
Hypoparathyroidism presenting with late onset seizures - a report of two cases from rural India

Abstract
Hypoparathyroidism leading to hypocalcaemia is an important treatable cause of recurrent seizures. Neurological manifestations due to hypoparathyroidism include: seizures, paresthesia, depression, psychosis, extra pyramidal symptoms, and features of raised intracranial pressure. Seizures may be the presenting symptoms preceding other signs of hypocalcaemia. Primary hypoparathyroidism presenting for the first time as seizures in the elderly is quite rare. Here we report two cases of hypoparathyroidism presenting with seizures in the elderly as the sole manifestation of hypocalcaemia. The goal of this report is to seek attention to such an uncommon reversible treatable cause of seizures and to consider hypoparathyroidism in the work up of these patients.
Keywords
Introduction
Hypoparathyroidism occurs when too little parathyroid hormone (PTH) is released from the parathyroid glands, or the released PTH does not work properly[1]. Common clinical manifestations of hypoparathyroidism include tingling and numbness, carpopedal spasm, neurocognitive dysfunction and seizures. Hypoparathyroidism and pseudohypoparathyroidism (due to deficient end-organ response to PTH) are the most familiar reasons for pathological basal ganglia calcification.
Invariably, hypoparathyroidism presents with a spectrum of clinical features as above, here we report two cases with late onset seizures being the only manifestation of primary hypoparathyroidism which is quite uncommon in literature.
Case reports
Case 1
A 62-year-old right-handed male farmer presented to emergency department with 3 episodes of abnormal movement in the form of generalized muscles stiffness with eyes-rolling and associated with frothing, tongue bite and incontinence in a 2-h cycle. Each episode lasted for approximately 5 min and at the interval of 30-40 min. He had similar two episodes one year and one episode 15 days back. These were not preceded by any aura and each attack lasts for two to three minutes with postictal confusion for 15 to 20 min. He reported no history of neck surgery, muscle cramps, parasthesia, and psychiatric illness. He had cataract extraction surgery on the right eye 4 years back. Family and social history were unremarkable.
He was treated by some local practitioner with antiepileptic drugs (phenytoin 300 mg and clobazam 20 mg/day) without basic investigations. His vital signs, weight and height were normal with no dysmorphic features (no dry hairs or brittle nails). Neurological examination, including higher mental functions, cranial nerves, motor, sensory, reflexes and coordination examination were normal. There was no carpopedal spasm or any other signs of tetany like Chvostek’s or Trousseau’s sign. Computerized tomography scan head was done showing bilateral basal ganglia calcification and many punctiform calcifications between cortical and subcortical parts [Figure 1].
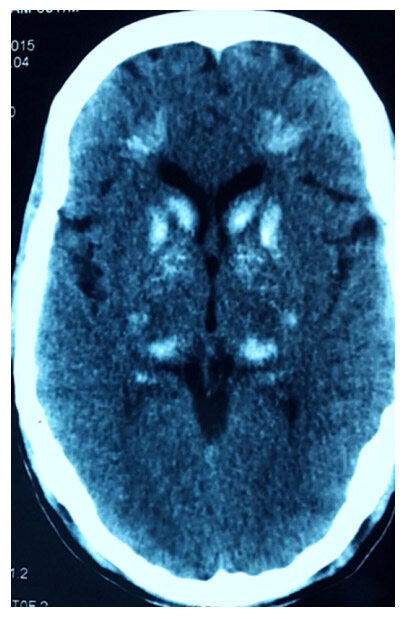
Figure 1. NCCT head scan was done which showed bilateral basal ganglia calcification and many punctiform calcifications between cortical and subcortical parts
Electroencephalography (EEG) was normal. The laboratory investigations included complete blood count, erythrocyte sedimentation rate, routine chemistries, liver, and renal function test were normal. He was found to have a serum calcium level of 6.3 mg/dL (normal range 8.0-10.4 mg/dL) with a serum PTH level of 3.8 pg/mL (normal range 15-68 pg/mL), serum phosphorus 5.9 mg/dL (normal range 2.5-4.5 mg/dL) and magnesium 1.4 mg/dL (normal range 1.3-2.5 mg/dL).
