aneurysm-induced mice and found that Ang 1-7 decreased the frequency of morality and IA rupture. The authors believe that Ang 1-7 acts through a Mas receptor-dependent pathway as Ang 1-7 administration did not decrease the frequency of aneurysm rupture in Mas KO mice.[60] To investigate the applicability of Ang 1-7 as a therapeutic option, immunostaining was performed on human cerebral aneurysms to confirm Mas receptor presence. Immunostaining for Mas receptors was found to be positive in unruptured and ruptured aneurysms. The expression of Mas receptors was also identified in the intima and media layers of control human cerebral arteries.[60] These data suggest Angiotensin 1-7 mediated targeting of the Mas receptor pathway may be an efficacious noninvasive treatment modality.
Aoki et al.[27] found that in aneurysm-induced rats, the activation of NF-κB in the arterial wall of earlier stages of aneurysmal development corresponded with the expression of the downstream pro-inflammatory genes, vascular cell adhesion molecule-1 (VCAM-1) and MCP-1. The group explored the inhibitory effects of NF-κB through the use of a synthesized decoy oligodeoxynucleotide (ODN) in a rat model. Investigators found that the facilitation of ODN 1-week following aneurysmal induction inhibited VCAM-1 and MCP-1 expression and overall, reduced aneurysm size and IEL disruption.[27] In a follow-up study, the authors used chimeric decoy ODNs against both NF-κB and proinflammatory transcription factor Ets-1.[61] Aoki et al.[61] found that chimeric decoy ODNs reduced IA size and thickened the walls of existing IAs in a rat model. Rats treated with chimeric decoy ODNs also showed a reduction in MCP-1 expression and macrophage infiltration into the aneurysm wall. If nuclease resistant ODNs can be administered transorally or transvenously, these findings suggest that NF-κB and Ets-1 are both potential therapeutic targets in human IAs.[61]
Additionally, Aoki et al.[19] tested the effects of tolylsam, a selective inhibitor of the gelatinases MMP-2, MMP-9, and MMP-12, in a rat model. Facilitation of tolylsam did inhibit aneurysm progression, as the rate of advanced aneurysms in the tolylsam group was lower; however, the incidence of aneurysm development in the tolylsam group and control group was not different. The authors concluded that the tolylsam may delay aneurysm progression rather than formation.[19]
Granulocytes were found to be present in the cerebral aneurysm wall.[17] Specifically, Ishibashi et al.[17] reaffirmed mast cells’ role in aneurysm pathogenesis by administering in a rat model the mast cell inhibitor, tranilast [N-(3,4-dimethoxycinnamoyl) anthranilic acid; Kissei Pharmaceutical, Nagano, Japan] and emedastine difumarate, [1-(2-ethoxyethyl)-2-(hexahydro-4-methyl- 1H-1,4-diazepin-1-yl)-benzimidazole difumarate; Kowa, Tokyo, Japan]. The facilitation of these inhibitors proved mast cells’ contribution to IEL and media degeneration as both of these processes were suppressed.[17] Ultimately, the facilitation of these inhibitors reduced the size of the induced aneurysms and the thinning of the vessel’s media layer. Inhibitors of mast cell degranulation have shown to be useful in the treatment of other inflammatory processes such as allergies.[17] As a result, the authors believe mast cell inhibitors may prove to be a practical and safe future anti-inflammatory treatment for IAs.[17]
TNF-α-TNFR1 cascade inhibition is a strategy that has been recently explored by Aoki et al.[25] Anti-TNF-α antibody or soluble TNF receptor has been successfully used in the treatment of an inflammatory disease, such as rheumatoid arthritis.[25] The authors note that the use of these inhibitors in the treatment of aneurysms would be likely efficacious; however, the expense of these drugs makes an alternative inhibitor of the TNF-α-TNFR1 signaling cascade desirable.[25] In addition, increased levels of TNF-α in unruptured and ruptured IAs in an in vivo model were reconfirmed by Starke et al.[62] This follow-up study focused on the therapeutic strategy of the TNF-α-TNFR1 signaling cascade by analyzing TNF-α knockout mice and mice administered an inhibitor of TNF-α synthesis, 3,6’dithiothalidomide (DTH). Both groups showed a lower incidence of aneurysm formation and rupture when compared with control mice.[62] The study also found that the inhibitor DTH led to an increase in aneurysm stabilization and consequently, a decrease in aneurysm rupture upon formation.[62] Although DTH is an inhibitor of TNF-α synthesis, the authors noted that the inhibitor’s actions are not fully understood as other properties of the inhibitor may explain aneurysm stabilization and prevention of rupture.[62]
Conclusion
Intensive investigation has implicated the inflammation in the complex pathophysiological processes that underlie IA development, progression, and rupture. Ongoing research shows how these inflammatory mechanisms can be clinically accessed and therapeutically modulated. Although advances in microsurgical and endovascular management of IA will inevitably lead to lower procedural complication rates, the need for a safe and effective noninvasive therapeutic strategy to prevent aneurysmal SAH will remain. Overall, these data suggest potential alternative medical treatment strategies for patients with human cerebral aneurysms.
