
Neuroprotection by minocycline in murine traumatic spinal cord injury: analyses of matrix metalloproteinases


Abstract
Aim: Minocycline has neuroprotective activities in several models of neurological disorders including spinal cord injury (SCI) where it prevents axonal loss and improves functional recovery. There are still gaps of knowledge on minocycline in SCI including whether it ameliorates neuronal loss at the focal site of trauma, and whether minocycline reduces the activity of matrix metalloproteinases (MMPs), a family of enzymes implicated in the pathophysiology of SCI. This study addressed these gaps.
Methods: Mice were treated with either minocycline or vehicle control after a spinal cord contusion. MMPs were compared between the two groups using real time polymerase chain reaction and zymography. Immunohistochemistry was used to examine microglial activation and neuronal cell death.
Results: While several MMP members were elevated in the spinal cord following injury, treatment with minocycline did not affect their expression. Importantly, minocycline reduced the loss of neurons in the epicenter of damage to the spinal cord and in segments caudal and rostral to the injury.
Conclusion: Despite the inability of minocycline to alter MMPs, the results of neuroprotection at the lesion site support the continued testing of minocycline as a neuroprotective medication in experimental and clinical SCI.
Keywords
Introduction
Minocycline, a synthetic derivative of tetracycline, has been used for the treatment of acne for several decades.It has been proposed as a promising neuroprotective agent in multiple central nervous system (CNS) pathologies including stroke, Huntington’s disease, amyotrophic lateral sclerosis and multiple sclerosis[1-3]. Minocycline has also been demonstrated to have beneficial effects in the treatment of acute spinal cord injury (SCI), a condition for which therapeutic treatments are urgently needed. Indeed, a phase 2 clinical trial administering minocycline in acute human spinal cord injury was completed in Canada with promising early results[4] and a phase 3 clinical trial is ongoing (ClinicalTrials.gov Identifier: NCT01828203). Encouragingly, the efficacy of minocycline in reducing tissue damage and in ameliorating functional impairment in SCI has been demonstrated despite variations in the models used, the species tested and the treatment paradigm[5-7]. The mechanism of action of minocycline in neurological diseases appears not to be related to its antimicrobial activity, but rather to immunomodulation, blockade of excitotoxicity and inhibition of cell death pathways[1,2,5-7], all of which have been implicated in the pathophysiology of SCI.
Minocycline has also been reported to inhibit the production and activity[8-10] of a family of proteolytic enzymes known as the matrix metalloproteinases (MMPs). A large number of MMPs have been identified and, collectively, the MMPs are capable of degrading all components of the extracellular matrix (ECM). As such, MMPs have roles in several developmental processes that involve ECM turnover as well as in wound healing throughout life. However, the aberrant expression of these molecules is detrimental to several diseases of the CNS, including SCI[11]. We have previously shown that several MMPs were upregulated after SCI in mice; in particular, we provided evidence that MMP-12 expression after SCI was linked to increased blood spinal barrier permeability, microglial activation, macrophage infiltration, and worsened functional outcome following injury[12]. In addition, Hsu et al.[13] and Noble et al.[14] demonstrated that MMP-9 limited functional recovery in the short-term following SCI in rats, although MMP-2 had an opposite outcome 7-14 days after injury.
Given that minocycline is currently in a phase 3 trial in traumatic SCI (ClinicalTrials.gov Identifier: NCT01828203), it is important to have a comprehensive picture of its spectrum of activity. Thus, in this study, we determined whether or not the efficacy of minocycline in SCI was due in part to its ability to inhibit the expression or activity of MMPs. In addition, we examined the response of microglia/macrophages following SCI and the impact of minocycline treatment. Finally, we addressed whether minocycline could protect against the death of neurons at the site of traumatic impact itself, since previous work has examined axons of passage and higher order neurons that project through the spinal cord[5]. These studies are relevant to the potential application of minocycline as a neuroprotective agent after SCI.
Methods
Surgery and minocycline treatment
CD1 outbred mice were anesthetized, the spinal cord exposed, and extradural mechanical compression of the cord with a modified aneurysm clip was achieved as previously described[5]. In brief, surgery consisted of anesthetizing mice with a mixture of ketamine/xylazine (200 and 10 mg/kg, respectively) intraperitoneally. Animals were subsequently immobilized in a stereotactic frame. An incision was made in the skin and the muscle and tissue overlying the vertebral column were blunt dissected away. Using the spiny process of T2 as a landmark, a laminectomy was performed at the level of T3/T4 and the spinal cord was exposed. A rigid hook was used to clear a path underneath the cord so that a modified aneurysm clip with a closing force of 8 g could be applied. Extradural compression of the cord was achieved by allowing the clip to slam shut on the cord producing mechanical trauma. The clip was maintained in position for one minute producing damage that also had ischemic components. Following injury the clip was removed and the wound was closed using nylon suture. Mice recovered in a room maintained at 27 ºC where they were kept thereafter for the determined survival time. Manual expression of the bladder was required twice daily and the food and water were placed directly in the cage to allow ready access.
