
The role of ubiquitinated TDP-43 in amyotrophic lateral sclerosis


Abstract
Deposition of intracellular ubiquitin inclusion in motor neurons is one of the leading pathogenic mechanisms of amyotrophic lateral sclerosis (ALS). The transactive response DNA binding protein-43 (TDP-43) is the main component of intracellular ubiquitin inclusion bodies in pathological deposits. TDP-43 is mainly distributed in the nucleus of neurons, and participates in nuclear RNA transcription, alternative splicing and mRNA stability regulation. The tardbp, as a coding gene, provides instructions for making TDP-43. After post-translational modification, the pathological TDP-43 induces pathological deposition in cells and is associated with neurodegenerative diseases, which is similar to tau in Alzheimer’s disease and alpha-synuclein in Parkinson’s disease. The pathogenic tardbp mutation can affect the localization of reverse transcription in the cell. This review summarizes the mechanisms underlying the pathogenesis of ALS by ubiquitination of TDP-43 protein.
Keywords
Introduction
Amyotrophic lateral sclerosis (ALS) is a disease of progressive degeneration of motor neurons with an insidious onset. It is fatal due to progressive weaking of respiratory muscles. Lack of effective treatment has frustrated the medical community, and the underlying mechanism of ALS remains undetermined. Following the discovery of superoxidase dismutase 1 (SOD1) mutation in familial ALS[1], TAR DNA-binding protein 43 (tdp43/tardbp) inclusions have been found in ALS to be related to familial ALS[2] (fALS). Moreover, TDP-43 protein, as an intracellular ubiquitin inclusion, has also been identified in sporadic ALS patients[3,4]. The understanding of the pathogenic mechanism of ALS has been gradually changed by the discovery of tdp43 mutation. In 2008, one observational study in France showed that 4% of familial ALS patients had a tdp43 mutation[2]. It was also found in sporadic ALS patients, which bolstered knowledge of the pathological mechanism of ALS[2]. The abnormal intracellular ubiquitin inclusion was confirmed to be the cause of neuronal cell death[3,5]. Therefore, the highly ubiquitinated phosphorylated TDP-43 protein is closely related to the development of ALS. This review illustrates the development of TDP-43 in ALS and its underlying mechanism.
Normal TDP-43 and its function
Normal TDP-43 is a protein with a length of 414 amino acids. It contains 2 RNA identity motifs (RRM1 and RRM2, respectively) at the N terminal[6,7] and hydrophobic sequence in the C-terminus[7]. The presence of the RRMs is a distinguishing feature of heterogeneous nuclear ribonucleoprotein proteins (hnRNP) and, in general, these regions are known to mediate RNA recognition as well as protein-protein interactions. The first RRM (RRM-1) is necessary and sufficient to bind specific RNA or DNA sequences[8]. TDP-43 is encoded by tardbp which mainly distributed in the nucleus. Normal TDP-43 does not form inclusion bodies and is mainly involved in nuclear RNA transcription, mRNA precursor shear and mRNA stability regulation. Recruitment of full-length TDP-43 into cytoplasmic deposition formed inclusion bodies. Next, TDP-43 combines with RNA/DNA and regulates RNA metabolism at multiple levels, including transcription, RNA splicing, and mRNA stability. In TDP-43 knockout HeLa cells and HEK293 cell cultures, tdp43 can regulate add2 gene expression by increasing Add2 mRNA stability, which is closely related to synaptic aggregation, synaptic remodeling and stability[7,9]. UTRs at the 3’ terminal of TDP-43 protein is a specific mRNAs that is associated with its distribution and aggregation in cells. TDP-43 takes charge of bidirectional transcription between the nucleus and cytoplasm. Its solubility and localization are closely related to its nuclear localization signal. In addition, it also affects the function of the protein[8].
By monitoring the levels of TDP-43 oligomers in vitro, TDP-43 has been found to communicate through the axon in the cell by synaptic and microencapsulated uptake[10]. With the help of nuclear magnetic resonance, simulation and microscopy, a sub-region has been found cooperatively but transiently to be folded into an α-Helical form that mediates TDP-43 phase separation[11]. It has also been illustrated that the low-complexity of TDP-43 in liquid-liquid and phase-separated in vitro granules demonstrates ALS-associated variants that disrupts interactions within the granules[11]. It is stabilized by extending its helices-structure and promotes binding between the molecules[11]. Stable helical conformation was adopted by residues Pro320-Leu340 in an isolated peptide of Met311-Gln360, while it is indeed lacking of any stable secondary structure. An observation at the glycine rich region of C-terminal domain is relatively conservative and related to the concentration and separation of molecules[12]. The concentration of TDP-43 molecules depends on its α-Helical structure of the protein C-terminal fractures (CTF)[11].
TDP-43 belongs to the family of the hnRNPs and is highly conserved among metazoans in both sequence and function[12]. In human cells, overexpression of TDP-43 decreases exon 9 recognition and specifically causes exon skipping in vitro[13].
