
Steroids and plasma exchange in Isaacs' syndrome with anti-Caspr2 antibodies


Abstract
Isaacs’ syndrome is a disease characterized by nerve hyperexcitability. The patients are commonly treated with symptomatic therapies and immunomodulatory approaches, but no clinical trials are available to date. Here, we report the case of an anti-Caspr2-positive patient, presenting with continuous muscle twitches and diffuse muscle pain. He experienced a nearly complete clinical response to intravenous high-dose steroids combined with plasma exchange, sustained for at least 1 year. Our experience suggests that methylprednisolone 1000 mg/day × 5 days and consecutive tapering followed by plasma exchange may be efficient and well tolerated in patient with Isaacs’ syndrome due to anti-Caspr2 antibodies.
Keywords
Introduction
Isaacs’ syndrome (“acquired neuromyotonia”) is a disease characterized by peripheral nerve hyperexcitability and spontaneous and continuous skeletal muscle overactivity presenting as twitching and painful cramps, often accompanied by stiffness, pseudomyotonia, pseudotetany and weakness[1]. The commonest acquired form is autoimmune, caused by antibodies against nerve voltage-gated potassium channels (VGKC). Patients are commonly treated with symptomatic therapies (carbamazepine, phenytoin, lamotrigine or valproate) and immunomodulatory approaches, but no clinical trial is available to date and the optimal treatment approach is unknown[1]. Here, we report the case of a patient with Isaacs’ syndrome tested positive for anti-contactin-associated protein-2 (Caspr2) antibodies.
Case report
A 74-year-old Italian man, with unremarkable family history and without any significant comorbidities, reported diffuse muscle pain for the last three weeks. In the previous year he had occasionally presented muscle cramps. The pain had gradually worsened so that required the use of morphine intravenously and carbamazepine (400 mg/day) and pregabalin (225 mg/day) oral administration. Neurological examination revealed continuous muscle twitches in upper and lower limbs and in the facial muscles [Video 1]. The remaining examination was unremarkable.
[Video 1]
Routine laboratory testing showed increased creatine kinase (CK) levels (579 U/L; normal < 190), mild anemia (haemoglobin ≈ 10 g/dL), increased erythrocyte sedimentation rate (74 mm/h; normal < 40) and C-reactive protein levels (13.6 mg/dL; normal < 0.5). Serological studies were negative for HIV, syphilis and Borrelia. Autoantibodies associated with rheumatic diseases, acetylcholine receptor and MuSK antibodies, anti-gangliosides, anti-Hu/Yo/Ri/CV2/Ma2, anti-amphiphisin and anti-GAD antibodies were not detected in the patient’s serum. Cerebrospinal fluid examination (CSF) showed moderately increased protein levels (73 mg/dL; reference range 15-40 mg/dL), without any signs of intrathecal immunoglobulin synthesis.
Total-spine and brain magnetic resonance imaging (MRI) and electroencephalography were normal. An extensive screening search for malignancies, including chest/abdomen computed tomography, gastroscopy, colonoscopy, and tumour markers measurement resulted negative, whereas a segmental Crohn’s disease was diagnosed. Sensory and motor evoked potentials and nerve conduction studies were normal, whereas electromyography (EMG) showed continuous spontaneous activity in all the examined muscles [Figure 1], suggesting the diagnosis of Isaacs’ syndrome. At last, this diagnosis was confirmed by the finding of anti-Caspr2 antibodies in the patient’s serum.