One hour after surgery, mice were randomized and injected intraperitoneally with either a solution of minocycline at a dose of 50 mg/kg or saline vehicle control. Subsequent injections were given at 24-h intervals until sacrifice. Following the second injection of 50 mg/kg, the dose of minocycline was reduced to 25 mg/kg for all remaining treatments. This dose regimen is identical to that used in our previous study[5] that demonstrated behavioral recovery by 3 days post-injury in the minocycline compared to vehicle group.
Analyses of MMPs
TaqMan real time PCR was used to profile MMP mRNA levels as detailed[15]. Minocycline and vehicle treated mice were sacrificed 1, 2 and 5 days after injury (4/group) and 4 mice served as uninjured controls. Spinal cords were removed and a 1-cm segment including the injury site was homogenized in Trizol reagent and total RNA extracted. One microgram of RNA was reverse transcribed to make cDNA. Each PCR reaction contained the equivalent of 5 ng of reverse transcribed RNA. The 18S rRNA gene was used as an endogenous internal control to account for differences in the extraction of original tissue and reverse transcription of total RNA. Primer sequences for the TaqMan assays were previously described elsewhere[15], and statistical validity for the multiple TaqMan targets has been corroborated.
Gelatin zymography of one cm segments of spinal cord was described previously[16] as was the method of in situ zymography[17]. Note that while gelatin zymography is a technique reliant on the activity of MMP-2 and -9 to degrade gelatin in-gel, it is ultimately proportional to, and thus a reflection of, the amount of MMP-2 and -9 in the test samples.
While the above in-gel gelatin zymography is a manifestation of protein content, the net gelatinolytic activity (i.e. overall enzyme activity in a milieu that also contains enzyme inhibitors) in an intact non-fixed tissue specimen can be evaluated using in situ zymography. For this method spinal cord injury was induced in 14 mice; 7 animals were injected with 50 mg/kg of minocycline 1 h and 24 h after, while the other 7 received saline vehicle at the same time points. One hour after the second injection, mice were given an overdose of ketamine/xylazine intraperitoneally and the spinal cord was carefully removed and directly frozen in isopentane at -70 ºC. Twenty µm thick longitudinal sections were cut on a cryostat, mounted on glass slides and stored at -80 ºC. Sections were thawed and incubated in a humid chamber in 100 mL (50 µL/s) of reaction buffer containing 100 mg/mL of FITC-labeled dye-quenched-gelatin (EnzCheck collagenase kit, Molecular probes, Eugene, OR) for 3 h at 37 ºC[16,17]. At the end of the incubation period slides were rinsed in PBS, fixed and then mounted. Prepared slides were observed using fluorescence microscopy. Sections incubated without dye-quenched-gelatin did not exhibit fluorescence and served as negative controls. Samples from SCI animals were analyzed in a blinded manner using a qualitative rating scale from 0 to 4, with zero representing no fluorescence and a score of 4 depicting maximal in situ zymography signal.
Immunohistochemistry
Spinal cord compression was performed in 36 mice as described above. Mice were randomly treated with either minocycline or vehicle and survived for 2 or 5 days (n = 9/group). Animals were given an overdose of ketamine/xylazine and the spinal cords were excised and post-fixed in 10% neutral buffered formalin and subsequently embedded in paraffin wax. Six micrometer thick sections were cut on a microtome and collected at a frequency of 1:10. Three series were obtained for immunohistochemical analysis of microglia, neurons and apoptotic cells using the antibodies Iba1, NeuN (Chemicon) and the Apoptag® Fluorescein In Situ Apoptosis Detection Kit (Tunel, Chemicon). lba1 and NeuN immunolabeling to detect microglia and neurons, respectively, was performed as previously described[12]. For NeuN labeling, sections had to undergo antigen retrieval by boiling in 10 mmol/L sodium citrate buffer (pH 6.5) for 10 min. Slides were incubated with mouse anti-NeuN (1:50, Chemicon) overnight at 4 ºC. Biotinylated anti-rabbit IgG was used for the secondary antibody and staining was visualized with ABC using DAB as the substrate.
For blinded Iba1 analyses, the degree of microglial/macrophage activation was determined by examining the morphology and density of the Iba1 labeled cells. Considerations were made for the size, shape and relative density of Iba1 labeled cells. Briefly, Iba1-stained sections were scored for microglial/macrophage activation using a scale from 0-4 where 0 was normal cord and 4 was the presence of highly activated microglia/macrophages. For NeuN labeled tissue, longitudinal sections containing central canal were identified as well as sections about 180 µm away on either side of the center section (total of 3 sections/mouse). The number of positively stained cells was manually counted from 5 regions of each section corresponding to lesion area, +1 mm rostral, +2 mm rostral, +1 mm caudal and +2 mm caudal. Unbiased stereology was not used in the blinded counts. Data were analyzed using univariate analysis of variance with scheffe post-hoc comparisons. Similarly, for Tunel labeled tissue, the total number of labeled cells in a 1-cm segment of cord, from a section containing central canal and including the lesion epicenter, was counted. In this case, data was analyzed using unpaired t-tests at 2 and 5 days.
