
Interactions between neurotropic pathogens, neuroinflammatory pathways, and autophagic neural cell death


Abstract
In recent times, there has been a significant increase in studies focusing on immunological functions of autophagy, however, knowledge of its roles and regulations in the central nervous system remains unclear. Present reviews highlight the molecular cross talk between host cell autophagy with inflammatory pathways in the context of neuro-infections. Intracellular pathogens might have an ability to manipulate the autophagy regulation process. An augmented autophagy and inflammation at the site of infection is traditionally considered host protective. Moreover, host cell autophagy might also facilitate pathogen survivability and multiplication in the brain environment. Consequently, an excessive autophagy and neuroinflammatory process do put surrounding healthy brain tissue at risk of pathogen invasion. The question arises, whether there are any known direct interactions of intracellular neurotropic pathogens with this degradative pathway that favour intracerebral pathogen survival and growth? It is worth exploring any such cooperation between pathogen factors and altered immune pathways that modulate autophagy regulatory genes causing massive neuronal damage. A detailed understanding of molecular mechanisms in microbial pathogenesis, neuroinflammatory and neuronal autophagy pathways might identify novel therapeutic targets and diagnostic biomarkers.
Keywords
Introduction
Neurotropism of certain microbial pathogens could lead to neurological health problems in humans. It is suggested that chronic infections in the central nervous system (CNS) might be associated with progressive neurodegeneration and/or neurobehavioral abnormalities[1-4]. Prolonged CNS infections with neurotropic pathogens, along with other underlying conditions such as autoimmune responses, vascular diseases, head injury, cerebral edema, changes in neurotransmitter concentrations, nutritional deficiencies, heavy metal poisoning, and effects of environmental toxins, might play roles in the pathogenesis of neurodegenerative and/or neurobehavioral diseases[5-8]. However, the role of any CNS infections in specific pathogenesis of neuronal cell death has been not yet been conclusively established.
Intracellular pathogens can invade host cells, protecting them from the host’s systematic immune response[9]. However, they face a serious challenge within the infected cell, posed as competent hosts possess innate immune mechanisms specifically tailored to eradicate foreign invaders. To combat this, infectious agents follow one or more strategies in order to avoid host immune attack. These sophisticated strategies include: lysis and escape from the phagocytic compartment; modifying the phagosome microenvironment; surviving in acidic compartment. Some pathogenic agents might use more than one of the above strategies in order to successfully survive within the host cell[9].
Presently, it is important to highlight interactions between the neurotropic pathogens and the host’s immune system in the brain micro environment to understand the mechanism of pathogen survival mechanisms, and also the roles of host and pathogen factors in the pathogenesis of neurodegenerative and/or neurobehavioral diseases. Thus, it is interesting to revisit and update on innate immunity in the CNS, and also to further discuss the neuroinflammatory pathways which contribute to massive neurodegeneration in CNS infections by many different neuropathogens.
Recent studies have shown that pathogen induced autophagy negatively influences its replication, as shown in the case of Japanese encephalitis virus (JEV) infected mouse neuronal cells; autophagy has been found to delay virus-induced cell death[10]. On the other hand, for some neuropathogens (Toxoplasma gondii and other related parasites), an intrinsic role for autophagy has been identified in persistent infections[11].
Besides autophagy being an intrinsic cellular defense mechanism against invading microorganisms, it can also appear to be linked to a number of other human disorders, such as autoimmune diseases, sterile inflammation, and even neoplasms[12]. Moreover, knowledge of its roles and regulations in the central nervous system remains unclear[13].
The main purpose of the present review is to find out possible interactions of neurotropic intracellular pathogens with neuronal cell autophagic process favoring the pathogen(s) for their intracerebral survival, as well as multiplication in CNS infectious diseases.
Immune responses of the central nervous system
The innate immunity of the CNS includes complex signaling pathways and a network of cells. Previously the brain was considered to be an immunologically privileged site in the human body. However, it is now known to have the ability to synthesize and release various active mediators, and a few pro-inflammatory molecules. The human brain also has the capacity to respond to any injury or insult, employing anti-inflammatory and/or pro-homeostatic mechanisms. Researchers at the University of Virginia, School of Medicine have discovered the presence of lymphatic vessels in the brain which connect the brain to body’s immune system[14].
A major part of CNS immunity revolves around the temporal relationship between cellular injury and the inflammatory response. Acute inflammation is the immediate response that occurs upon commencement of any injury or insult. The blood components, such as polymorphonuclear leukocytes from peripheral circulation, have access to the CNS when the blood brain barrier (BBB) is compromised. In addition, with the latest discovery of lymphatic vessels between the meninges and skull bones, inflammatory mediators may have easier access to brain tissues. Even though these cells act as a defense system, they could contribute to cellular damage when excessively deployed in the interstitial space. Inflammation in the brain becomes chronic, and often pathological, when usually acute inflammation ceases in a short period and does not contribute to repair.
The microglia activation mechanism is an important determinant in the protection of the neural parenchyma in response to various infections, neuroinflammation, stroke, tumors, trauma, and neurodegenerative diseases[15]. Through a variety of mechanisms, activated microglia are the first cells in the CNS to respond to neuronal damage; they are usually able to exert two opposing functions both promoting neuronal regeneration, and killing neurons[15,16]. Exerting either function is largely determined by the particular conditions that evoke microglial activation[16]. However, the specific nature of any such constructive or destructive mechanisms remains nebulous.
Activated microglial cells release a number of cytotoxic molecules in vitro, (i.e. proteases, reactive oxygen intermediates, NO, cytokines, arachidonic-acid derivatives, excitatory amino acids, and quinolinic acid)[17-19]. HIV-infected mononuclear cells are known to produce low molecular weight neurotoxins, possibly causing neuronal damage via N-methyl-D-aspartate receptors[20]. The cytokine tumor necrosis factor-α (TNF-α) in the CNS produced by microglial cells could cause bystander damage during the demyelination process. Moreover, the free oxygen radicals released by microglia have a direct toxic effect, as evident in co-cultures of neurons and microglia[21]. Mostly information on the cytotoxic properties of activated microglia are obtained from in vitro cultures, and has not yet been replicated in vivo. Furthermore, the cytotoxic properties of microglial cells are subject to considerable species variation; NO production is established for rat microglia, but in humans astrocytes might contribute to NO synthesis in addition to microglia[22].