In relation to its function, TDP-43 has a variety of diverse roles including gene transcription, RNA splicing, RNA shuttling and translation, and microRNA biogenesis[14]. TDP-43 regulates various processes of transcription through RNA and DNA binding. Moreover, recent reports have shown that the protein interacts with the 3-UTRs of specific mRNAs[15]. In a previous study, the depletion of TDP-43 through RNA interference removes splicing inhibition caused by unfavorable (UG)mU(n) sequences, indicating that TDP-43 exerts a potent inhibitory effect in vivo[15]. More recently, new evidence that TDP-43 protein continuously shuttles between the nucleus and cytoplasm in a transcription-dependent manner has been reported[16]. The functional TDP-43 plays a dominant role in exon 9 splicing regulatory elements and shows that TDP-43 related splice site has determined the evolution of positive splicing regulatory elements in contrast to this inhibition. On the other hand, TDP 43 was also found to repress cryptic exon splicing in order to promote cell survival[14]. TDP-43-dependent splicing defects, revealing TDP-43 extensively regulated cryptic splicing, are a significant overlap in genes that undergo TDP-43-dependent cryptic splicing repression[14].
The origin of pathological TDP-43
Pathological TDP-43 mediated neuronal death is mainly caused by neurotoxicity and loss of TDP-43 function[17]. Phosphorylation and ubiquitination of TDP-43, the major features of pathological TDP-43, have not been detected in the normal brain. Phosphorylated and/or ubiquitinated TDP-43 have been found in the brain and spinal cord of patients with ALS[4]. In human studies, not all of TDP-43 inclusion bodies have been ubiquitinated, especially in the early stage of ALS, which suggests that ubiquitination is an advanced metabolic phenomenon in ALS disease[18]. The phosphorylated TDP-43 exists more commonly with serine (Ser) 379, Ser 403 + Ser 404, Ser 409 +, and Ser 410, which mainly between Ser 409-410. Most of the inclusion bodies of TDP-43 and TDP-43 25-kDa, as the degradation fragments of TDP 43, are detected in the phosphorylated form[4]. Therefore, the process of phosphorylation is likely preceded by ubiquitination. However, it is still unclear which form plays a more determinant role in mediating TDP-43 induced neurodegeneration.
In pathological conditions, TDP-43 protein degrades into two degradation fragments at the C-terminal[19]. By selectively expressing mutations in neurons and glial cells, the pathological TDP-43 protein is more commonly found to concentrate in neuronal cells, which may cause it failure to regulate synaptic plasticity and neuronal death[8,20]. Thus, mistakenly accumulated TDP-43 in motor neurons might be the initial mechanism of ALS onset[20].
The fragments of TDP-43 protein can induce TDP-43 deposition, which is related to the ubiquitin proteasome system[5,21]. The overexpression of the full-length protein of TDP-43 and its aggregation can be detected in high expression of tardbp CTFs cells. Although both TDP-43 and TDP-43 fragments would be affected by the ubiquitin proteasome system, CTFs fragments are more likely to foster transcription without stopping due to the absence of two nuclear localization signals. In addition, Cdc48TS, as an enhancer of neurotoxicity, promotes the deposition of pathological TDP-43[22]. Thus, any influence on the ubiquitin proteasome system may increase the expression of ubiquitination of TDP-43.
Pathogenesis of TDP-43
In Drosophila motor neurons, the high expression of TDP-43 caused axonal swelling and impaired mobility. Moreover, impairment was more severe in the motor neurons of A315T mutant phenotype[23]. The TDP-43 peptide segments can form in vitro in both the wild-type and A315T mutant, which are misfolded like precipitation as seen using an electron microscope[23]. Therefore, the main pathogenesis of ALS may be caused by the abnormal accumulation of TDP-43 in motor neurons and secondary atrophy of neurons or glial cells.
According to the findings in polymerization kinetics study, the pathological C terminus had a prion-like domain structure. This prion domain is present in most of the pathogenic mutations of TDP-43[23,24]. The pathological C terminus is intrinsically disordered only with some nascent secondary structures in aqueous solutions, but processes the capacity to assemble into dynamic oligomers rich in β-sheet structures. There structures interact with nucleic acid, which triggers rapid aggregation for most mutants[25]. Although RNA-binding protein prion-like domains have no homology or sequence similarity to the human prion protein that forms infectious protein aggregates in new variant Creutzfeldt-Jakob disease, many of these proteins have been identified as the major components of cytoplasmic inclusions associated with subtypes of ALS and frontotemporal dementia (FTD). In addition, the pathogenic mutations caused by tardbp are concentrated in the glycine enriched region of the C terminus[23,26]. So far, over 60 mutations in tardbp have been found to cause fALS and FTD[24]; they are listed in Table 1 as extracted from the ClinVar database. Tardbp mutations in the nucleus might disrupt the formation of alpha helices, or their ability to stabilize[11]. Mutations in the spiral region affect molecular binding, concentration and the separation phase. The aggregation of pathological TDP-43 is due to the overexpression and stacking of TDP-43 proteins. The TDP-43 prion-like domain appears to have an energy landscape, which allows the assembly of the wild-type sequence into dynamic oligomers only under very limited conditions. ALS-causing point mutations are sufficient to remodel it into a more favorable formation of amyloid and its irreversible aggregation, thus supporting the emerging view that such pathologic aggregation may occur via the exaggeration of functionally important assemblies[24].