Acknowledgments
We would like to thank Ms. Teresa Ruggle for her assistance with the preparation of the figure.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Juvela S, Poussa K, Lehto H, Porras M. Natural history of unruptured intracranial aneurysms: a long-term follow-up study. Stroke 2013;44:2414-21.
2. Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013;35:93-112.
3. Juvela S. Prevalence of and risk factors for intracranial aneurysms. Lancet Neurol 2011;10:595-7.
4. Korja M, Lehto H, Juvela S. Response to letter regarding article, "lifelong rupture risk of intracranial aneurysms depends on risk factors: a prospective finnish cohort study". Stroke 2014;45:e211.
5. Chalouhi N, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Koch WJ, Dumont AS. Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab 2012;32:1659-76.
6. Hudson JS, Hoyne DS, Hasan DM. Inflammation and human cerebral aneurysms: current and future treatment prospects. Future Neurol 2013;8.
7. Chalouhi N, Hoh BL, Hasan D. Review of cerebral aneurysm formation, growth, and rupture. Stroke 2013;44:3613-22.
8. Tronc F, Mallat Z, Lehoux S, Wassef M, Esposito B, Tedgui A. Role of matrix metalloproteinases in blood flow-induced arterial enlargement: interaction with NO. Arterioscler Thromb Vasc Biol 2000;20:E120-6.
9. Killer-Oberpfalzer M, Aichholzer M, Weis S, Richling B, Jones R, Virmani R, Cruise GM. Histological analysis of clipped human intracranial aneurysms and parent arteries with short-term follow-up. Cardiovasc Pathol 2012;21:299-306.
10. Kosierkiewicz TA, Factor SM, Dickson DW. Immunocytochemical studies of atherosclerotic lesions of cerebral berry aneurysms. J Neuropathol Exp Neurol 1994;53:399-406.
11. Starke RM, Chalouhi N, Ding D, Raper DM, Mckisic MS, Owens GK, Hasan DM, Medel R, Dumont AS. Vascular smooth muscle cells in cerebral aneurysm pathogenesis. Transl Stroke Res 2014;5:338-46.
12. Nakajima N, Nagahiro S, Sano T, Satomi J, Satoh K. Phenotypic modulation of smooth muscle cells in human cerebral aneurysmal walls. Acta Neuropathol 2000;100:475-80.
13. Nixon AM, Gunel M, Sumpio BE. The critical role of hemodynamics in the development of cerebral vascular disease. J Neurosurg 2010;112:1240-53.
14. Nerem RM, Harrison DG, Taylor WR, Alexander RW. Hemodynamics and vascular endothelial biology. J Cardiovasc Pharmacol 1993;21 Suppl 1:S6-10.
15. Aoki T, Nishimura M, Matsuoka T, Yamamoto K, Furuyashiki T, Kataoka H, Kitaoka S, Ishibashi R, Ishibazawa A, Miyamoto S, Morishita R, Ando J, Hashimoto N, Nozaki K, Narumiya S. PGE(2) -EP(2) signalling in endothelium is activated by haemodynamic stress and induces cerebral aneurysm through an amplifying loop via NF-κB. Br J Pharmacol 2011;163:1237-49.
16. Hoh BL, Hosaka K, Downes DP, Nowicki KW, Wilmer EN, Velat GJ, Scott EW. Stromal cell-derived factor-1 promoted angiogenesis and inflammatory cell infiltration in aneurysm walls. J Neurosurg 2014;120:73-86.
17. Ishibashi R, Aoki T, Nishimura M, Hashimoto N, Miyamoto S. Contribution of mast cells to cerebral aneurysm formation. Curr Neurovasc Res 2010;7:113-24.
18. Sadamasa N, Nozaki K, Hashimoto N. Disruption of gene for inducible nitric oxide synthase reduces progression of cerebral aneurysms. Stroke 2003;34:2980-4.
19. Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke 2007;38:162-9.
20. Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, Jaaskelainen J. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke 2004;35:2287-93.
21. Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N, Lawton MT, Young WL, Liang EI, Nuki Y, Hashimoto T. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke 2011;42:173-8.
22. Hasan D, Chalouhi N, Jabbour P, Hashimoto T. Macrophage imbalance (M1 vs. M2) and upregulation of mast cells in wall of ruptured human cerebral aneurysms: preliminary results. J Neuroinflammation 2012;9:222.
23. Chalouhi N, Points L, Pierce GL, Ballas Z, Jabbour P, Hasan D. Localized increase of chemokines in the lumen of human cerebral aneurysms. Stroke 2013;44:2594-7.
24. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Egashira K, Hashimoto N. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke 2009;40:942-51.
25. Aoki T, Fukuda M, Nishimura M, Nozaki K, Narumiya S. Critical role of TNF-alpha-TNFR1 signaling in intracranial aneurysm formation. Acta Neuropathol Commun 2014;2:34.
26. Ali MS, Starke RM, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Owens GK, Koch WJ, Greig NH, Dumont AS. TNF-α induces phenotypic modulation in cerebral vascular smooth muscle cells: implications for cerebral aneurysm pathology. J Cereb Blood Flow Metab 2013;33:1564-73.
27. Aoki T, Kataoka H, Shimamura M, Nakagami H, Wakayama K, Moriwaki T, Ishibashi R, Nozaki K, Morishita R, Hashimoto N. NF-kappaB is a key mediator of cerebral aneurysm formation. Circulation 2007;116:2830-40.
28. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Morishita R, Hashimoto N. Reduced collagen biosynthesis is the hallmark of cerebral aneurysm: contribution of interleukin-1beta and nuclear factor-kappaB. Arterioscler Thromb Vasc Biol 2009;29:1080-6.
29. Nahed BV, Bydon M, Ozturk AK, Bilguvar K, Bayrakli F, Gunel M. Genetics of intracranial aneurysms. Neurosurgery 2007;60:213-25.
30. Rinkel GJ, Djibuti M, Algra A, van Gijn J. Prevalence and risk of rupture of intracranial aneurysms: a systematic review. Stroke 1998;29:251-6.
31. Bilguvar K, Yasuno K, Niemelä M, Ruigrok YM, von Und Zu Fraunberg M, van Duijn CM, van den Berg LH, Mane S, Mason CE, Choi M, Gaal E, Bayri Y, Kolb L, Arlier Z, Ravuri S, Ronkainen A, Tajima A, Laakso A, Hata A, Kasuya H, Koivisto T, Rinne J, Ohman J, Breteler MM, Wijmenga C, State MW, Rinkel GJ, Hernesniemi J, Jaaskelainen JE, Palotie A, Inoue I, Lifton RP, Gunel M. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet 2008;40:1472-7.
32. Yasuno K, Bilguvar K, Bijlenga P, Low SK, Krischek B, Auburger G, Simon M, Krex D, Arlier Z, Nayak N, Ruigrok YM, Niemela M, Tajima A, von und zu Fraunberg M, Doczi T, Wirjatijasa F, Hata A, Blasco J, Oszvald A, Kasuya H, Zilani G, Schoch B, Singh P, Stuer C, Risselada R, Beck J, Sola T, Ricciardi F, Aromaa A, Illig T, Schreiber S, van Duijn CM, van den Berg LH, Perret C, Proust C, Roder C, Ozturk AK, Gaal E, Berg D, Geisen C, Friedrich CM, Summers P, Frangi AF, State MW, Wichmann HE, Breteler MM, Wijmenga C, Mane S, Peltonen L, Elio V, Sturkenboom MC, Lawford P, Byrne J, Macho J, Sandalcioglu EI, Meyer B, Raabe A, Steinmetz H, Rufenacht D, Jaaskelainen JE, Hernesniemi J, Rinkel GJ, Zembutsu H, Inoue I, Palotie A, Cambien F, Nakamura Y, Lifton RP, Gunel M. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat Genet 2010;42:420-5.
33. Alg VS, Sofat R, Houlden H, Werring DJ. Genetic risk factors for intracranial aneurysms: a meta-analysis in more than 116,000 individuals. Neurology 2013;80:2154-65.
34. Bakir-Gungor B, Sezerman OU. The identification of pathway markers in intracranial aneurysm using genome-wide association data from two different populations. PLoS One 2013;8:e57022.