Results
Expression profile of MMP transcripts in minocycline and vehicle treated SCI animals
The expression profile of 20 MMPs was examined by TaqMan PCR in mouse spinal cord following injury. Figure 1 shows that transcripts encoding MMP-3, -7, -10, -11, -12, -13, -19, -20 and -21 were elevated after spinal cord injury compared to uninjured controls, while those of MMP-23 and -24 were reduced. Compared to vehicle treated SCI samples, minocycline administration had no significant effect on altering the MMP transcripts at 1, 2 and 5 days after injury.
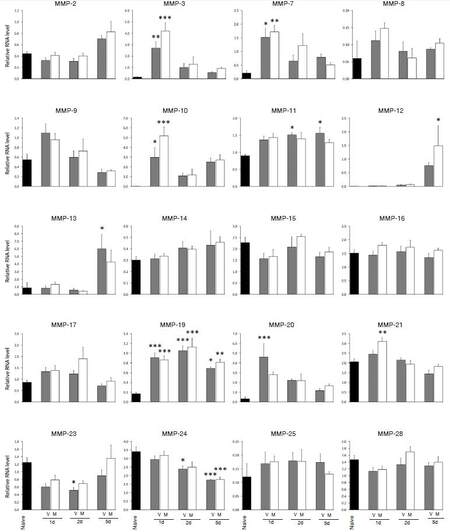
Figure 1. No impact of minocycline treatment on the profile of MMPs after SCI. While the expression of several MMPs was altered after SCI, none of these MMPs were significantly different between the vehicle and minocycline groups. Values are mean ± SEM of 4 samples from vehicle (V) or minocycline (M) group, at 1, 2 or 5 days after injury. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to naïve controls. All analyses were by one-way ANOVA with Tukey's multiple comparisons. MMPs: matrix metalloproteinases; SCI: spinal cord injury
We also measured transcripts encoding physiological antagonists of MMPs, the 4 tissue inhibitors of metalloproteinases (TIMPs). Figure 2 shows that the expression of TIMP-1, but not TIMP-2, -3 and -4, was upregulated by injury. The levels of all four TIMPs in the minocycline treated cords were not altered from those of the vehicle treatment groups, with the exception of TIMP-4 that was raised by minocycline at 1 day of injury.
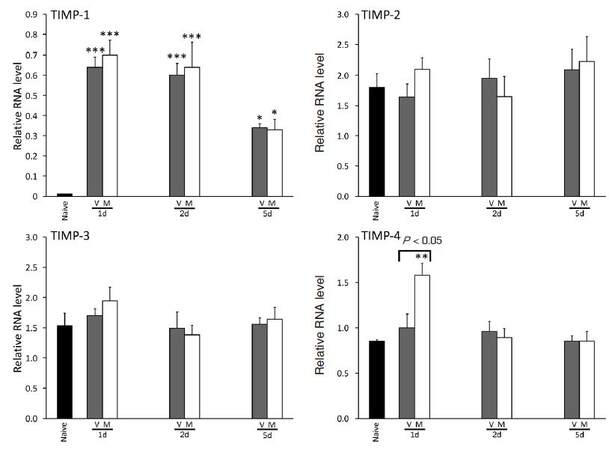
Figure 2. TIMPs remain largely unaltered by minocycline treatment after SCI. TIMP-1 was the only inhibitor that was altered (upregulated) by injury compared to naïve controls, and there was no effect of minocycline treatment with the exception of an increase of TIMP-4 at 1 day after SCI. Values are mean ± SEM of 4 samples from vehicle (V) or minocycline (M) group, at 1, 2 or 5 days after injury. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to naïve controls (one-way ANOVA with Tukey's multiple comparisons). TIMPs: tissue inhibitors of metalloproteinases; SCI: spinal cord injury
Gelatin zymography shows that minocycline does not alter MMP-2 and -9 protein content
Measurement of transcripts encoding MMPs provides a broad overview of the changes occurring to all known MMP members, since several individual MMPs continue to be difficult to measure using Western blot or activity assays. A reproducible and commonly used method for protein levels involves the determination of MMP-2 and MMP-9 by the method of gelatin zymography. Although we did not detect the elevation of their transcripts after SCI [Figure 1], MMP-9 protein can be made outside of the CNS, including in neutrophils, and then deposited into the lesion site. For these reasons, we examined the levels of MMP-2 and -9 protein using gelatin zymography. The pro-MMP-9 protein was minimally expressed in control uninjured spinal cord tissue [Figure 3A]. Injury resulted in an upregulation of both the pro- and active forms of MMP-9 one day after injury. Minocycline treatment had no obvious effect on the injury-induced MMP-9. The pro-MMP-2 species did not elevate profoundly after injury or in the presence of minocycline. The quantification of the gelatin zymograms is displayed in Figure 3B and confirms the lack of effect of minocycline on MMP-2 and -9 protein expression after SCI.