Case 2
A 65-year-old right-handed female housewife presented with history of episodic eye-rolling with generalized tonic-clonic movements of limbs and was associated with incontinence of urine and tongue bite, for the duration of 6 months. She has 6-7 attacks/month and each attack last for 3-4 min without aura with postictal state for 30 min. There was no past or family history of epilepsy. She was on antiepileptic drugs carbamezapine 800 mg/day and clobazam 20 mg/day started by local practitioner. The patient did not have any symptoms of hypocalcemia. Her general examination was normal and there were no focal neurological signs.
Signs of hypocalcaemia were elicited by inflating a cuff above systolic blood pressure over the arm for 2 min, provoking flexion of the wrist and metacarpophalangeal joints, hyperextension of the fingers, and flexion of the thumb (carpopedal spasm - Trousseau’s sign), and by tapping the face just anterior to the ear, provoking twitching of the ipsilateral facial muscles (Chovstek’s sign). Computerized tomography (CT) scan of the head revealed bilateral basal ganglia and cerebellar calcification [Figures 2 and 3]. She was investigated for hypoparathyroidism. Her serum calcium level was 5.9 mg/dL with a serum PTH level of 2.8 pg/mL and serum phosphorus 7.2 mg/dL. EEG showed runs of higher amplitude theta and delta activity with very marked response during hyperventilation.
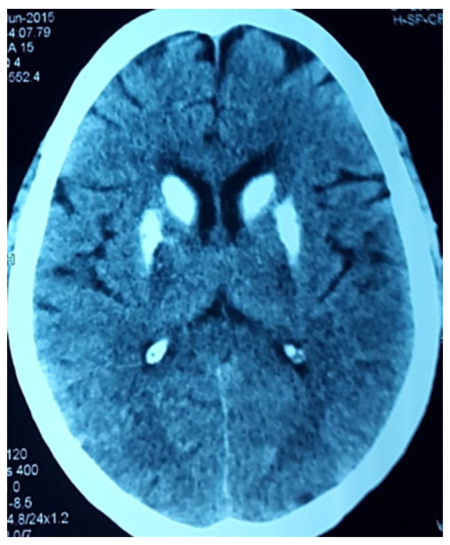
Figure 2. Computerized tomography scan of the head revealed bilateral basal ganglia
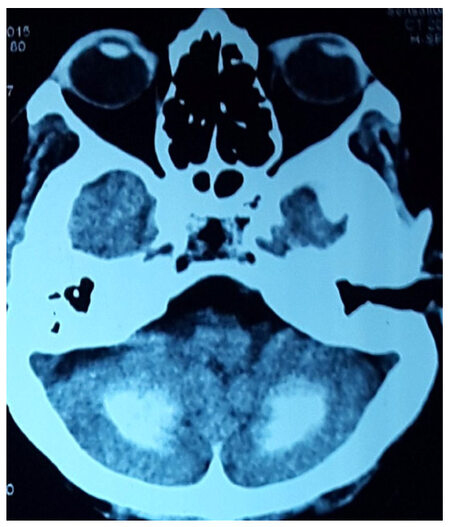
Figure 3. Computerized tomography scan of cerebellar calcification
Because of the presence of bilateral basal ganglia and cerebellar calcification on CT, the patients were investigated for hypoparathyroidism. Both patients had hypocalcemia, hyperphosphatemia with low serum PTH levels. The biochemical profile favored the diagnosis of primary hypoparathyroidism. Both patients were started on daily supplementation of 0.5 µg of calcitriol and 1000 mg of elemental calcium (calcium carbonate). The patients were discharged with oral calcium and vitamin D. The biochemical profile normalized, seizures were controlled, and eventually antiepileptic drugs were withdrawn. They did not experience any further seizures in the next 12 months.
Discussion
Hypoparathyroidism can be caused by congenital disorders (like Di George syndrome, mitochondrial cytopathies), receptor insensitivity (pseudohypoparathyroidism type Ia–c, II), surgery, autoimmune disorders (familial autoimmune polyglandular syndrome type I), or hemochromatosis, or can be idiopathic[2,3].
Acquired chronic hypoparathyroidism is generally the after effect of unintentional surgical removal of all the parathyroid glands. Even rarer causes of acquired chronic hypoparathyroidism include radiation-induced damage and glandular damage in patients with hemochromatosis or hemosiderosis after repeated blood transfusions. Secondary hypoparathyroidism is a physiological state in which PTH levels are low in response to a primary process that causes hypercalcemia.