List of all TARDBP related mutations in ALS condition
| Name | Gene(s) | Condition(s) | Clinical significance | Variation ID | Allele ID |
|---|---|---|---|---|---|
| NM_007375.3(TARDBP):c.-126G>T | TARDBP | FTD, ALS, dominant | Uncertain significance | 291727 | 275977 |
| NM_007375.3(TARDBP):c.-122G>A | TARDBP | FTD | Benign | 291728 | 275978 |
| NM_007375.3(TARDBP):c.-117G>A | TARDBP | FTD, ALS, dominant | Uncertain significance | 291729 | 275982 |
| NM_007375.3(TARDBP):c.-110C>T | TARDBP | FTD, ALS, dominant | Uncertain significance | 291730 | 275839 |
| NM_007375.3(TARDBP):c.-77G>A | TARDBP | FTD, ALS, dominant | Uncertain significance | 291731 | 275812 |
| NM_007375.3(TARDBP):c.-42C>T | TARDBP | FTD, ALS, dominant | Uncertain significance | 291732 | 275983 |
| NM_007375.3(TARDBP):c.-12-10_-12-9delTT | TARDBP | FTD, ALS, dominant | Likely benign | 291733 | 275813 |
| NM_007375.3(TARDBP):c.87C>T (p.Ser29=) | TARDBP | Not provided | Uncertain significance | 444152 | 437792 |
| NM_007375.3(TARDBP):c.198T>C (p.Ala66=) | TARDBP | FTD not specified/ALS, dominant | Benign/likely benign | 291734 | 275840 |
| NM_007375.3(TARDBP):c.238+9C>T | TARDBP | FTD, ALS, dominant | Likely benign | 291735 | 275850 |
| NM_007375.3(TARDBP):c.239-15G>A | TARDBP | FTD, ALS, dominant | Uncertain significance | 291736 | 275814 |
| NM_007375.3(TARDBP):c.499A>G (p.Met167Val) | TARDBP | FTD, ALS, dominant | Uncertain significance | 291737 | 275988 |
| NM_007375.3(TARDBP):c.506A>G (p.Asp169Gly) | TARDBP | ALS type 10 | Conflicting interpretations of pathogenicity | 5233 | 20272 |
| NM_007375.3(TARDBP):c.675A>G (p.Pro225=) | TARDBP | FTD, ALS, dominant | Likely benign | 291738 | 275815 |
| NM_007375.3(TARDBP):c.720G>A (p.Ala240=) | TARDBP | FTD, ALS, dominant | Uncertain significance | 291739 | 275851 |
| NM_007375.3(TARDBP):c.859G>A (p.Gly287Ser) | TARDBP | ALS type 10/motor neuron disease | Conflicting interpretations of pathogenicity | 21483 | 34335 |
| NM_007375.3(TARDBP):c.869G>C (p.Gly290Ala) | TARDBP | ALS type 10 | Pathogenic | 5231 | 20270 |
| NM_007375.3(TARDBP):c.881G>T (p.Gly294Val) | TARDBP | ALS type 10 | Pathogenic | 21484 | 34336 |
| NM_007375.3(TARDBP):c.881G>C (p.Gly294Ala) | TARDBP | ALS type 10 | Pathogenic | 5230 | 20269 |
| NM_007375.3(TARDBP):c.883G>A (p.Gly295Ser) | TARDBP | ALS type 10 | Pathogenic | 21485 | 34337 |
| NM_007375.3(TARDBP):c.892G>A (p.Gly298Ser) | TARDBP | ALS type 10 | Pathogenic | 5232 | 20271 |
| NM_007375.3(TARDBP):c.943G>A (p.Ala315Thr) | TARDBP | ALS type 10 | Pathogenic | 5236 | 20275 |
| NM_007375.3(TARDBP):c.945G>A (p.Ala315=) | TARDBP | Not provided | Uncertain significance | 374720 | 361606 |
| NM_007375.3(TARDBP):c.991C>A (p.Gln331Lys) | TARDBP | ALS type 10 | Pathogenic | 5229 | 20268 |
| NM_007375.3(TARDBP):c.1009A>G (p.Met337Val) | TARDBP | ALS type 10 | Pathogenic | 5228 | 20267 |
| NM_007375.3(TARDBP):c.1028A>G (p.Gln343Arg) | TARDBP | ALS type 10 | Pathogenic | 5235 | 20274 |
| NM_007375.3(TARDBP):c.1042G>T (p.Gly348Cys) | TARDBP | ALS type 10/not provided | Pathogenic | 5234 | 20273 |
| NM_007375.3(TARDBP):c.1043G>T (p.Gly348Val) | TARDBP | Motor neuron disease | Pathogenic | 266064 | 260865 |
| NM_007375.3(TARDBP):c.1098C>G (p.Ala366=) | TARDBP | FTD not specified/ALS, Dominant | Benign/likely benign | 291740 | 275852 |
| NM_007375.3(TARDBP):c.1122T>G (p.Tyr374Ter) | TARDBP | Motor neuron disease | Uncertain significance | 266065 | 260866 |
| NM_007375.3(TARDBP):c.1144G>A (p.Ala382Thr) | TARDBP | ALS type 10/FTD with TDP43 inclusions, TARDBP-related/not provided | Pathogenic/likely pathogenic | 21474 | 34326 |
| NM_007375.3(TARDBP):c.1150G>C (p.Gly384Arg) | TARDBP | ALS type 10 | Pathogenic | 190399 | 188225 |
| NM_007375.3(TARDBP):c.1153T>G (p.Trp385Gly) | TARDBP | ALS type 10 | Pathogenic | 190400 | 188226 |
| NM_007375.3(TARDBP):c.*83T>C | TARDBP | ALS type 10 | Pathogenic | 21465 | 34317 |
| NM_007375.3(TARDBP):c.*129T>C | TARDBP | FTD, ALS, dominant | Uncertain significance | 291741 | 276082 |