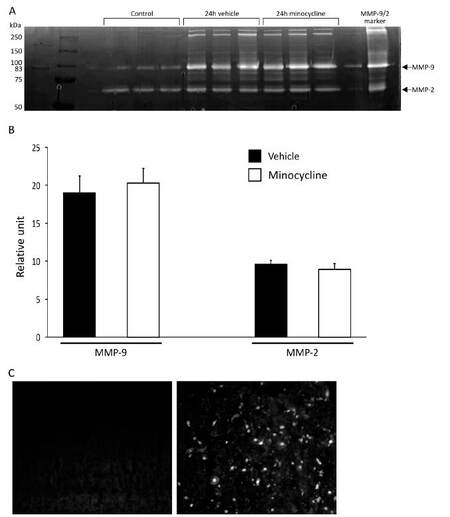
Figure 3. Increased protein levels of MMP-9 after SCI are unaltered by treatment with minocycline. A: Gelatin zymogram displaying pro-MMP-2 and -9 protein species (designated in figure by MMP-2 and MMP-9, respectively) in spinal cords from uninjured control mice; after injury, additional bands running between pro-MMP-2 and pro-MMP-9 represent the activated forms of MMP-9. Minocycline treatment did not markedly change the expression pattern of MMP-2 or -9 levels after SCI (A) and this was corroborated by quantification of band densities through densitometry (n = 6 each) from the zymogram (B); C: in situ zymography of a longitudinal section from non-injured (left, slide has been over-exposed to confirm the lack of signal) or vehicle-treated mouse (right, 2 days after injury). In situ zymography signal at the lesion site (one longitudinal section containing the central canal per mouse, n = 7 per group) is not different between vehicle- and minocycline-treated mice in blinded analyses (see text). MMPs: matrix metalloproteinases; SCI: spinal cord injury
In situ zymography reveals no alterations of net proteolytic activity by minocycline
Although gelatin zymography [Figure 3A] is based on the degradation of gelatin in-gel by MMP-2 and -9, it is a reflection of the content of these gelatinases (MMP-2 or -9) rather than their net enzymatic activity, given that all enzyme co-factors are supplied in optimal amounts for manifestation of catalysis in-gel. In addition, while the mRNA for MMPs are elevated in SCI [Figure 1], so are the transcripts for the TIMP inhibitors [Figure 2], thus making it relevant to address the balance of proteolytic activity in specimens. To determine the net proteolytic activity existent in the injured cord, in situ zymography was used, whereby the gelatin-FITC substrate was overlaid onto longitudinal unfixed sections encompassing the injured area[17], and where if there is relative abundance of enzyme over inhibitors, then there would be net proteolysis of the gelatin-FITC. In normal uninjured cord, no fluorescence signal was evident. Twenty-four hours following SCI, varying grades of signals were observed [Figure 3C]. Blinded analyses across 14 injured specimens concluded that there was no difference in net proteolysis around the lesion site between vehicle and minocycline samples (1.6 ± 0.3 and 2.0 ± 0.5 graded scores, respectively) (mean ± SEM, P > 0.05, Mann-Whitney non-parametric test).
Minocycline reduces microglial reactivity at the site of injury
Iba1 immunoreactivity was performed on spinal cord sections to assess the degree of microglial/macrophage reactivity. Figure 4 displays Iba1 labeling in the spinal cord of normal uninjured mice (A, B), or in vehicle (C, D) or minocycline treated (E, F) mice after SCI. The Iba1 antibody does not discriminate between microglia and macrophages, so positive cells are collectively referred to as microglia/macrophages. The density of Iba1 positive cells adjacent to the epicenter of injury was prominently increased after SCI (C, D) compared to normal uninjured conditions, and this appeared qualitatively to be reduced in the minocycline treated samples (E, F). Furthermore, the morphology of microglia/macrophages in vehicle treated mice (D) was indicative of highly activated cells with amoeboid morphology and thick stubby processes, contrasting the highly ramified morphology of microglia in the normal cord [Figure 4B]. In contrast, minocycline treated mice had microglia/macrophage morphology that was intermediate between these extremes (F). As the morphological transformation of microglia from ramified to amoeboid forms is indicative of their increasing state of activation, these results suggest that minocycline reduced microglia/macrophage activation after injury.