35. de Rooij NK, Linn FH, van der Plas JA, Algra A, Rinkel GJ. Incidence of subarachnoid haemorrhage: a systematic review with emphasis on region, age, gender and time trends. J Neurol Neurosurg Psychiatry 2007;78:1365-72.
36. Kurki MI, Gaál EI, Kettunen J, Lappalainen T, Menelaou A, Anttila V, van 't Hof FN, von Und Zu Fraunberg M, Helisalmi S, Hiltunen M, Lehto H, L aakso A, Kivisaari R, Koivisto T, Ronkainen A, Rinne J, Kiemeney LA, Vermeulen SH, Kaunisto MA, Eriksson JG, Aromaa A, Perola M, Lehtimaki T, Raitakari OT, Salomaa V, Gunel M, Dermitzakis ET, Ruigrok YM, Rinkel GJ, Niemela M, Hernesniemi J, Ripatti S, de Bakker PI, Palotie A, Jaaskelainen JE. High risk population isolate reveals low frequency variants predisposing to intracranial aneurysms. PLoS Genet 2014;10:e1004134.
37. Zhang Y, Lang Q, Li J, Xie F, Wan B, Yu L. Identification and characterization of human LYPD6, a new member of the Ly-6 superfamily. Mol Biol Rep 2010;37:2055-62.
38. Nigro P, Abe J, Berk BC. Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal 2011;15:1405-14.
39. Murakami K, Tanaka M, Usui T, Kawabata D, Shiomi A, Iguchi-Hashimoto M, Shimizu M, Yukawa N, Yoshifuji H, Nojima T, Ohmura K, Fujii T, Umehara H, Mimori T. Follistatin-related protein/ follistatin-like 1 evokes an innate immune response via CD14 and toll-like receptor 4. FEBS Lett 2012;586:319-24.
40. Liu L, Aerbajinai W, Ahmed SM, Rodgers GP, Angers S, Parent CA. Radil controls neutrophil adhesion and motility through ß2-integrin activation. Mol Biol Cell 2012;23:4751-65.
41. Stefansson B, Ohama T, Daugherty AE, Brautigan DL. Protein phosphatase 6 regulatory subunits composed of ankyrin repeat domains. Biochemistry 2008;47:1442-51.
42. Stefansson B, Brautigan DL. Protein phosphatase 6 subunit with conserved Sit4-associated protein domain targets IkappaBepsilon. J Biol Chem 2006;281:22624-34.
43. Nakaoka H, Tajima A, Yoneyama T, Hosomichi K, Kasuya H, Mizutani T, Inoue I. Gene expression profiling reveals distinct molecular signatures associated with the rupture of intracranial aneurysm. Stroke 2014;45:2239-45.
44. Nayak L, Lin Z, Jain MK. "Go with the flow": how Krüppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid Redox Signal 2011;15:1449-61.
45. Weinsheimer SM, Xu H, Achrol AS, Stamova B, McCulloch CE, Pawlikowska L, Tian Y, Ko NU, Lawton MT, Steinberg GK, Chang SD, Jickling G, Ander BP, Kim H, Sharp FR, Young WL. Gene expression profiling of blood in brain arteriovenous malformation patients. Transl Stroke Res 2011;2:575-87.
46. Giusti B, Rossi L, Lapini I, Magi A, Pratesi G, Lavitrano M, Biasi GM, Pulli R, Pratesi C, Abbate R. Gene expression profiling of peripheral blood in patients with abdominal aortic aneurysm. Eur J Vasc Endovasc Surg 2009;38:104-12.
47. Pera J, Korostynski M, Golda S, Piechota M, Dzbek J, Krzyszkowski T, Dziedzic T, Moskala M, Przewlocki R, Szczudlik A, Slowik A. Gene expression profiling of blood in ruptured intracranial aneurysms: in search of biomarkers. J Cereb Blood Flow Metab 2013;33:1025-31.
48. Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater 2014;13:125-38.
49. Hasan DM, Mahaney KB, Magnotta VA, Kung DK, Lawton MT, Hashimoto T, Winn HR, Saloner D, Martin A, Gahramanov S, Dosa E, Neuwelt E, Young WL. Macrophage imaging within human cerebral aneurysms wall using ferumoxytol-enhanced MRI: a pilot study. Arterioscler Thromb Vasc Biol 2012;32:1032-8.