| NM_007375.3(TARDBP):c.*159A>C | TARDBP | FTD, ALS, dominant | Uncertain significance | 291742 | 275989 |
| NM_007375.3(TARDBP):c.*208G>A | TARDBP | FTD, ALS, dominant | Likely benign | 291743 | 275853 |
| NM_007375.3(TARDBP):c.*214T>C | TARDBP | FTD, ALS, dominant | Uncertain significance | 291744 | 275816 |
| NM_007375.3(TARDBP):c.*505delA | TARDBP | FTD, ALS, dominant | Uncertain significance | 291745 | 275994 |
| NM_007375.3(TARDBP):c.*666G>A | TARDBP | FTD, ALS, dominant | Uncertain significance | 291746 | 275995 |
| NM_007375.3(TARDBP):c.*697G>A | TARDBP | ALS type 10/FTD with TDP43 inclusions, TARDBP-related | Pathogenic | 5239 | 20278 |
| NM_007375.3(TARDBP):c.*842G>A | TARDBP | FTD, ALS, dominant | Uncertain significance | 291747 | 275996 |
| NM_007375.3(TARDBP):c.*862G>T | TARDBP | FTD, ALS, dominant | Uncertain significance | 291748 | 275855 |
| NM_007375.3(TARDBP):c.*963C>T | TARDBP | FTD, ALS, dominant | Uncertain significance | 291749 | 275856 |
| NM_007375.3(TARDBP):c.*1008T>G | TARDBP | FTD, ALS, dominant | Likely benign | 291750 | 275998 |
| NM_007375.3(TARDBP):c.*1081C>T | TARDBP | FTD, ALS, dominant | Likely benign | 291751 | 276021 |
| NM_007375.3(TARDBP):c.*1084A>T | TARDBP | FTD, ALS, dominant | Uncertain significance | 291752 | 276022 |
| NM_007375.3(TARDBP):c.*1597_*1600delTGTT | TARDBP | FTD, ALS, dominant | Uncertain significance | 291753 | 275859 |
| NM_007375.3(TARDBP):c.*1622A>T | TARDBP | FTD, ALS, dominant | Uncertain significance | 291754 | 275860 |
| NM_007375.3(TARDBP):c.*1623T>A | TARDBP | FTD, ALS, dominant | Uncertain significance | 291756 | 276025 |
| NM_007375.3(TARDBP):c.*1633delT | TARDBP | FTD, ALS, dominant | Uncertain significance | 291755 | 275867 |
| NM_007375.3(TARDBP):c.*1795A>G | TARDBP | FTD, ALS, dominant | Uncertain significance | 291757 | 275817 |
| NM_007375.3(TARDBP):c.*2005T>C | TARDBP | FTD, ALS, dominant | Uncertain significance | 291758 | 276105 |
| NM_007375.3(TARDBP):c.*2029C>T | TARDBP | FTD, ALS, dominant | Uncertain significance | 291759 | 275868 |
| NM_007375.3(TARDBP):c.*2046T>G | TARDBP | FTD, ALS, dominant | Uncertain significance | 291760 | 276026 |
| NM_007375.3(TARDBP):c.*2154G>T | TARDBP | FTD, ALS, dominant | Uncertain significance | 291761 | 276108 |
| NM_007375.3(TARDBP):c.*2252A>G | TARDBP | FTD, ALS, dominant | Uncertain significance | 291762 | 275872 |
| NM_007375.3(TARDBP):c.*2294_*2295insGTTTT | MASP2|TARDBP | FTD, MASP2 deficiency/ALS, dominant | Benign | 291763 | 276114 |
| NM_007375.3(TARDBP):c.*2331A>G | MASP2|TARDBP | FTD, MASP2 deficiency/ALS, dominant | Likely benign | 291764 | 275874 |
| NM_007375.3(TARDBP):c.*2334G>A | TARDBP | FTD, ALS, dominant | Uncertain significance | 291765 | 276127 |
| NM_007375.3(TARDBP):c.*2360C>T | TARDBP | FTD, ALS, dominant | Uncertain significance | 291766 | 276128 |
| NM_007375.3(TARDBP):c.*2538delC | TARDBP | FTD, ALS, dominant | Uncertain significance | 291767 | 275875 |
| NM_007375.3(TARDBP):c.*2740G>A | TARDBP | FTD, ALS, dominant | Likely benign | 291768 | 276138 |
| NM_007375.3(TARDBP):c.*2750G>A | TARDBP | FTD, ALS, dominant | Uncertain significance | 291769 | 275827 |
| NM_007375.3(TARDBP):c.*2773A>G | TARDBP | FTD, ALS, dominant | Uncertain significance | 291770 | 276140 |
| NM_007375.3(TARDBP):c.*2829dupT | TARDBP | FTD, ALS, dominant | Uncertain significance | 291771 | 275835 |
| NM_006610.3(MASP2):c.*225T>C | MASP2|TARDBP | FTD, MASP2 deficiency/ALS, dominant | Benign | 291772 | 275885 |
| NG_008734.1:g.19080G>A | MASP2|TARDBP | FTD, ALS, dominant | Likely benign | 368798 | 353027 |
| NM_006610.3(MASP2):c.1617T>C (p.Asn539=) | MASP2|TARDBP | FTD, MASP2 deficiency/ALS, dominant | Likely benign | 291779 | 275924 |
TDP-43 oligomers may further delay the release from each other[11], resulting in the TDP-43 oligomerization in the nucleus, which is a possible mechanism of disruption of TDP-43[24]. Aging or inhibition of protein degradation may increase the toxicity of TDP-43 in glial cells and cause neuropathological changes.