Autophagy and its regulating factors under physiologic conditions
Autophagy is a cellular process that facilitates delivery of cytoplasmic constituents of eukaryotic cells for lysosomal degradation for nutrients recycling, and survival during starvation[23]. Physiologically, autophagy removes damaged or obsolete intracellular organelles. Meanwhile, it protects the body against microbial invasion by eliminating intracellular pathogens[24].
Autophagy is induced by both metabolic and immune signals, comprised of pathogen recognition and proinflammatory cytokine mediated stimulation[25].
There are three different types of autophagy known to occur in mammalian cells. (1) Macroautophagy-which relies on cytosolic double-membrane vesicle (autophagosomes) formation de novo, to sequester and transport cargo to the lysosome. (2) Chaperone-mediated autophagy-where individual unfolded proteins are transported directly across the lysosomal membrane. (3) Microautophagy- involving the direct uptake of cargo through invagination of the lysosomal membrane. All these types of autophagy ultimately lead to degradation of cargo and release of the breakdown products back into the cytosol for reuse by the cell[26].
The degradative autophagy pathway is activated upon starvation, mediated through a protein kinase (Tor)[27]. This kinase might also inhibit the autophagy pathway, either by acting in a signal transduction cascade through various downstream effectors, or by causing the ATG13 hyperphosphorylation[28]. Phosphorylated ATG13 has a lower affinity for the ATG1 kinase, and thereby a reduced interaction might inhibit the autophagy process[29]. The Gcn2, along with its targets, eIF2α, and the Gcn4 transcriptional transactivator proteins in the downstream, might also play a role in induction of autophagy[30].
Previously, studies on autophagy have been focused on its occurrences and association with nutrient deprivation, as well as age-associated issues. However, recently, there has been a significant increase in studies focusing on immunological functions of autophagy[12]. In the context of infection, immunity, and neurodegeneration, autophagy seems to have a pivotal role in neuronal homeostasis[31]. Its dysfunction has been linked to several neurodegenerative diseases such as Parkinson’s, Huntington’s, and Alzheimer’s diseases. As the first line of defense in brain, the autophagic pathway is known to be involved in both physiological and pathological processes. However, its immunological role in the CNS environment is not yet clearly studied.
Interplay between inhibitory cytokines and the autophagy process has recently been reported, which reveals a novel mechanism by which autophagy could control the immune response. Interactions between autophagy and the regulatory cytokines IL-10, transforming growth factor-β, and IL-27 are evident from earlier studies[12]. IL-37 and IL-35 are two newly discovered anti-inflammatory cytokines. IL-37 inhibits the production of pro-inflammatory cytokines in response to inflammation[32]. IL-35 suppresses T cell proliferation predominantly, and thereby inhibits its effector functions[33]. The possible interactions between above two anti-inflammatory cytokines, and autophagy have also been recently[12]. However, no such interaction in the context of the CNS environment is discussed in literature.
Autophagy induction and its alteration following infection
Accumulating evidence demonstrates that autophagy plays a protective role against infectious diseases by diminishing intracellular pathogens, including bacteria, viruses, and parasites. The following section will summarize the interplay between autophagy induction and infection due to various microbial agents.
Several intracellular bacterial agents, such as Anaplasma phagocytophilum, Brucella abortus, Coxiella burnetii, Legionella pneumophila, and Staphylococcus aureus, have the potential to alter the autophagic pathway to their own advantage[9]. These bacterial agents might stimulate their uptake and internalization into autophagosomes by secretion of special effector molecules[34]. Moreover, these pathogens seem to be efficiently grown within auto-phagosome compartments. Additionally, survival of some of these microorganisms is experimentally inhibited by using some autophagy inhibitors, and even their survival is compromised when grown within cells of deficient or defective ATG5 gene[35,36].
Some studies have reported that some pathogenic bacteria also have ability to manipulate autophagy regulation processes at gene transcription level. For instance, Francisella tularensis, Yersinia enterocolitica, and Burkholderia cenocepacia can down-regulate the transcription of important autophagy-related (ATG) genes. Thus, they reduce the activity of autophagy at cellular level during infection. Conversely some pathogens up-regulate autophagy at gene level, which could augment inflammation at the site of infection. As previously discussed, prolonged inflammation could result in further injury to surrounding body tissues. In addition, the autophagic pathway is known to be exploited by RNA viruses (e.g. mouse hepatitis virus, rhinovirus and poliovirus), for promoting RNA replication with membrane scaffold[37]. In this review, the interactions between different CNS intracellular pathogens and autophagic genes, along with the resulting autophagic and inflammatory processes are being studied.
The presence of pathogenic antigens can induce autophagic genes through a stratified array of principal immunological processes, and therefore result in augmented autophagy and inflammation at the site of infection, which limits bacterial proliferation. However, as mentioned above, excessive autophagy and inflammatory process do put surrounding healthy brain tissue at risk.
A detailed understanding of molecular mechanisms in neuroinflammatory and neural cell death/autophagy dysfunction pathways might identify interesting targets for drug-discovery and biomarker identification in various CNS disorders. Also, there is potential for precise anti-inflammatory drugs in future that may interfere with these genetic mechanisms, allowing more tailored therapeutics.
Modulation of expression profiles of autophagic genes in response to infection
Listeria Monocytogenes is an intracellular Gram positive bacterium which can cause gastroenteritis. Upon its hematogenous dissemination it can breach BBB and thus cause meningitis, meningoencephalitis, and brain abscesses. An experiment using quantitative polymerase chain reaction (q-PCR) arrays containing 84 genes to analyse the expression profiles of autophagic genes in response to L. monocytogenes infection were carried out in rat brains. It was shown that 7 out of 84 genes were clearly modified with TNF, which functions to limit brain damage, being the most highly up-regulated gene after infection[38]. The expression of chemokine (C-X-C motif) receptor 4, and the insulin-like growth factor 1 that act as co-regulators of autophagy pathway, were down-regulated. Only the expression of core autophagy gene ATG12 was modulated by infection.