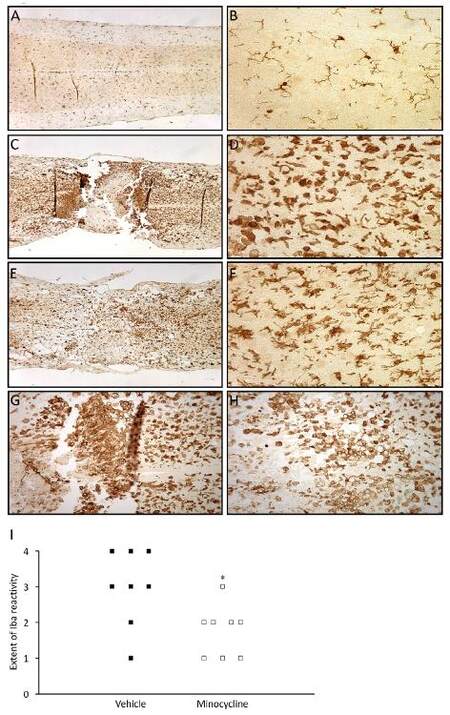
Figure 4. Microglial/macrophage activation and infiltration is reduced in minocycline treated mice. Microglia/macrophages were visualized using Iba1 immunostaining 5 days after injury. A comparison of representative sections from normal uninjured mice (A, B) and SCI mice treated with vehicle (C, D) reveals that there was increased density of cells in the SCI tissue, as well as a more amoeboid morphology characteristic of microglial/macrophage activation after SCI (D). In contrast, in minocycline treated mice (E, F), the density of microglia/macrophages appeared to be qualitatively reduced compared to vehicle controls. In minocycline treated mice (F), the microglia, while demonstrating morphological changes indicative of activation (i.e. shortened and thickened processes), did not progress as much as in the vehicle animals (D). Finally, within the epicenter of the injury in vehicle and minocycline treated animals (G and H respectively), there was an increased number of Iba1 labeled cells displaying an amoeboid morphology, with a greater density of these cells in the vehicle treated mice compared to the minocycline group. Iba1 stained sections were scored blinded for microglial/macrophage activation using a scale from 0-4, where 0 was normal cord and 4 indicated the presence of highly activated microglia/macrophages. There was a significant difference (*P < 0.05, Mann Whitney U test) in the morphology and density of Iba1 labeled cells in minocycline treated mice after SCI compared to vehicle (I, where each point represents one longitudinal section containing the central canal per mouse). SCI: spinal cord injury
We also determined the representation of microglia/macrophages at the epicenter of injury. We found that the density of Iba1 labeled cells was qualitatively lower in minocycline treated mice (H) than in vehicle controls (G), even though the morphology of cells, with the majority being amoeboid, did not differ between the 2 groups. To quantitate the extent of microglial/macrophage reactivity encompassing the lesion and remote areas, Iba1 immunoreactivity was scored by three independent observers blinded to treatment according to previously published methods[12]. Agreement between observers was good in large part and the identical result of 2 reviewers was noted as the score for a particular section. The blinded assessments [Figure 4I] indicated that there was a significant difference in Iba1 immunoreactivity between minocycline and vehicle treated mice 5 days after injury (P < 0.05, Mann Whitney U test).
Minocycline decreases apoptotic cell death as revealed by Tunel labeling
The number of Tunel positive cells was counted in sections of spinal cords taken from mice at 2 and 5 days after injury. The data shows cell death occurring at 2 and 5 days,and that minocycline treatment reduced the number of Tunel positive cells at the latter time point [Figure 5]. We did not address whether the Tunel positive cells were oligodendrocytes and/or neurons, and whether there is preferential rescue of one cell type versus another by minocycline.
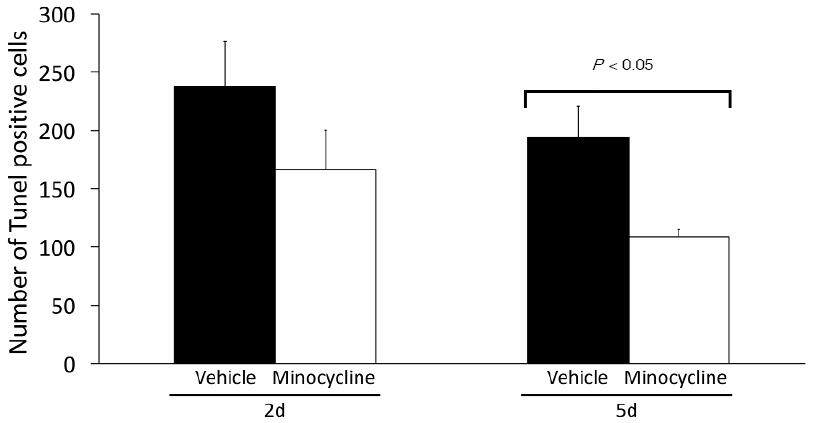
Figure 5. Minocycline treatment reduces the number of cells undergoing apoptotic cell death. Tunel positive cells were counted in sections from vehicle and minocycline treated mice harvested at 2 and 5 days post SCI. Cell death was evident 2 days after injury and was still ongoing at 5 days. The number of Tunel positive cells counted from the minocycline group was significantly lower than the vehicle group at 5 days post SCI (Student's t-test, *P < 0.05, n = 9/group, one longitudinal section containing the central canal per mouse). SCI: spinal cord injury
Minocycline spares neurons at the lesion site after spinal cord injury
Figure 6 displays NeuN labeling in the normal uninjured spinal cord (A, B), in vehicle treated cord at 5 days after injury (C, D), and in minocycline treated mice at comparable points (E, F). While neurons were lost after SCI, there appears to be more neurons preserved in the minocycline versus the vehicle group. To confirm this, blinded counts were taken from the lesion epicenter and areas both rostral and caudal to the injury site. Univariate analysis of variance revealed that there was a significant area effect at 2 and 5 days (P < 0.001 for both), where SCI resulted in reduced neuron numbers, with the greatest loss occurring at the lesion site and with more spared neurons as the distance from the epicenter increased rostrally and caudally (lesion site significantly different from areas 1 and 2 mm rostral and caudal from the lesion epicenter, P < 0.001) [Figure 6]. Furthermore, while the same pattern of loss was present in minocycline treated animals, there was greater preservation of neurons after SCI when compared to vehicle controls (P < 0.001 for both 2 and 5 days) [Figure 6].