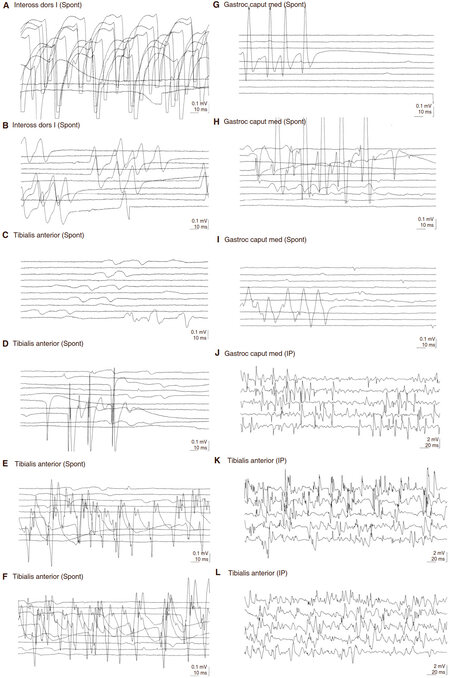
Figure 1. Continuous spontaneous activity in all the examined muscle districts. Electromyography showing abnormal repetitive spontaneous activity of the muscle fibers (doublet, triplet, or multiple single-unit discharges with high, irregular intraburst frequency) in all the muscle groups (reported in the figure). (A-E) Right side; (F-G) left side. Spont: spontaneous activity
Therefore, the patient was treated with high-dose corticosteroids (intravenous methylprednisolone 1000 mg/day × 5 days and consecutive tapering to prednisone 25 mg/day) followed by 5 sessions (about 2 h every other day) of plasma exchange (PEX), with immediate remission of the muscle twitches and pain at the end of the corticosteroid cycle and after the first PEX session [Video 2]. EMG confirmed this excellent improvement [Video 2]. At discharge, CK levels were normal. One year after discharge, the disease was still well controlled with low-dose oral corticosteroids (prednisone 25 mg/day) and symptomatic therapy with carbamazepine and pregabalin. Five years after discharge, the disease is excellently controlled by azathioprine 125 mg/day, which was prescribed as a corticosteroid-sparing treatment. Prednisone administration was interrupted. An extensive screening search for malignancies was repeated and was still normal.
[Video 2]
Discussion
Neuromyotonia is a syndrome of spontaneously occurring muscle activity of peripheral nerve origin, which can be triggered by voluntary or induced muscle contraction[2]. It is one among several causes of visible myokymia. The abnormal activity is characterized electromyographically by doublet, triplet or multiplet single unit discharges that have a high intraburst frequency, the frequency of the bursts themselves being irregular[2]. Our patient fulfilled these EMG criteria [Figure 1].
Anti-VGKC autoantibodies have been identified in patients with acquired neuromyotonia, limbic encephalitis, or a combination of both (Morvan’s syndrome). However, recent studies have shown that in fact the great majority of “anti-VGKC” autoantibodies are not directed against the potassium channel subunits but against three proteins that are complexed with these channels, including the cell-adhesion molecule Caspr2[3]. VGKC-antibodies define neurological conditions that are usually immunotherapy-responsive, but patients with anti-Caspr2 antibodies could have an increased risk of an underlying tumour and a poor prognosis[3].
Caspr2 antibodies bind to the juxtaparanodal regions of myelinated fibers in brain and peripheral nerve[4]. Patients with anti-Caspr2 antibodies may show both peripheral and central nervous system features[3,4]. Neuropathic pain may be a significant and rather specific manifestation of anti-Caspr2 autoimmunity; hyperexcitability of nociceptive pathways has been implicated[5]. Peripheral neuromuscular hyperexcitability and pain were the main clinical features in our patient; no clinical or MRI signs of limbic encephalitis were noted. Additional work is required to explain different clinical phenotypes in patients with autoantibodies against Caspr2.
Isaacs’ syndrome has been described in combination with other autoimmune disorders[6], including myasthenia gravis[7], or associated with a variety of neoplasms, including thymoma and lymphoma[8]. It can be diagnosed several years before a neoplasm is discovered[8]. In our case, an extensive screening was negative for neoplasms, but revealed a segmental Crohn’s disease (to our knowledge, this is the first report of this association). Patients with Isaacs’ syndrome[9,10] usually improve after treatment of an underlying cancer or with symptomatic treatment, although evidence is based on case reports[1]. Carbamazepine, phenytoin, lamotrigine and sodium valproate can be used, if necessary in combination[1]. In patients whose symptoms are debilitating or refractory to symptomatic therapy, immunomodulatory therapies should be tried[1]. There are no trials of long-term oral immunosuppression. Prednisolone, with or without azathioprine or methotrexate, has been used in some patients[1]. Single case studies suggest that plasma exchange and intravenous immunoglobulins may produce some clinical improvement[1]. Of note, all the case studies reporting the effects of the above mentioned immunomodulatory approaches predated the discovery of anti-Caspr2 antibodies[3]. Therefore, the immunological serotype of these patients was unknown. Further studies are strongly needed to clarify if the serotype influence the optimal therapeutic approach.