TDP-43 C-terminus encodes a prion-like domain, widely presented in RNA-binding proteins like a prion-like domain. C-terminus is essential for solubility and cellular localization, because its deletion results in the formation of large nuclear and cytoplasmic aggregates[14]. Disruption of the RNA-recognition domain required for RNA and DNA binding, however, alters nuclear distribution by decreasing TDP-43 presence in the nucleoplasm.
The assembly of the wild-type sequence into dynamic oligomers was only seen under very limited conditions; ALS-causing point mutations are sufficient to remodel it to favor the amyloid formation or irreversible aggregation, thus supporting the emerging view that pathologic aggregation may occur via the exaggeration of functionally important assemblies[24]. Furthermore, the coupled capacity of TDP-43 in aggregation and membrane interaction may critically account for its high neurotoxicity[27].
In addition, the proteinopathy of D169G and K263E mutants at the RRM domain of TDP-43 could form the basis of ALS, including the increased solvent-accessible surface area, conformational flexibility as well as unfolding of TDP-43, and the altered RNA conformation in TDP-43-RNA complex. These changes also brought the enhanced aggregation propensity in the cytoplasm[28]. These novel findings were important to illustrate the mechanism in the structural and functional aspects of ALS development.
Redistribution of intracellular TDP-43
The abnormal TDP-43 fragments would be re-distributed in the extracellular region of the nucleus[6]. More studies suggested that TDP-43 solubility and localization are particularly sensitive to disruptions that extend beyond the newly found nuclear localization signal and depend on a combination of factors that are closely connected to the functional properties of this protein[14]. TDP-43 fragmentation accelerates the formation of inclusion body and cell mRNA processing. The N-terminus fragment is highly distinctive, which promotes aggregation of the C-terminus structure[6]. When overexpression of TDP-43 and its C- terminal fragments in HEK293T cells, fragments of TDP-43 protein and TDP35 are recruited and removed into the cytoplasmic inclusion bodies[8]. TDP-35 participates in the aggregation of mRNA precursors, which makes the transformation of proteins into polymers easier. The insoluble fraction of ALS acts as a seed of TDP-43 aggregation when it is introduced in SH-SY5Y cells, and subsequently transmitted to other co-cultured cells[19,29]. Such extracellular accumulation, in a potentially more harmful way, is similar to the prion infections[30,31]. In addition, normal TDP-43 distribution in nucleus is not toxic to the cell, while only tardbp mutants cause redistribution in the extracellular region of the nucleus with neurotoxicity[3]. In a clinical study, previous work showed that accumulation of pathological TDP-43 or FUS coexist with misfolded HuWtSOD1 in patient motor neurons, and can trigger its misfolding in cultured cells[31]. In vitro, immunocytochemistry and immunoprecipitation were used to demonstrate that TDP-43 or FUS-induced misfolded HuWtSOD1 can propagate from cell-to-cell via conditioned media, and seed cytotoxic misfolding of endogenous HuWtSOD1 in the recipient cells in a prion-like fashion[31]. While siRNA in recipient cells and incubation of conditioned media with misfolded SOD1-specific antibodies could inhibit intercellular transmission in vitro[30], intercellular spread of SOD1 misfolding is not accompanied by transmission of TDP-43 or FUS pathology.
The spreading of pathological TDP-43
In vivo, TDP-43 protein is found in secreted exosomes from Neuro2a cells and primary neurons but not astrocytes and microglia[32]. The pathological TDP-43 protein aggregation and autophagy inhibition promote exosomal secretion of TDP-43[29]. The levels of exosomal full length TDP-43 and C-terminal fragment species are upregulated in the brain of ALS patients. If Neuro2a cells are exposed to the cerebrospinal fluid of ALS patients, the deposition of intracellular pathological TDP-43 proteins would be take place. In addition, Neuro2a cells can have TDP-43 deposition by regulating silent genes or inhibiting exocrine secretion. Upregulation of exocrine secretion in the TDP-43A315T mutant transgenic mice exacerbates the disease process. Exosome secretion is considered a key pathway for clearance of pathological TDP-43[29].