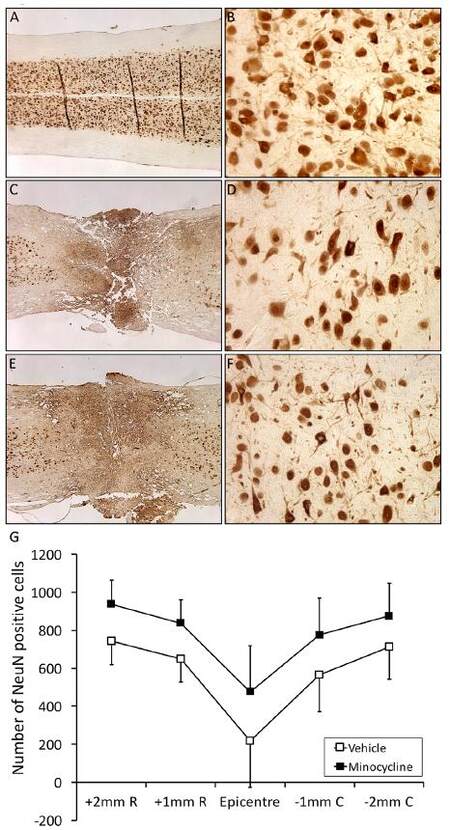
Figure 6. Minocycline treatment increases neuronal survival at the site of SCI. Panels A and B are representative pictures taken from normal uninjured cord immunostained with the neuronal marker, NeuN. Spinal cord compression produces injury at the lesion site that is characterized by a qualitative decrease in the number of NeuN positive cells (C and D, day 5). Minocycline treatment appears qualitatively to preserve some of the neurons within the vicinity of the lesion site (E and F), which was verified by blinded counts of NeuN-positive cells at the lesion epicenter as well as in areas 1 and 2 mm rostral (R) and caudal (C) to the lesion (G, mean ± SD). Panel G shows the data from day 2, and a similar pattern was found at day 5 after injury (data not shown); there were 9 mice per group, where one longitudinal section containing the central canal per mouse was examined. Univariate analysis of variance with scheffe post-hoc comparisons revealed that the difference associated with area (-2, -1, 0, +1 or +2) was significant (P < 0.001), with the number of cells remaining in the lesion epicenter being reduced compared to adjacent regions. Furthermore, there was a significant group effect with minocycline treated animals having more remaining neurons than vehicle treated controls (P < 0.001). SCI: spinal cord injury
Discussion
MMPs are implicated in neural cell death. The application of MMP-1 and -9 to neurons in culture results in their demise[17]. In animal models of SCI, MMP-12 transcripts were upregulated 189 fold over basal levels, and functional recovery was significantly improved in MMP-12 null mice compared to wild-type controls[12]. Similarly, a role for MMP-9 in SCI was demonstrated in a study by Noble et al.[14] who showed that MMP-9 was upregulated following a contusion injury and that this was correlated with a reduction in blood spinal barrier integrity and the recruitment of inflammatory neutrophils. These authors also showed that functional recovery from SCI was significantly improved in MMP-9 null mice compared to wild-type controls. In correspondence with these results, inhibitors of MMPs applied acutely following SCI improved histological and functional outcomes[14]. Nonetheless, it must be borne in mind that MMPs also have beneficial functions in the CNS, and that the prolonged usage of MMP inhibitors can impair recovery[18,19].
Minocycline has documented MMP inhibitory activities[8-10]. In a model of ischemia in mice, minocycline reduced injury in wild-type, but not MMP-9 null mice, implicating MMP-9 as a target of minocycline activity in that condition[20]. Thus, it was reasonable to address whether or not the application of minocycline to mice subjected to SCI would result in reduced expression or activity of MMPs at lesion sites. Our overall results, however, do not support a mode of action of minocycline in acute SCI involving MMPs. We did not find reduced expression of transcripts encoding most MMPs in the spinal cord of mice afflicted with SCI and given minocycline. Also, MMP-2 and -9 protein levels were unaltered in the spinal cord of mice administered minocycline. Furthermore, we used in situ zymography to determine net proteolytic activity in spinal cord sections and we found no alterations in minocycline versus vehicle treated controls. It is probable that the acute administration of minocycline did not result in concentrations that were high enough to alter MMP activity and levels.
Despite the lack of effect of minocycline on MMPs, the medication did produce neuroprotective effects exemplified by the NeuN data. Previously we have demonstrated that minocycline produces robust neuroprotection in this model of spinal cord compression in the mouse, significantly decreasing lesion size and improving recovery in hindlimb function as assessed by the BBB scale in open field testing and the inclined plane task[5]. Furthermore, in that study, there were indications of white matter sparing determined by Bielchowsky silver stain and retrograde labeling of brain stem neurons by the fluorogold tracer[5]. The current data confirms that there is preservation of the gray matter at the level of the impact injury as well. While several groups have reported improved outcomes from spinal cord injury when minocycline is administered[5-7], these studies have not comprehensively evaluated the survival of neurons throughout the gray matter at the level of injury. Taken together, the existing body of research indicates that minocycline has a global neuroprotective effect.