During primary infection, intracellular L. monocytogenes along with infected host cells are usually targeted for degradation by autophagy. Once entering human body, L. monocytogenes are endocytosed into a vacuole, and listeriolysin O (LLO) is secreted to perforate the vacuole membrane, and subsequently gain access into the host cell cytosol. Subsequently, the product of the actA gene induces the polymerization of host actin, which eventually forms a tail, propelling bacterial movement within host cytosol for cell-to-cell spread. In this study of utilizing wild-type L. monocytogenes to infect mouse embryonic fibroblast cells, it is demonstrated that induction of protective autophagy pathway depends on expression of LLO, suggesting that vacuole permeabilization is a prerequisite for autophagy. On top of that, a mutant L. monocytogenes strain deficient of bacterial phospholipase production, has been degraded due to autophagy. It suggests that phospholipases plays a role in evasion of autophagy. Hence, it is hypothesized that the therapeutic target of actA as well as genes coding for phospholipases, might enhance autophagy, resulting in eradication of intracellular L. monocytogenes. Thus, identification of genes modulated upon infection in brain cells, as well as the mechanism of resulting autophagy, may confer a new strategy for therapeutic intervention in infectious disease[39-41].
Pathogen adaptations to host cell autophagy
Infectious agents which successfully parasitize their target cells have evolved to develop multiple strategies to dampen autophagy-dependent activation of host immune responses. So far, the types of identified microbial adaptation mechanisms can be broadly classified as strategies to: (1) prevent autophagy induction; (2) avoid pathogen recognition by the autophagic machinery; (3) prevent the autophagosome maturation into autolysosome; and (4) utilize components or functions of the autophagic mechanism to facilitate intracellular survival, multiplication, and release of intracellular microbes out of the infected cell[42].
Herpes simplex viruses (HSVs) are able to inhibit autophagy in neurons, and subsequently confer neuro-virulence by three main mechanisms, including the blockage of autophagy-stimulatory protein kinase resource (PKR) signaling, blockage of Beclin-1, or via the activation of mTOR signaling. In HSV-1, the utilization of a single viral virulence protein ICP34.5 to block the Beclin-1 dependent autophagy has been shown to be essential for HSV-1 encephalitis[43]. Of interest, it has been suggested that the inefficient fusion between lysosomes and autophagosomes in HSV-infected cells is caused by oxidative stress, which is reported to be associated with neurodegeneration[44].
Coxsackievirus has been shown to utilize autophagosomes for replication, in which it limits autophagosome and lysosome fusion by increasing light chain 3 (LC3) cleavage. It has been hypothesized that this is achieved by increasing a protein-activated signaling cascade, known as the calpain-dependent pathway[45].
A proposed mechanism of L. monocytogenes meningitis is that this intracellular pathogen camouflages and avoids recognition, subsequently rapidly replicating in the host cytosol by assembling host cell proteins to the bacterial cell surface via ActA ot InlK proteins. Also, L. monocytogenes phospholipase C is used to avoid autophagy by decreasing autophagic flux, diminishing host PI3P stores, and inhibiting the maturation of preautophagosomal structures[46].
It is reported with evidence that autophagy plays a pivotal role as a major defense mechanism in host cells, not only in the brain, but in other cells as well. Recently, a number of reports have successfully elaborated the scope of the autophagic process in immunity, being evolved from an antimicrobial defense mechanism to a complex immunological process that plays a major role in adaptive immunity, innate immunity, and inflammation. In the support of the importance of autophagy as a key defense mechanism, it is now clear that highly evolved intracellular pathogens possess specialized anti-autophagic adaptations to block or hinder their elimination. Looking forward, it will be necessary to further understand how different microbes manipulate autophagy, and their interaction with host autophagy mechanisms, to provide a potential source for the development of antimicrobial treatment modalities that antagonize the pro-microbe responses, at the same time promoting the anti-microbe functions of autophagy.
Autophagy induction and intracellular replication in viral central nervous system infections
There is a growing list of viruses which have been studied for relating their interactions with autophagy. For numerous viruses, there is a link between infection and autophagy. Certain viruses are responsible for the induction of an autophagic event, while others are involved in the intermediate processes of autophagy. Research has shown that autophagy not only plays a role in JEV and dengue infection, but also positively regulates the virus replication[47]. It is a well-known fact that energy is required for any cellular replication. In dengue viral infection, autophagy is shown to be connected to lipid metabolism in the virus, thus providing free fatty acids for synthesis of ATP[36]. This strongly suggests that the principle role of autophagy in dengue virus replication is for the regulating lipid metabolism. In contrast to dengue virus, the autophagic vacuole shows no evidence of being the site of replication for JEV. However, this finding is not consistent with the experiments conducted by Jin et al.[48] and Wang et al.[49], who stated that the accumulation of the autophagosome and the autophagosome-lysosome fusion are essential to promote JEV replication[47-49]. In addition to initiating autophagy, viral intrusion also affects the intermediate step of the event. Evidence which supports this statement is the accumulation of p62 protein (which is degraded by autophagy process) in simian immunodeficiency virus (SIV)-infected brain tissues[50].