To study the potential propagation of TDP-43, a HEK293 cell culture model was used, which supports the propagated misfolding of HuWtSOD1. It showed significant protein expression in cells transfected with TDP-43 constructs, but no expression in the incubated cells, indicating that the conditioned media contains no active residual lipofectamine reagent and that the transfection-encoded TDP-43 protein does not transmit to recipient HEK293 cell cultures[31]. Thus, pathological TDP-43 is cleared via the proteasome, which reduces efficient clearance of mis-folded SOD1.
Recently, novel intracytoplasmic inclusions immunoreactive for phosphorylated transactivation response TDP-43 (p-TDP 43) were found in anterior horn cells in a case of a sporadic amyotrophic lateral sclerosis (sALS) patient. His spinal cord showed severe degeneration involving the anterior and lateral funiculi, whereas the posterior funiculus was preserved. Most neurons in the anterior horn and Clarke’s column were markedly lost, while some remaining anterior horn cells had round and densely eosinophilic or amphophilic intracytoplasmic inclusions[33]. They were immune-reactive for ubiquitin, p-TDP-43, cystatin C and transferrin[33]. On confocal laser microscopy, cystatin C was found to consistently surround p-TDP-43 within the inclusions. It was a key finding that these unique inclusions may have been formed under a specific condition whereby p-TDP-43 and cystatin C interacted with each other[32].
Removal of pathological TDP-43
Although the tardbp gene mutation is uncommon, other gene mutations related to ALS can also lead to abnormal intracellular TDP-43 levels[29,34]. Therefore, it is necessary to analyze the clearing pathway of pathological TDP-43. The published study has reported such process not only involved the dynamic transportation by exocrine secretion and small vesicles, but also by autophagy as well[26]. With overcoming the confounding effects of aggregation and toxicity, pathogenic mutations that significantly shorten TDP-43 half-life were found in a single-cell optical method. A novel autophagic flux assay combined with an in silico screen identified compounds that effectively stimulate autophagy in neurons though enhancement of TDP-43 clearance and reduction of its mis-localization. On the other hand, the induction of autophagy can improve scavenging ability of TDP-43, enhancing the primary neuronal survival capacity[26]. TDP-43 causes differential pathology in neuronal versus glial cells in the mouse brain by increasing the TDP-43 clearance which can slow down the progression of ALS[26].
The accumulation of intracellular TDP-43 is also related to the impairment of TDP-43 CTFs fragment clearance. The autophagy mediated TDP-43 CTFs fragmentation is caused by the failure of the phagocytosis of membrane vesicles[26], which suggested that autophagy also affected the transcription capacity of TDP-43 proteins. In addition, several studies have illustrated that TDP-43 concentration may increase toxicity in HeLa cells, suggesting that the autophagy system and the ubiquitin proteasome system may affect transcription of TDP-43[34].
TDP-43 mitochondrial localization inhibitory peptide can also abolish cytoplasmic TDP-43 accumulation, restore mitochondrial function, prevent neuronal loss, and alleviate motor-coordinative and cognitive deficits in adult hemizygous TDP-43M337V mice[35].
Targeting TDP-43 as a potential treatment for ALS
Due to the fact that ALS patients demonstrate the inability of the cell’s protein garbage disposal system to “pull out” and destroy TDP-43, a therapy targeting TDP-43 removal shows promise in clinical treatment. In a pilot study, researchers delivered parkin genes to neurons which slowed down ALS pathologies linked to TDP-43[36]. In another animal model, increased expression of UPF1, the master regulator of a nonsense-mediated decay pathway, can significantly protect mammalian motor neurons from TDP-43 mediated toxicity. UPF1 has shown promising results in animal models of ALS involving TDP-43 dysfunction and provides a rationale for developing gene-based therapies for ALS indicating the efficacy of a UPF1-based therapy in animal models of TDP-43 induced ALS pioneered in this laboratory[37]. Similarly, overexpression of the mammalian Sis1 homologue, DNAJB1, relieves TDP-43 mediated toxicity in primary rodent cortical neurons, suggesting that Sis1 and its homologues may have neuroprotective effects in ALS[38].
In ALS disease progression, TDP-43 is ubiquitinated, hyper-phosphorylated, and cleaved to form intranuclear and cytosolic aggregates. There is an overall shift in its localization from the nucleus to the cytoplasm and axons [Figure 1]. Over 60 dominant missense mutations have been defined in TDP-43, which may have an increased propensity to cleavage and may be resistant to degradation. More stimulation studies in this mechanism show that TDP-43 antibodies could be one potential strategy for disease intervention.