The essential capacity of PTH is to keep up the extracellular fluid (ECF) calcium concentration within a normal narrow range. The hormone acts directly on bone and kidney and indirectly on the intestine through its effects on synthesis of 1,25(OH)2D3 to increase serum calcium concentrations; in turn, PTH generation is firmly directed by the concentration of serum ionized calcium. Any tendency toward hypocalcemia, is counteracted by an increased secretion of PTH. This in turn: (1) increases the rate of dissolution of bone mineral, thereby increasing the flow of calcium from bone into blood; (2) reduces the renal clearance of calcium, returning more of the calcium filtered at the glomerulus into ECF; and (3) increases the efficiency of calcium absorption in the intestine by stimulating the production of 1,25(OH)2D3.
Hypocalcemia produces hyperexcitability of nerve fibers with spontaneous and repetitive discharges. As a result, patients have perioral and distal numbness and parasthesiae, carpopedal spasm and diffuse muscle cramps. Latent tetany can be elicited by hyperventilation, by tapping the facial muscles (Chovstek’s sign) or by occluding venous return from arm, resulting in carpopedal spasm (Trousseau’s sign). In severe cases laryngeal muscle spasm and convulsions may develop. Respiratory arrest may also occur. Chronic hypoparathyroidism is associated with markedly abnormal skeletal microstructure, despite marked increases in bone mineral density.
Increased intracranial pressure, papilledema, and convulsions can also be present, and must be differentiated from severe tetany muscular spasms[4,5]. Rare extrapyramidal signs including parkinsonism and cerebellar signs have been reported. Mental changes include irritability, depression, and psychosis. The QT interval on the electrocardiogram is prolonged, in contrast to its shortening with hypercalcemia. Arrhythmias occur, and digitalis effectiveness may be reduced. Intestinal cramps and chronic malabsorption may also occur.
Seizures are a frequent complication: they have been reported in 20%-25% of patients with acute hypocalcemia and in 30%-70% of patients with idiopathic hypoparathyroidism[6]. Epileptic seizures may happen at any age and usually generalized tonic clonic with loss of consciousness. Non-convulsive status has been also reported. Seizures may be the presenting symptoms preceding other signs of hypocalcemia such as chorea and tetany. Several types of partial motor seizures including jacksonian seizures may also be observed. If not treated the results of these convulsions may be very serious. Seizures are thought to occur due to hypocalcemia and intracranial calcification that occur in vascular and perivascular locations[7,8].
Early EEG changes associated with hypocalcemia include evolution from alpha through theta and delta dominance. Runs of higher amplitude theta activity also appear and a very marked response to over breathing occurs. Other EEG findings (generalized spikes, sharp-waves burst of delta activity with sharp components). Generalized paroxysmal discharges and absence status epileptic have also been reported[9]. No correlation was found between calcium level and EEG changes and these changes typically revert to normal with correction of the serum calcium levels.
Eaton et al.[10] in 1939 first described basal ganglia calcification (BGC) in association with chronic hypoparathyroidism. Pathogenesis is obscure, but its occurrence with hypocalcemia signifies an important role of increased calcium-phosphorus complex formation. Radiological studies have found that calcification encompassing cerebral veins most habitually happen in the lentiform (putamen and globus pallidus) and the caudate cores of the basal ganglia; however, the factors that predispose individuals to basal ganglia calcification have not been identified[11]. Other areas affected by BCG include the thalamus, dentate nuclei, cerebral cortex, gray-white junctions, and the cerebellum[10,12]. Such intracranial calcification occurs in 0.3% to 1.5% of patients with hypoparathyroidism, and is often detected incidentally[13,14]. Treatment involves replacement with vitamin D or 1,25(OH)2D3 (calcitriol) combined with a high oral calcium intake. For many patients, vitamin D in doses of 40,000-120,000 U/day (1-3 mg/day) combined with ≥ 1 g elemental calcium is satisfactory. Many physicians now use 0.5-1 mg of calcitriol in management of such patients, especially if they are difficult to control. It is critical to monitor therapy closely since overtreatment may result in hypercalciuria, hypercalcemia, renal stones, and nephrocalcinosis. Modi et al.[15] reported among 70 patients, seizures were present in 64.3% of patients with idiopathic hypoparathyroidism and they responded to antiepileptic drugs (AEDs) and calcium/1a(OH)D3 during the follow-up and it was possible to withdraw AEDs in 71% of patients.