It is also vital to find out if viral-induced autophagy will result in changes in the number of autophagosomes, which signifies the initiation of autophagy. Intriguingly, in the SIV-infected neurons, there is a significant reduction in the autophagosomes, in addition to changes in the distribution of the autophagosomes in the neurons[50]. The decrease in the number of autophagosomes may be due to the loss of neurons. This observation also implies the lack of initiation of autophagy in SIV infection. Further studies to test for the autophagic flux need to be carried out in order to determine how these two interact. Analysis showed that in SIV-infected microglia, there was loss of the neuronal processes and a decrease in the number of autophagosomes in the remaining processes; however the number of autophagosomes in the soma of the neurons remained constant. These findings demonstrate two reasonings: lack of initiation of the autophagy, and the increased clearance of the autophagosomes during the intermediate process. Further studies are required to analyze the importance of autophagosomal distribution in the pathogenesis of viral infection in microglia. It is interesting to note that rapamycin pre-treatment (an autophagy inducer) protected against the neurotoxic effects of the SIV, although the pre-treatment failed to recover the number of neurons to baseline level[50]. This indicates that autophagy might be one of the many pathways which serves to protect neurons in viral infections.
Increased hepatitis C virus replication and neurotoxicity with elevated autophagy
Even though hepatitis C virus (HCV) is a hepatotropic virus, this infection is also considered a systemic disease with extra-hepatic manifestations. Up to 50% of patients with chronic HCV infection have neuropsychiatric disorders. HCV RNA is detected in CD68+ cells of the CNS (macrophages/microglial) from 8 cases, suggesting direct HCV neuro invasion[51]. It is hypothesized that presence of lymphatic vessels in the brain might be the main pathway allowing infected peripheral blood monocytes to cross the BBB, and to serve as a precursors of the CNS microglia in addition to TNF-α and IL- 8 secreted by microglial cells infected with HCV. These two proinflammatory cytokines are toxic to neurons. They mediate a local inflammatory response and are highly associated with neuropsychiatric disorders. Transcription factor NF-κB is reported to be involved in cytokine gene expression. Therefore, blocking NF-κB can be a therapeutic approach to controlling HCV mediated neuroinflammation[52].
According to the above study, viral core proteins have been observed to activate microglial cells, astrocytes, and macrophages of patients infected with HCV. It has been shown that activation of microglial cells and subsequent diffusion of pro-inflammatory cytokines into the brain occurs as a result of changes in permeability of the BBB due to induced apoptosis of the brain microvascular endothelial cells in which the HCV virus replicates[53].
Particularly, HCV proteins (core and non- structural) have been demonstrated to be involved in neurotoxicity in two ways.
(1) Direct exposure of primary human neurons to core proteins of HCV causes neurite retraction, leading to suppressed neuronal autophagy in the brain. It has been demonstrated that the level of lLC3 is a marker of autophagosome formation. LC3 is highly expressed in human fetal neurons (HFN) exposed to Gal protein, while LC-II expression is significantly reduced in HCV core- exposed HFNs. It is suggested that HCV core proteins have the capability to inhibit LC3-I to LC3-II conversion, thereby reducing autophagosome formation in HCV core-exposed HFNs. Moreover, Gal-exposed neural cells demonstrated that HCV core proteins are active at directly at the neuronal membrane, contributing to the death of neurons by modulating the autophagy pathway[54].
(2) HCV proteins activate by both toll-like receptor 2 (TLR2) signaling and extracellular signal-related kinase (ERK). Prolonged TLR2 mediated activation of ERK has been found to result in neuronal injury/neurotoxicity.
In addition, a recent study reported that HCV triggers an unfolded protein response (UPR) and subsequently activates autophagy. Viral infection is often known to cause stress to endoplasmic reticulum (ER), and UPR (a signaling network) is specifically activated to restore ER homeostasis, refold misfolded proteins, and trigger the initiation of forming autophagosomes[55]. Consequently, it promotes the replication of HCV. However, under circumstances where accumulated unfolded and misfolded proteins in ER lumen are unresolvable, UPR can lead to apoptosis, resulting in chronic disorders such as neurodegeneration. In contrast, has also been reported that deficiency of ATG7 can reduce the synthesis of infectious HCV virion particles without significant effects on the viral protein expressions and/or RNA biosynthesis[56].
Moreover, according to the study, interference and subsequently loss of UPR-autophagy by gene silencing activates the innate immune response. It has been demonstrated that stable silencing of ATG5, or a UPR-activated transcriptional factor, CCAAT/enhancer binding protein homologous protein (CHOP) could further upregulate HCV pathogen-associated molecular pattern (PAMP)-triggered interferon-beta (IFNB) promoter activity and IFNB mRNA level. In addition, attenuated UPR-autophagy also remarkably elevated the downstream innate immunity pathways to inhibit replication of HCV in a paracrine fashion. Most importantly, it also demonstrated that a UPR-autophagy suppressing antiviral innate immune response can occur independently of this virus infection. Thus, interference in the UPR-autophagic pathway by gene modification exhibits therapeutic potential in suppressing viral replication[57].
Proviral role of autophagy in Japanese encephalitis virus and dengue virus infection
JEV is a mosquito-borne enveloped flavivirus with a positive-sense RNA genome, which causes acute encephalitis with high mortality in humans. Autophagy has been shown to be induced in human natural killer cells infected with an attenuated (RP-2ms) JEV strain, especially at the later stage, and to a lesser extent with a virulent (RP-9) strain[47]. In this study, the induction of autophagy by rapamycin was shown to enhance JEV replication, whereas 3-methyadenine mediated inhibition of autophagy reduced viral replication for both the strains of JEV. In addition, knockdown of ATG5 or Beclin 1 expression in cells also reduced JEV replication, suggesting a proviral role of autophagy in JEV multiplication. It has been shown that following endocytosis, the internalized JEV particles are targeted to preautolysosomal vacuoles (amphisomes) for viral uncoating. It is also hypothesized that an enhanced autophagy would increase the synthesis of viral RNA, leading to a raised viral protein expression level and yield of virus. In short, autophagy positively regulates JEV replication[47].
Lately, the efficient dengue virus replication has been shown to be facilitated by an autophagy-dependent lipid droplets processing. Free fatty acids release, augmented cellular beta- oxidation, and ATP generation have provided energy and nutrient sources for viral production. It is noteworthy that dengue virus is also one of the members of Flaviviridae family[58]. In other words, therapeutic potential which inhibit autophagy upon infection has bacteriostatic effect too[59].