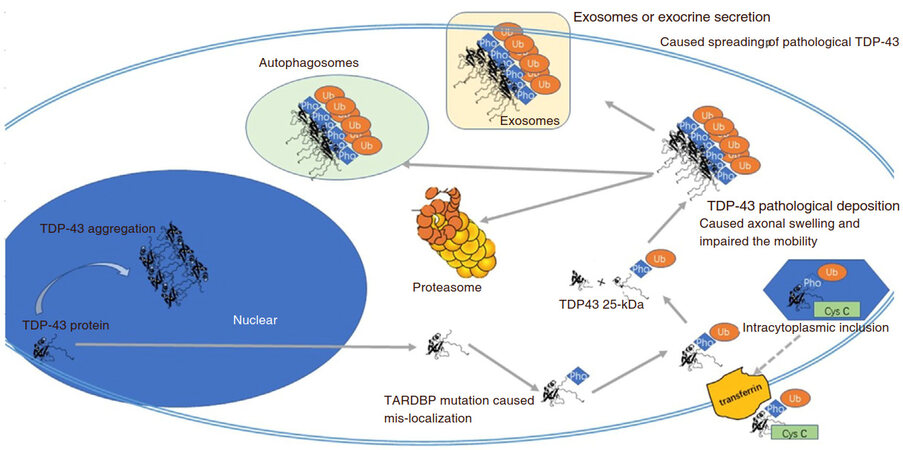
Figure 1. The role of ubiquitinated tdp43 that forms aggregates in amyotrophic lateral sclerosis
In summary, the pathogenic mechanism of ubiquitinated TDP-43 in ALS, including the origin and redistribution of pathological TDP-43, has been studied intensively in the past ten years. Currently, phosphorylation and ubiquitination of TDP-43 have been identified and recognized to be the source of pathological protein aggregation, inclusion bodies formation and abnormal exosome secretion. Similar to prion propagation and autophagy, these findings may help understand the relationship between ubiquitinated TDP-43 and ALS pathogenesis. More research is needed on the metabolic pathways of neurotoxic TDP-43 fragments.
Declarations
Authors’ contributionsDesigned this study: Dong Y, Chen Y
Participated in material review and draft the manuscript: Dong Y
Revised the manuscript: Chen Y
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Siddique T, Figlewicz DA, Pericak-Vance MA, Haines JL, Rouleau G, Jeffers AJ, Sapp P, Hung WY, Bebout J, McKenna-Yasek D, Deng G, Horvitz HR, Gusella JF, Brown RH, Roses AD. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N Engl J Med 1991;324:1381-4.
2. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. Tardbp mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008;40:572-4.
3. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. Tdp-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commu 2006;351:602-11.
4. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated tdp-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130-3.
5. Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 2008;121:3778-85.
6. Shodai A, Morimura T, Ido A, Uchida T, Ayaki T, Takahashi R, Kitazawa S, Suzuki S, Shirouzu M, Kigawa T, Muto Y, Yokoyama S, Takahashi R, Kitahara R, Ito H, Fujiwara N, Urushitani M. Aberrant assembly of RNA recognition motif-1 links to pathogenic conversion of tar DNA-binding protein of 43 kda (tdp-43). J Biol Chem 2013;288:14886-905.
7. Morgan BR, Zitzewitz JA, Massi F. Structural rearrangement upon fragmentation of the stability core of the ALS-linked protein TDP-43. Biophys J 2017;113:540-9.
8. Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 2009;276:36337-43.
9. Costessi L, Porro F, Iaconcig A, Muro AF. TDP-43 regulates b-adducin (add2) transcript stability. RNA Biol 2014;11:1280-90.
10. Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, Li D, Thal DR, Walther P, Ludolph AC, Danzer KM, Weishaupt JH. Tdp-43 is intercellularly transmitted across axon terminals. J Cell Biol 2015;211:897-911.
11. Conicella AE, Zerze GH, Mittal J, Fawzi NL. Als mutations disrupt phase separation mediated by α-helical structure in the tdp-43 low-complexity c-terminal domain. Structure 2016;24:1537-49.
12. Ayala YM, Pantano S, D'Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE. Human, drosophila, and c.Elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol 2005;348:575-88.
13. Buratti E, Brindisi A, Pagani F, Baralle FE. Nuclear factor tdp-43 binds to the polymorphic tg repeats in cftr intron 8 and causes skipping of exon 9: a functional link with disease penetrance. Am J Hum Genet 2004;74:1322-5.
14. Ayala YM, Zago P, Ambrogio AD, Xu YF, Petrucelli L, Buratti E, Baralle F. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 2008;121:3778-85.
15. Ayala YM, Pagani F, Baralle FE. Tdp43 depletion rescues aberrant cftr exon 9 skipping. FEBS Lett 2006;580:1339-44.
16. Tan Q, Yalamanchili HK, Park J, De Maio A, Lu HC, Wan YW, White JJ, Bondar VV, Sayegh LS, Liu X, Gao Y, Sillitoe RV, Orr HT, Liu Z, Zoghbi HY. Extensive cryptic splicing upon loss of rbm17 and tdp43 in neurodegeneration models. Hum Mol Genet 2016;25:5083-93.
17. Gendron TF, Petrucelli L. Rodent models of tdp-43 proteinopathy: investigating the mechanisms of tdp-43-mediated neurodegeneration. J Mol Neurosci 2011;45:486-99.
18. Herhaus L, Dikic I. Expanding the ubiquitin code through post-translational modification. EMBO Rep 2015;16:1071-83.
19. Buratti E, Baralle FE. The molecular links between tdp-43 dysfunction and neurodegeneration. Adv Genet 2009;66:1-34.
20. Yan S, Wang CE, Wei W, Gaertig MA, Lai L, Li S, Li XJ. Tdp-43 causes differential pathology in neuronal versus glial cells in the mouse brain. Hum Mol Genet 2014;23:2678-93.