Our report suggests that methylprednisolone 1000 mg/day × 5 days and consecutive tapering followed by plasma exchange can be effective and well tolerated in patient with Isaacs’ syndrome due to anti-Caspr2 antibodies. Since the incidence of this condition is rare, controlled clinical studies are not likely to be conducted. Therefore, it is important to report single observations; more cases will be necessary to confirm (or not) the positive effect of this immunomodulatory schedule in anti-Caspr2 Isaacs’ syndrome.
Declarations
AcknowledgmentsThe authors are grateful to Dr. Valeria Calsolaro (Neurology Imaging Unit, Imperial College, London & Department of Clinical and Experimental Medicine, University of Pisa, Italy) for her language revision.
Authors’ contributionsPrepared the manuscript: Orsucci D
Revised the manuscript: Mazzoni M
Clinically studied the patient: Moscato G, Napolitano A
Performed the electromyographic examinations: Cafforio G
Performed the plasma exchange procedure: Margelli M
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentThe manuscript does not contain patient identifiable data.
Ethics approvalNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Skeie GO, Apostolski S, Evoli A, Gilhus NE, Illa I, Harms L, Hilton-Jones D, Melms A, Verschuuren J, Horge HW; European Federation of Neurological Societies. Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol 2010;17:893-902.
2. Newsom-Davis J, Mills KR. Immunological associations of acquired neuromyotonia (Isaacs' syndrome). Report of five cases and literature review. Brain 1993;116:453-69.
3. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, Peles E, Buckley C, Lang B, Vincent A. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734-48.
4. Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A, Martinez-Hernandez E, Wilson C, Jacobs D, Lai M, Walker RW, Graus F, Bataller L, Illa I, Markx S, Strauss KA, Peles E, Scherer SS, Dalmau J. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011;69:303-11.
5. Klein CJ, Lennon VA, Aston PA, McKeon A, Pittock SJ. Chronic pain as a manifestation of potassium channel-complex autoimmunity. Neurology 2012;79:1136-44.
6. Myers KA, Baker SK. Late-onset seropositive Isaacs' syndrome after Guillain-Barre syndrome. Neuromuscul Disord 2009;19:288-90.
7. Lang B, Vincent A. Autoimmune disorders of the neuromuscular junction. Curr Opin Pharmacol 2009;9:336-40.
8. Rana SS, Ramanathan RS, Small G, Adamovich B. Paraneoplastic Isaacs' syndrome: a case series and review of the literature. J Clin Neuromuscul Dis 2012;13:228-33.
9. Hart IK, Waters C, Vincent A, Newland C, Beeson D, Pongs O, Morris C, Newsom-Davis J. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997;41:238-46.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Orsucci D, Cafforio G, Moscato G, Napolitano A, Margelli M, Mazzoni M. Steroids and plasma exchange in Isaacs' syndrome with anti-Caspr2 antibodies. Neurosciences 2018;5:7. http://dx.doi.org/10.20517/2347-8659.2017.67
AMA Style
Orsucci D, Cafforio G, Moscato G, Napolitano A, Margelli M, Mazzoni M. Steroids and plasma exchange in Isaacs' syndrome with anti-Caspr2 antibodies. Neuroimmunology and Neuroinflammation. 2018; 5: 7. http://dx.doi.org/10.20517/2347-8659.2017.67
Chicago/Turabian Style
Orsucci, Daniele, Gianfranco Cafforio, Gianluca Moscato, Alessandro Napolitano, Marco Margelli, Monica Mazzoni. 2018. "Steroids and plasma exchange in Isaacs' syndrome with anti-Caspr2 antibodies" Neuroimmunology and Neuroinflammation. 5: 7. http://dx.doi.org/10.20517/2347-8659.2017.67
ACS Style
Orsucci, D.; Cafforio G.; Moscato G.; Napolitano A.; Margelli M.; Mazzoni M. Steroids and plasma exchange in Isaacs' syndrome with anti-Caspr2 antibodies. Neurosciences. 2018, 5, 7. http://dx.doi.org/10.20517/2347-8659.2017.67
About This Article
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.