Contradictory findings of interaction between autophagy and HIV 1 virus in central nervous system infection
The autophagy in the brains of the HIV patients do not have any direct effect on neurons. However, neuronal dysfunction due to inflammation and the massive involvements of macrophages in HIV-induced encephalitis is apparent[60]. Wilcox et al.[61] reported a positive correlation of autophagy and apoptosis in the viral infected regions of neonatal brains, compared to adult brains which showed a negative relation in autophagy and apoptosis. This statement is supported by the observation made, that knock-out of ATG7 in neonates resulted in decreased apoptosis[61]. Through this disconnection in the observations, it can be inferred that autophagic processes may be age dependent, particularly in the developing brains.
HIV destabilizes autophagy to facilitate self-replication, affecting genes essential for HIV replication (ATG7, MAP1LC3B, ATG12 and ATG16L2 involved in nucleation, and elongation of autophagosomes; CLN3 and LAPTM5 involved in lysosomal functioning) which are identified using small RNAi screening[62,63]. Studies elsewhere also demonstrated that replication of HIV is inhibited due to autophagy-associated silencing of genes: BECN1 and ATG5 in macrophages and BECN1 and ATG7 in monocytes. BECN1 encodes for Beclin1 which is involved in regulation of autophagy in human body. HIV elicits autophagy activation but blocks the process of late proteolysis[62].
Furthermore, in recent studies, HIV has been shown to inhibit autophagy in HIV-uninfected bystander cells. Also HIV Tat is known to inhibit interferon-gamma (IFN-γ) induced autophagy in macrophages that are uninfected by inhibiting STAT1 phosphorylation. As a consequence, immune effector mechanism for targeted intracellular pathogens destruction is attenuated, and HIV patients are more susceptible to infections such as tuberculosis and toxoplasmosis[62]. In contrast to the effect of Tat on on non-infected macrophages, HIV Env protein enhances apoptotic death of uninfected neurons and CD4+T cells, through a mechanism that involves accumulation of BECN1 as well as autophagy induction[62].
In the postmortem examination of the frontal cortices of both HIV-infected patients and non-infected individuals, autophagic markers were assessed by Western blotting and microscopy (confocal/electron). Autophagic proteins Beclin1, autophagy-related genes ATG5, ATG7 and LC-II were observed to be significantly activated in brains from HIV-1 encephalitis cases. Autophagosome development needs Beclin1 for nucleation, whereas covalent binding of ATG5 to ATG12 regulates vesicle elongation, following a pathway catalyzed by ATG7 and ATG10[64]. In other words, the level of autophagy is likely increased in encephalitis caused by HIV. Additionally, autophagic protein levels and autophagosome formation are reported to be increased in the neurons of those treated with CXCR4- or CCR-tropic HIV-1 gp 120. In contrast, no increase in the extent of autophagic death is observed in the brains of non-encephalitic HIV patients compared to HIV-uninfected subjects. Therefore, the above study suggested that although autophagy could help neurons to sustain survival, the enhancement in autophagic death contributes to encephalitis and so also to cognitive impairment. Hence, the above study suggested that by combining drugs that reduce autophagic activity alongside anti-retroviral medications, the neurological deficits associated with HIV-1 infection could be prevented or reversed, since dysregulation of autophagy is highly correlated with pathogenesis of neurological HIV infection. Experiments in the future are needed to investigate further whether the process of autophagy leads to completion in the brains, or if there is any accumulation of autophagic proteins without autophagy-related degradation[62,65].
Sindbis virus infection of central nervous system induces autophagy
In this study, it has been demonstrated that Sindibis virus (SIN) infection induces autophagy in virally infected neurons in vivo, and viral antigen colocalizes with autophagosomes in neurons. It is important to note that ATG gene ATG5 encodes an essential component of ATG5-ATG12-ATG16 conjugation system, and is required for formation of autophagosomes[66].
The role of the autophagy gene ATG5 is very essential for providing protection against infection with lethal SIN in the mouse CNS. Inactivating ATG5 in SIN-infected murine neurons results in a late viral protein clearance, an increased accumulation of adaptor protein p62, and enhanced neuronal death, however the viral replication levels remain unaffected. In vitro, the cellular protein p62 interacts with capsid protein of SIN and is required for capsid targeting to the autophagosome. Genetic deficiency of p62 blocks the targeting of viral capsids to autophagosomes, accelerates viral capsid aggregation, and thereby increases virus-induced cell death without hampering virus multiplication. In other words, increased SIN-induced animal mortality is not due to a direct role of neuronal ATG5 in the control of viral replication or regulation of innate immune signaling. Rather, the disruption of neuronal ATG5 function leading to neuronal death was actually associated with impaired SIN protein clearance. In this study, one plausible explanation is that in post mitotic cells such as neurons, the failure to properly clear viral proteins by autophagy results in cellular toxicity and increased animal lethality[67].
Transmissible spongiform encephalopathies associated pathological implications of autophagy
Though the autophagy process of cell death has been identified in scrapie (experimental infections) for quite some time this has only recently been reevaluated as a possible cell death process in prion diseases[68]. However, apoptosis is generally assumed to be the cause of neuronal death in transmissible spongiform encephalopathies (TSE)[69,70]. These pathologies of TSE, and many other neurodegenerative conditions, are characterized by the accumulation of aggregated and misfolded proteins in the brain; autophagy may be playing a protective role by removing such “toxic” protein aggregates[71,72].
The scrapie responsive gene 1 (Scrg1) encodes a cysteine-rich protein that is highly conserved and expressed primarily in the CNS. This protein is targeted to the Golgi apparatus as well as large dense-core secretory granules in neuronal cells[68]. It has been recently demonstrated that the Scrg1 protein is induced widely in scrapie-infected murine neurons, suggesting that Scrg1 plays a role in the neuronal death and/or the host response to stress. The consistent association of autophagic structures (typical of scrapie) with Scrg1 is in agreement with the recruitment of Golgi-specific proteins during this process of degradation, and therefore it is suggested that Scrg1 might be used as a specific probe to identify the process of neuronal autophagy in TSE[68].