One reason that minocycline is such an attractive pharmacological agent for neurological disorders is that it has multiple mechanisms of action targeting separate pathological processes simultaneously. Besides reducing inflammatory activity of microglia and T cells[1], the over-activation of which contributes to neural cell death, minocycline reduces toxicity to oligodendrocytes and neurons through potentially direct mechanisms. In this regard, minocycline inhibits the activity of caspases[21,22] and the release of cytochrome c from mitochondria[23], which are both apoptosis-inducing events. Minocycline reduces signaling of the p38 mitogen activated kinase pathway[6,24], and it prevents the activation of poly (ADP-ribose) polymerase[25,26], actions that contribute to the alleviation of neural cell death. This drug has also been shown to decrease apoptosis of oligodendrocytes through a mechanism involving the inhibition of proNGF production by microglia[27]. Minocycline has been reported to reduce glutamate excitotoxicity[28,29], to detoxify free radicals that contribute to neurotoxicity[30,31] and to inhibit lipid peroxidation[32].
A limitation of the present study is that we did not perform neurobehavioral studies to accompany the histological and MMP results. However, the dose regimen employed is identical to that used in our previous study[5] that demonstrated behavioral recovery by 3 days post-injury in the minocycline compared to vehicle group. Another limitation is that for the majority of MMPs examined in the current study, only gene expression and not protein amount or activity was measured. However, protein levels of MMPs-2 and 9 were measured using gel zymography, and net proteolytic activity for these two molecules were examined with in situ zymography. Despite this limitation, the transcript expression pattern of MMPs provides valuable information in understanding the mechanisms by which minocycline may exert its effects after SCI.
In conclusion, the novel findings are that minocycline confers protection to neurons at the site of SCI, and that this does not involve the alteration of most MMPs. Given the neurotoxicity that can be inflicted by MMPs acutely after SCI, and the apparent lack of minocycline effect on most MMPs and TIMPs in this study, our results suggest that the combined treatment of minocycline and a specific MMP inhibitor may result in greater recovery than either treatment alone. Nonetheless, even without MMP inhibitory activity acutely after SCI, the myriad of mechanisms attributed to minocycline as aforementioned would position minocycline for further study as a neuroprotective medication after SCI in humans.
Declarations
Authors’ contributionsConducted experiments, and provided results: T. Rice, J.E.A. Larsen, H. Li, P.H. Larsen, R.K. Nuttall
Co-wrote the first draft: T. Rice, J.E.A. Larsen
Edited the manuscript: T. Rice, R.K. Nuttall, S. Casha, J. Hurlbert, D.R. Edwards
Co-supervised aspects of the project: S. Casha, J. Hurlbert, D.R. Edwards
Provided overall supervision of the project and edited and finalized the manuscript: V.W. Yong
Financial support and sponsorshipThis work was supported by an operating grant from the Canadian Institutes of Health Research to VWY (MOP 133477).
Conflicts of interestThe authors have no conflicts of interest to declare.
Patient consentNot applicable.
Ethics approvalAll studies involving animals were reviewed and approved by ethics committee at the University of Calgary, Canada.
Copyright© The Author(s) 2017.
REFERENCES
1. Yong VW, Wells JEA, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol 2004;3:744-51.
2. Stirling DP, Koochesfahani KM, Steeves JD, Tetzlaff W. Minocycline as a neuroprotective agent. Neuroscientist 2005;11:308-22.
3. Kwon BK, Okon E, Hillyer J, Mann C, Baptiste D, Weaver LC, Fehlings MG, Tetzlaff W. A systematic review of non-invasive pharmacologic neuroprotective treatments for acute spinal cord injury. J Neurotrauma 2011;28:1545-88.
4. Casha S, Zygun D, McGowan MD, Bains I, Yong VW, Hurlbert RJ. Results of a phase II placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain 2012;135:1224-36.
5. Wells JE, Hurlbert RJ, Fehlings MG, Yong VW. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain 2003;126:1628-37.
6. Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci 2004;24:2182-90.
7. Festoff BW, Ameenuddin S, Arnold PM, Wong A, Santacruz KS, Citron BA. Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. J Neurochem 2006;97:1314-26.
8. Brundula V, Rewcastle NB, Metz LM, Bernard CC, Yong VW. Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain 2002;125:1297-308.
9. Ryan ME, Usman A, Ramamurthy NS, Golub LM, Greenwald RA. Excessive matrix metalloproteinase activity in diabetis: inhibition by tetracycline analogues with zinc reactivity. Curr Med Chem 2001;8:305-16.
10. Paemen L, Martens E, Norga K, Masure S, Roets E, Hoogmartens J, Opdenakker G. The gelatinase inhibitory activity of tetracyclines and chemically modified tetracycline analogues as measured by a novel microtiter assay for inhibitors. Biochem Pharmacol 1996;52:105-11.
11. Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2001;2:502-11.
12. Wells JE, Rice TK, Nuttall RK, Edwards DR, Zekki H, Rivest S, Yong VW. An adverse role for matrix metalloproteinase 12 after spinal cord injury in mice. J Neurosci 2003;23:10107-15.