21. Okamoto K, Fujita Y, Mizuno Y. Pathology of protein synthesis and degradation systems in als. Neuropathology 2010;30:189-93.
22. Tank EM, True HL. Disease-associated mutant ubiquitin causes proteasomal impairment and enhances the toxicity of protein aggregates. PLoS Genet 2009;5:e1000382.
23. Barmada SJ. Cytoplasmic mislocalization of tdp-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 2010;30:639-49.
24. Lim L, Wei Y, Lu Y, Song J. Als-causing mutations significantly perturb the self-assembly and interaction with nucleic acid of the intrinsically disordered prion-like domain of tdp-43. PLoS Biol 2016;14:e1002338.
25. Barmada SJ, Finkbeiner S. Pathogenic tardbp mutations in amyotrophic lateral sclerosis and frontotemporal dementia: disease-associated pathways. Rev Neurosci 2010;21:251-72.
26. Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, Tsvetkov A, Pleiss M, Li X, Peisach D, Shaw C, Chandran S, Finkbeiner S. Autophagy induction enhances tdp43 turnover and survival in neuronal als models. Nat Chem Biol 2014;10:677-85.
27. Kim PY, Tan O, Liu B, Trahair T, Liu T, Haber M, Norris MD, Marshall GM, Cheung BB. High tdp43 expression is required for trim16-induced inhibition of cancer cell growth and correlated with good prognosis of neuroblastoma and breast cancer patients. Cancer Lett 2016;374:315-23.
28. Bhandare VV, Ramaswamy A. The proteinopathy of d169g and k263e mutants at the RNA recognition motif (RRM) domain of tar DNA-binding protein (tdp43) causing neurological disorders: a computational study. J Biomol Struct Dyn 2017; doi: 10.1080/07391102.2017.1310670.
29. Iguchi Y, Eid L, Parent M, Soucy G, Bareil C, Riku Y, Kawai K, Takagi S, Yoshida M, Katsuno M, Sobue G, Julien JP. Exosome secretion is a key pathway for clearance of pathological tdp-43. Brain 2016;139:3187-201.
30. Pokrishevsky E, Grad LI, Yousefi M, Wang J, Mackenzie IR, Cashman NR. Aberrant localization of fus and tdp43 is associated with misfolding of sod1 in amyotrophic lateral sclerosis. PLoS One 2014;7:e35050.
31. Pokrishevsky E, Grad LI, Cashman NR. TDP-43 or FUS-induced misfolded human wild-type sod1 can propagate intercellularly in a prion-like fashion. Sci Rep 2016;6:22155.
32. Gros-Louis F, Gaspar C, Rouleau GA. Sporadic and hereditary amyotrophic lateral sclerosis. Biochim Biophys Acta 2015;1852:679-84.
33. Shintaku M, Kaneda D, Oyanagi K. Novel intracytoplasmic inclusions immunoreactive for phosphorylated-TDP43 and cystatin c in anterior horn cells in a case of sporadic amyotrophic lateral sclerosis. Neuropathology 2017;37:526-34.
34. Braun RJ. Ubiquitin-dependent proteolysis in yeast cells expressing neurotoxic proteins. Front Mol Neurosci 2015;8:8.
35. Wang W, Arakawa H, Wang L, Okolo O, Siedlak SL, Jiang Y, Gao J, Xie F, Petersen RB, Wang X. Motor-coordinative and cognitive dysfunction caused by mutant TDP-43 could be reversed by inhibiting its mitochondrial localization. Mol Ther 2017;25:127-39.
36. Hebron ML, Lonskaya I, Sharpe K, Weerasinghe PP, Algarzae NK, Shekoyan AR, Moussa CE. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J Biol Chem 2013;288:4103-15.
37. Jackson KL, Dayton RD, Orchard EA, Ju S, Ringe D, Petsko GA, Maquat LE, Klein RL. Preservation of forelimb function by UPF1 gene therapy in a rat model of TDP-43-induced motor paralysis. Gene Ther 2015;22:20-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Dong Y, Chen Y. The role of ubiquitinated TDP-43 in amyotrophic lateral sclerosis. Neurosciences 2018;5:5. http://dx.doi.org/10.20517/2347-8659.2017.47
AMA Style
Dong Y, Chen Y. The role of ubiquitinated TDP-43 in amyotrophic lateral sclerosis. Neuroimmunology and Neuroinflammation. 2018; 5: 5. http://dx.doi.org/10.20517/2347-8659.2017.47
Chicago/Turabian Style
Dong, Yi, Yan Chen. 2018. "The role of ubiquitinated TDP-43 in amyotrophic lateral sclerosis" Neuroimmunology and Neuroinflammation. 5: 5. http://dx.doi.org/10.20517/2347-8659.2017.47
ACS Style
Dong, Y.; Chen Y. The role of ubiquitinated TDP-43 in amyotrophic lateral sclerosis. Neurosciences. 2018, 5, 5. http://dx.doi.org/10.20517/2347-8659.2017.47
About This Article
Copyright

Data & Comments
Data


 Cite This Article 19 clicks
Cite This Article 19 clicks

 Like This Article 29
likes
Like This Article 29
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.