Regulation of autophagy in peripheral neurons
For decades, autophagy has been closely associated with both nutrient acquisition and pathogen destruction. In the apparent coevolution with pathogens, adaptive immune protection mechanisms of higher eukaryotic organisms such as human beings have also learned to exploit autophagy. Autophagy has evolved from a nutrient providing pathway to one which aids in higher functions in the innate, adaptive and cell-intrinsic immunological pathways.
Hence, it is not a surprise that autophagic pathways do indeed cross talk with innate immunity mechanisms, contributing to the combat of pathogenic infiltration. Though research into how pathogens obtain nutrients remains in its infancy, it has always been speculated that pathogens require autophagy as one of the many sources of intracellular nutrients, to survive and propagate in their host cells. All aspects considered, the critical questions that remain unanswered are, which nutrients do pathogens acquire from autophagy, and what is the degree to which they rely on these nutrients.
Neuronal autophagy is regulated uniquely, andis also adapted to a great extent along with local axonal physiology. Moreover, the detailed mechanism for neuronal autophagy might be significantly different to classically induced autophagy. In recent times, several studies have shown the importance of autophagy in various neurodegenerative conditions; studies have also identified autophagic process to be a potential target in drug discovery. Therefore, further understanding of the process of autophagic death of neuronal cells would eventually aid in novel drug target identification and rational designing of drug screening protocols in order to combat various neurodegenerative conditions[73].
It is a good idea to identify some therapeutic agents that can control autophagy during infection, particularly when massive autophagic neural cell death causes the major pathology that might be induced possibly by some pathogen factors. Therapeutic manipulation of the interactions between autophagy and inhibitory cytokines might represent a novel method of regulating the immune response, and thereby a more facilitated clinical translation[12].
Only very limited information is available on possible therapeutics to inhibit autophagy in terms of providing protection, particularly in the case of excessive autophagic death in neurodegerative disorders. Inhibiting transglutaminase 2 (TG2), which is a multifunctional protein having implications in diverse pathophysiological processes, might offer a novel therapeutic approach for managing excessive autophagy[74]. Inhibition of the enhancer of zeste homolog 2 (EZH2) gene expression has also been shown to inhibit autophagy significantly, as reported in a study on human ovarian cancer where EZH2 expression could reverse the cisplatin resistance by inhibiting autophagy[75]. Downregulation of the methionine synthase reductase (MTRR) gene might also be an approach to inhibit the PI3K/Akt autophagy pathway; in a recent study, MTRR silencing could significantly increase cisplatin-induced apoptosis and reduce the autophagy induced by cisplatin in SKOV3/DDP cells[76]. The glucose regulated protein 78 (GRP78) is also known to affect autophagy and apoptosis; particularly in ovarian cancer cells. GRP78 is reported to have a regulatory role in expressions of Beclin1, Bcl-2 and CHOP, thereby affecting the sensitivity to cisplatin in ovarian carcinoma, which may be a new method for ovarian carcinoma treatment through improvement of the sensitivity to cisplatin. However, there is no study done to validate such hypotheses regarding the possible role of inhibiting excessive neurodegeneration due to autophagy in minimizing the bulk self-digestion of the neuronal cells.
Conclusion and recommendation
The presence of antigenic stimuli of pathogens can induce autophagic genes through a stratified array of principal immunologic processes, and therefore result in augmented autophagy and inflammation at the site of infection, which is considered to be protective to the host. However, an excessive auto-degeneration of the neuronal cells can be harmful. The question arises, whether there are any known direct interactions of intracellular pathogens (having neurotropism) with this degradative pathway that favor the pathogens for intracerebral survival and growth? It is worth exploring if there is any cooperation between pathogen factors altering immune inflammatory pathways, thereby influencing host cell autophagy regulatory genes that cause massive neuronal damage in intracranial infections as hypothesized presently [Figure 1]. Targeting some key pathways with respect to infectious causes of neurodegeneration will be the need of tomorrow’s new drug discovery that may or may not include the targeting of autophagy for minimizing brain matter degeneration.
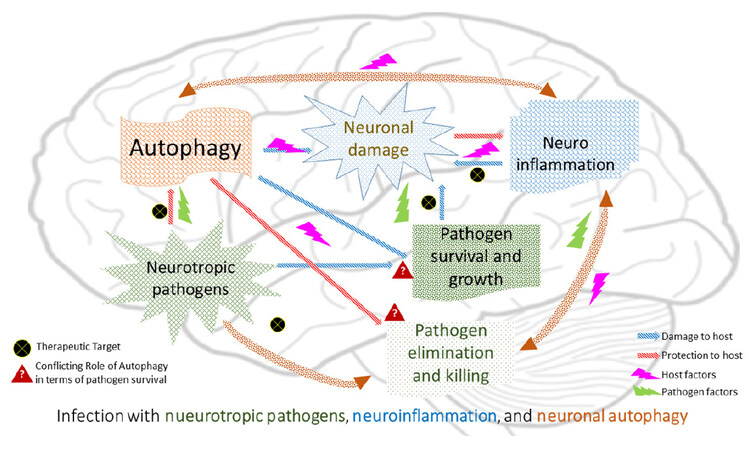
Figure 1. Schematic presentation depicting possible cooperation between pathogen factors altering immune inflammatory pathways in general influencing host cell autophagy regulatory genes (hypothetical) that cause a massive neuronal damage in certain intracranial infections
Declarations
AcknowledgmentsThe authors sincerely acknowledge Ms. Hollie Craig, from University of Edinburgh Medical School for her time and contribution in proof reading and polishing the English language in this manuscript.
Authors’ contributionsConceptualized the theme, designed the literature review process: Sahu PS
Contributed in the acquisition, analysis, and interpretation of information; drafted the manuscript; and approved the final version to be published: Sahu PS, Ter E
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Nicolson GL. Chronic bacterial and viral infections in neurodegenerative and neurobehavioral diseases. Lab Med 2008;39:291-9.
2. Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect or association? J Clin Invest 2003;111:163-9.
4. Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Investig 2005;115:1449-57.
5. Larsson HJ, Eaton WW, Madsen KM, Vestergaard M, Olesen AV, Agerbo E, Schendel D, Thorsen P, Mortensen PB. Risk factors for autism: perinatal factors, parental psychiatric history and socioeconomic status. Am J Epidemiol 2005;101:916-25.
6. Griffin WS. Inflammation and neurodegenerative diseases. Am J Clin Nutr 2006;83:S470-4.
7. Kern JK, Jones AM. Evidence of toxicity, oxidative stress and neuronal insult in autism. J Toxicol Environ Health B Crit Rev 2006;9:485-99.
8. Muravchick S, Levy RJ. Clinical implications of mitochondrial dysfunction. Anesthesiology 2006;105:819-37.
9. Pareja ME, Colombo MI. Autophagic clearance of bacterial pathogens: molecular recognition of intracellular microorganisms. Front Cell Infect Microbiol 2013;3:54.
10. Sharma M, Bhattacharyya S, Sharma KB, Chauhan S, Asthana S, Abdin MZ, Vrati S, Kalia M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J Gen Virol 2017;98:1027-39.
11. Di Cristina M, Dou Z, Lunghi M, Kannan G, Huynh MH, McGovern OL, Schultz TL, Schultz AJ, Miller AJ, Hayes BM, van der Linden W, Emiliani C, Bogyo M, Besteiro S, Coppens I, Carruthers VB. Toxoplasma depends on lysosomal consumption of autophagosomes for persistent infection. Nat Microbiol 2017;2:17096.
12. Wu TT, Li WM, Yao YM. Interactions between autophagy and inhibitory cytokines. Int J Biol Sci 2016;12:884-97.
13. Tooze SA, Schiavo G. Liaisons dangereuses: autophagy, neuronal survival and neurodegeneration. Curr Opin Neurobiol 2008;18:504-15.
14. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH, Kipnis J. Structural and functional features of central nervous system lymphatic vessels. Nature 2015;523:337-41.
15. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci 1996;19:312-8.
17. Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett 1987;223:284-8.
19. Banati RB, Rothe G, Valet G, Kreutzberg GW. Detection of lysosomal cysteine proteinases in microglia: flow cytometric measurement and histochemical localization of cathepsin B and L. Glia 1993;7:183-91.
20. Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 1990;250:1593-6.
21. Théry C, Chamak B, Mallat M. Cytotoxic effect of brain macrophages on developing. Eur J Neurosci 1991;3:1155-64.
22. Lee SC, Dickson DW, Liu W, Brosnan CF. Induction of nitric oxide synthase activity in human astrocytes by interleukin-1 beta and interferon-gamma. J Neuroimmunol 1993;46:19-24.
23. Keller CW, Lünemann JD. Autophagy and autophagy-related proteins in CNS autoimmunity. Front Immunol 2017;8:165.
24. Choy A, Roy CR. Autophagy and bacterial infection: an evolving arms race. Trends Microbiol 2013;21:451-6.
25. Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: consequences for human disease. Autophagy 2016;12:245-60.
26. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 2014;20:460-73.
28. Abeliovich H, Dunn WA Jr, Kim J, Klionsky DJ. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol 2000;151:1025-34.
29. Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000;150:1507-13.
30. Tallóczy Z, Jiang W, Virgin HW 4th, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A 2002;99:190-5.
31. Rosello A, Warnes G, Meier UC. Cell death pathways and autophagy in the central nervous system and its involvement in neurodegeneration, immunity and central nervous system infection: to die or not to die-that is the question. Clin Exp Immunol 2012;168:52-7.
32. Nold MF, Nold-Petry CA, Zepp JA, Palmer BE, Bufler P, Dinarello CA. IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol 2010;11:1014-22.
33. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007;450:566-9.
34. Amer AO, Swanson MS. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol 2005;7:765-78.
35. Romano PS, Gutierrez MG, Berón W, Rabinovitch M, Colombo MI. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell Microbiol 2007;9:891-909.
36. Mestre MB, Colombo MI. Staphylococcus aureus promotes autophagy by decreasing intracellular cAMP levels. Autophagy 2012;8:1865-7.
37. Wileman T. Aggresomes and pericentriolar sites of virus assembly: cellular defense or viral design? Annu Rev Microbiol 2007;61:149-67.
38. Virna S, Deckert M, Lütjen S, Soltek S, Foulds KE, Shen H, Körner H, Sedgwick JD, Schlüter D. TNF is important for pathogen control and limits brain damage in murine cerebral listeriosis. J Immunol 2006;177:3972-82.
39. Disson O, Lecuit M. Targeting of the central nervous system by Listeria monocytogenes. Virulence 2012;3:213-21.
40. Remuzgo-Martínez S, San Segundo D, Cruz CS, Beares I, Valdizáncd EM, Icardo JM, Ramos-Vivas J. Absence of core autophagy gene expression in an ex vivo central nervous system model infected with Listeria monocytogenes. Inmunología 2013;32:87-93.
41. Kishore R, Krishnamurthy P, Garikipati VN, Benedict C, Nickoloff E, Khan M, Johnson J, Gumpert AM, Koch WJ, Verma SK. Interleukin-10 inhibits chronic angiotensin II-induced pathological autophagy. J Mol Cell Cardiol 2015;89:203-13.
42. Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009;5:527-49.
43. Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007;1:23-35.
44. Santana S, Sastre I, Recuero M, Bullido MJ, Aldudo J. Oxidative stress enhances neurodegeneration markers induced by herpes simplex virus type 1 infection in human neuroblastoma cells. PLoS One 2013;8:e75842.
45. Yoon SY, Ha YE, Choi JE, Ahn J, Lee H, Kweon HS, Lee JY, Kim DH. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J Virol 2008;82:11976-8.
46. Mitchell G, Ge L, Huang Q, Chen C, Kianian S, Roberts MF, Schekman R, Portnoy DA. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect Immun 2015;83:2175-84.