13. Hsu JY, McKeon R, Goussev S, Werb Z, Lee JU, Trivedi A, Noble-Haeusslein LJ. Matrix metalloproteinase-2 facilitates wound healing events that promote functional recovery after spinal cord injury. J Neurosci 2006;26:9841-50.
14. Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci 2002;22:7526-35.
15. Nuttall RK, Pennington CJ, Taplin J, Wheal A, Yong VW, Forsyth PA, Edwards DR. Elevated membrane-type matrix metalloproteinases in gliomas revealed by profiling proteases and inhibitors in human cancer cells. Mol Cancer Res 2003;1:333-45.
16. Larsen PH, Wells JE, Stallcup WB, Opdenakker G, Yong VW. Matrix metalloproteinase-9 facilitates remyelination in part by processing the inhibitory NG2 proteoglycan. J Neurosci 2003;23:11127-35.
17. Xue M, Hollenberg MD, Yong VW. Combination of thrombin and matrix metalloproteinase-9 exacerbates neurotoxicity in cell culture and intracerebral hemorrhage in mice. J Neurosci 2006;26:10281-91.
18. Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci 2005;6:931-44.
19. Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med 2006;12:441-5.
20. Koistinaho M, Malm TM, Kettunen MI, Goldsteins G, Starckx S, Kauppinen RA, Opdenakker G, Koistinaho J. Minocycline protects against permanent cerebral ischemia in wild type but not in matrix metalloprotease-9-deficient mice. J Cereb Blood Flow Metab 2005;25:460-7.
21. Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 2000;6:797-801.
22. Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol 2002;52:54-61.
23. Zhu S, Stavrovskaya I, Drozda M, Kim B, Ona V, Li M, Sarang S, Liu A, Hartley D, Wu D, Gullans S, Ferrante R, Przedborski S, Kristal B, Friedlander R. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002;417:74-8.
24. Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A 2001;98:14669-74.
25. Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol 2002;52:54-61.
26. Alano CC, Kauppinen TM, Valls AV, Swanson RA. Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc Natl Acad Sci U S A 2006;103:9685-90.
27. Yune TY, Lee JY, Jung GY, Kim SJ, Jiang MH, Kim YC, Oh YJ, Markelonis GJ, Oh TH. Minocycline alleviates death of oligodendrocytes by inhibiting pro-nerve growth factor production in microglia after spinal cord injury. J Neurosci 2007;27:7751-61.
28. Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl D-aspartate neurotoxicity by inhibiting microglia. J Immunol 2001;166:7527-33.
29. Darman J, Backovic S, Dike S, Maragakis NJ, Krishnan C, Rothstein JD, Irani DN, Kerr DA. Viral-induced spinal motor neuron death is non-cell-autonomous and involves glutamate excitotoxicity. J Neurosci 2004;24:7566-75.
30. Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J Neurochem 2005;94:819-27.
31. Morimoto N, Shimazawa M, Yamashima T, Nagai H, Hara H. Minocycline inhibits oxidative stress and decreases in vitro and in vivo ischemic neuronal damage. Brain Res 2005;1044:8-15.
32. Sonmez E, Kabatas S, Ozen O, Karabay G, Turkoglu S, Ogus E, Yilmaz C, Caner H, Altinors N. Minocycline treatment inhibits lipid peroxidation, preserves spinal cord ultrastructure, and improves functional outcome after traumatic spinal cord injury in the rat. Spine (Phila Pa 1976) 2013;38:1253-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Rice T, Larsen JEA, Li H, Nuttall RK, Larsen PH, Casha S, Hurlbert J, Edwards DR, Yong VW. Neuroprotection by minocycline in murine traumatic spinal cord injury: analyses of matrix metalloproteinases. Neurosciences 2017;4:243-53. http://dx.doi.org/10.20517/2347-8659.2017.51
AMA Style
Rice T, Larsen JEA, Li H, Nuttall RK, Larsen PH, Casha S, Hurlbert J, Edwards DR, Yong VW. Neuroprotection by minocycline in murine traumatic spinal cord injury: analyses of matrix metalloproteinases. Neuroimmunology and Neuroinflammation. 2017; 4: 243-53. http://dx.doi.org/10.20517/2347-8659.2017.51
Chicago/Turabian Style
Rice, Tiffany, Jennifer E.A. Larsen, Hui Li, Robert K. Nuttall, Peter H. Larsen, Steven Casha, John Hurlbert, Dylan R. Edwards, V. Wee Yong. 2017. "Neuroprotection by minocycline in murine traumatic spinal cord injury: analyses of matrix metalloproteinases" Neuroimmunology and Neuroinflammation. 4: 243-53. http://dx.doi.org/10.20517/2347-8659.2017.51
ACS Style
Rice, T.; Larsen JEA.; Li H.; Nuttall RK.; Larsen PH.; Casha S.; Hurlbert J.; Edwards DR.; Yong VW. Neuroprotection by minocycline in murine traumatic spinal cord injury: analyses of matrix metalloproteinases. Neurosciences. 2017, 4, 243-53. http://dx.doi.org/10.20517/2347-8659.2017.51
About This Article
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.