50. Hasan DM, Amans M, Tihan T, Hess C, Guo Y, Cha S, Su H, Martin AJ, Lawton MT, Neuwelt EA, Saloner DA, Young WL. Ferumoxytol-enhanced MRI to image inflammation within human brain arteriovenous malformations: a pilot investigation. Transl Stroke Res 2012;3:166-73.
51. Hasan D, Chalouhi N, Jabbour P, Dumont AS, Kung DK, Magnotta VA, Young WL, Hashimoto T, Winn HR, Heistad D. Early change in ferumoxytol-enhanced magnetic resonance imaging signal suggests unstable human cerebral aneurysm: a pilot study. Stroke 2012;43:3258-65.
52. Hasan DM, Chalouhi N, Jabbour P, Dumont AS, Kung DK, Magnotta VA, Young WL, Hashimoto T, Richard Winn H, Heistad D. Evidence that acetylsalicylic acid attenuates inflammation in the walls of human cerebral aneurysms: preliminary results. J Am Heart Assoc 2013;2:e000019.
53. Chalouhi N, Jabbour P, Magnotta V, Hasan D. The emerging role of ferumoxytol-enhanced MRI in the management of cerebrovascular lesions. Molecules 2013;18:9670-83.
54. Hasan DM, Chalouhi N, Jabbour P, Magnotta VA, Kung DK, Young WL. Imaging aspirin effect on macrophages in the wall of human cerebral aneurysms using ferumoxytol-enhanced MRI: preliminary results. J Neuroradiol 2013;40:187-91.
55. Gounis MJ, Vedantham S, Weaver JP, Puri AS, Brooks CS, Wakhloo AK, Bogdanov AA, Jr. Myeloperoxidase in human intracranial aneurysms : preliminary evidence. Stroke 2014;45:1474-7.
56. DeLeo MJ 3rd, Gounis MJ, Hong B, Ford JC, Wakhloo AK, Bogdanov AA, Jr. Carotid artery brain aneurysm model: in vivo molecular enzyme-specific MR imaging of active inflammation in a pilot study. Radiology 2009;252:696-703.
57. Hasan DM, Mahaney KB, Brown RD Jr, Meissner I, Piepgras DG, Huston J, Capuano AW, Torner JC. Aspirin as a promising agent for decreasing incidence of cerebral aneurysm rupture. Stroke 2011;42:3156-62.
58. Hasan D, Hashimoto T, Kung D, Macdonald RL, Winn HR, Heistad D. Upregulation of cyclooxygenase-2 (COX-2) and microsomal prostaglandin E2 synthase-1 (mPGES-1) in wall of ruptured human cerebral aneurysms: preliminary results. Stroke 2012;43:1964-7.
59. Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 2007;292:C82-97.
60. Peña Silva RA, Kung DK, Mitchell IJ, Alenina N, Bader M, Santos RA, Faraci FM, Heistad DD, Hasan DM. Angiotensin 1-7 reduces mortality and rupture of intracranial aneurysms in mice. Hypertension 2014;64:362-8.
61. Aoki T, Kataoka H, Nishimura M, Ishibashi R, Morishita R, Miyamoto S. Regression of intracranial aneurysms by simultaneous inhibition of nuclear factor-κB and Ets with chimeric decoy oligodeoxynucleotide treatment. Neurosurgery 2012;70:1534-43.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Dooley SA, Hudson JS, Hasan DM. Inflammation in human cerebral aneurysms: pathogenesis, diagnostic imaging, genetics, and therapeutics. Neurosciences 2015;2:77-85. http://dx.doi.org/10.4103/2347-8659.154433
AMA Style
Dooley SA, Hudson JS, Hasan DM. Inflammation in human cerebral aneurysms: pathogenesis, diagnostic imaging, genetics, and therapeutics. Neuroimmunology and Neuroinflammation. 2015; 2: 77-85. http://dx.doi.org/10.4103/2347-8659.154433
Chicago/Turabian Style
Dooley, Sarah A., Joseph S. Hudson, David M. Hasan. 2015. "Inflammation in human cerebral aneurysms: pathogenesis, diagnostic imaging, genetics, and therapeutics" Neuroimmunology and Neuroinflammation. 2: 77-85. http://dx.doi.org/10.4103/2347-8659.154433
ACS Style
Dooley, SA.; Hudson JS.; Hasan DM. Inflammation in human cerebral aneurysms: pathogenesis, diagnostic imaging, genetics, and therapeutics. Neurosciences. 2015, 2, 77-85. http://dx.doi.org/10.4103/2347-8659.154433
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.