47. Li JK, Liang JJ, Liao CL, Lin YL. Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect 2012;14:159-68.
48. Jin R, Zhu W, Cao S, Chen R, Jin H, Liu Y, Wang S, Wang W, Xiao G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS One 2013;8:e52909.
49. Wang H, Sun HQ, Zhu X, Zhang L, Albanesi J, Levine B, Yin H. GABARAPs regulate PI4P-dependent autophagosome: lysosome fusion. Proc Natl Acad Sci U S A 2015;112:7015-20.
50. Alirezaei M, Kiosses WB, Fox HS. Decreased neuronal autophagy in HIV dementia: a mechanism of indirect neurotoxicity. Autophagy 2008;4:963-6.
51. Wilkinson J, Radkowski M, Laskus T. Hepatitis C virus neuroinvasion: identification of infected cells. J Virol 2009;83:1312-9.
52. Adinolfi LE, Nevola R, Lus G, Restivo L, Guerrera B, Romano C, Zampino R, Rinaldi L, Sellitto A, Giordano M, Marrone A. Chronic hepatitis C virus infection and neurological and psychiatric disorders: an overview. World J Gastroenterol 2015;21:2269-80.
53. Fletcher NF, Wilson GK, Murray J, Hu K, Lewis A, Reynolds GM, Stamataki Z, Meredith LW, Rowe IA, Luo G, Lopez-Ramirez MA, Baumert TF, Weksler B, Couraud PO, Kim KS, Romero IA, Jopling C, Morgello S, Balfe P, McKeating JA. Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology 2012;142:634-43.e6.
54. Vivithanaporn P, Maingat F, Lin LT, Na H, Richardson CD, Agrawal B, Cohen EA, Jhamandas JH, Power C. Hepatitis C virus core protein induces neuroimmune activation and potentiates Human Immunodeficiency virus-1 neurotoxicity. PLoS One 2010;5:e12856.
55. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007;8:519-29.
56. Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009;5:937-45.
57. Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest 2011;121:37-56.
58. Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010;8:422-32.
59. Remaut K, Oorschot V, Braeckmans K, Klumperman J, De Smedt SC. Lysosomal capturing of cytoplasmic injected nanoparticles by autophagy: an additional barrier to non-viral gene delivery. J Control Release 2014;195:29-36.
60. Spector SA, Zhou D. Autophagy: an overlooked mechanism of HIV-1 pathogenesis and neuro AIDS? Autophagy 2008;4:704-6.
61. Wilcox DR, Wadhwani NR, Longnecker R, Muller WJ. Differential reliance on autophagy for protection from HSV encephalitis between newborns and adults. PLoS Pathog 20158;11:e1004580.
62. Campbell GR, Spector SA. Inhibition of human immunodeficiency virus type-1 through autophagy. Curr Opin Microbiol 2013;16:349-54.
63. Campbell GR, Spector SA. Toll-like receptor 8 ligands activate a vitamin D mediated autophagic response that inhibits human immunodeficiency virus type 1. PLoS Pathog 2012;8:e1003017.
64. Meng C, Zhou Z, Jiang K, Yu S, Jia L, Wu Y, Liu Y, Meng S, Ding C. Newcastle disease virus triggers autophagy in U251 glioma cells to enhance virus replication. Arch Virol 2012;157:1011-8.
65. Zhou D, Masliah E, Spector SA. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J Infect Dis 2011;203:1647-57.
66. Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004;15:1101-11.
67. Orvedahl A, MacPherson S, Sumpter R Jr, Tallóczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010;7:115-27.
68. Dron M, Bailly Y, Beringue V, Haeberlé AM, Griffond B, Risold PY, Tovey MG, Laude H, Dandoy-Dron F. SCRG1, a potential marker of autophagy in transmissible spongiform encephalopathies. Autophagy 2006;2:58-60.
69. Boellaard JW, Kao M, Schlote W, Diringer H. Neuronal autophagy in experimental scrapie. Acta Neuropathol 1991;82:225-8.
70. Jeffrey M, Scott JR, Williams A, Fraser H. Ultrastructural features of spongiform encephalopathy transmitted to mice from three species of bovidae. Acta Neuropathol 1992;84:559-69.
71. Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004;306:990-5.
72. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004;36:585-95.
73. Yue Z, Friedman L, Komatsu M, Tanaka K. The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim Biophys Acta 2009;1793:1496-507.
74. Agnihotri N, Mehta K. Transglutaminase-2: evolution from pedestrian protein to a promising therapeutic target. Amino Acids 2017;49:425-39.
75. Sun Y, Jin L, Liu JH, Sui YX, Han LL, Shen XL. Interfering EZH2 expression reverses the cisplatin resistance in human ovarian cancer by inhibiting autophagy. Cancer Biother Radiopharm 2016;31:246-52.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sahu PS, Ter E. Interactions between neurotropic pathogens, neuroinflammatory pathways, and autophagic neural cell death. Neurosciences 2018;5:2. http://dx.doi.org/10.20517/2347-8659.2017.43
AMA Style
Sahu PS, Ter E. Interactions between neurotropic pathogens, neuroinflammatory pathways, and autophagic neural cell death. Neuroimmunology and Neuroinflammation. 2018; 5: 2. http://dx.doi.org/10.20517/2347-8659.2017.43
Chicago/Turabian Style
Sahu, Priyadarshi Soumyaranjan, Eunice Ter. 2018. "Interactions between neurotropic pathogens, neuroinflammatory pathways, and autophagic neural cell death" Neuroimmunology and Neuroinflammation. 5: 2. http://dx.doi.org/10.20517/2347-8659.2017.43
ACS Style
Sahu, PS.; Ter E. Interactions between neurotropic pathogens, neuroinflammatory pathways, and autophagic neural cell death. Neurosciences. 2018, 5, 2. http://dx.doi.org/10.20517/2347-8659.2017.43
About This Article
Copyright

Data & Comments
Data


 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.