In both of our patients’ basic investigations were not done before starting the antiepileptic drugs due to lack of facilities in this rural and remote part of country. Hypoparathyroidism is a rare yet treatable cause for seizures. For any patient having new onset seizures even in the elderly population basic investigations, including calcium profile, may uncover the hidden etiology.
The diagnosis of idiopathic hypoparathyroism is often missed if presentation is in elder age group. The objective of this report is to increase awareness of such a rare, reversible cause of seizures and to consider hypoparathyroidism in the work up of these patients.
Declarations
Authors’ contributionsBoth the case workup and treatment: Verma A
Manuscript writing: Kumar A
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentConsents from both patients were established prior to submission.
Ethics approvalAll treatment and study were performed in compliance with our institutional standard and the Declaration of Helsinki, No: 314 /UPUMS/Dean/2016-2017.
Copyright© The Author(s) 2018.
REFERENCES
2. Bilezikian JP, Khan A, Potts JT, Brandi ML, Clarke BL, Shoback D, Jüppner H, D'Amour P, Fox J, Rejnmark L, Mosekilde L, Rubin M, Dempster D, Gafni R, Collins MT, Sliney J, Sanders J. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Miner Res 2011;26:2317-37.
3. Chung SH, Chen SC, Chen WJ, Lee CC. Symmetric basal ganglia calcification in a 9-year-old child with MELAS. Neurology 2005;65:E11-9.
4. Levine M. Hypoparathyroidism and pseudohypoparathyroidism. In: DeGroot LJ, Jameson JL, editors. Endocrinology. Philadelphia: WB Saunders; 2001. pp. 1133-53.
5. Weiss-Guillet EM, Takala J, Jakob SM. Diagnosis and management of electrolyteemergencies. Best Pract Res Clin Endocrinol Metab 2003;17:623-51.
6. Nardonea R, Brigoc F, Trinkaa E. Acute symptomatic seizures caused by electrolyte disturbances. J Clin Neurol 2016;12:21-33.
7. Fulop M, Zeifer B. Case report: extensive brain calcification in hypoparathyroidism. Am J Med Sci 1991;302:292-5.
8. Usdin TB, Hoare SR, Wang T, Mezey E, Kowalak JA. TIP39: a new neuropeptide and PTH2-receptor agonist from hypothalamus. Nat Neurosci 1999;2:941-3.
10. Eaton LM, Camp JD, Love JG. Symmetric cerebral calcifications, particularly of the basal ganglia demonstrate roentgenographically; calcification of the cerebral blood vessels. Arch Neurol Psychiatry 1939;41:921-42.
11. Goswami R, Sharma R, Sreenivas V, Gupta N, Ganapathy A, Das S. Prevalence and progression of basal ganglia calcification and its pathogenic mechanism in patients with idiopathic hypoparathyroidism. Clin Endocrinol Oxf 2012;77:200-6.
12. Rizvi I, Ansari NA, Beg M. Widespread intracranial calcification, seizures, and extrapyramidal manifestations in a case of hypoparathyroidism. N Am J Med Sci 2012;4:369-72.
13. Verulashvili IV, Glonti LS, Miminoshvili DK, Maniia MN, Mdivani KS. Basal ganglia calcification: clinical manifestations and diagnostic evaluation. Georgian Med News 2006;140:39-43.
14. Basak R. A case report of basal ganglia calcification: a rare finding of hypoparathyroidism. Oman Med J 2009;24:220-2.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Verma A, Kumar A. Hypoparathyroidism presenting with late onset seizures - a report of two cases from rural India. Neurosciences 2018;5:20. http://dx.doi.org/10.20517/2347-8659.2017.65
AMA Style
Verma A, Kumar A. Hypoparathyroidism presenting with late onset seizures - a report of two cases from rural India. Neuroimmunology and Neuroinflammation. 2018; 5: 20. http://dx.doi.org/10.20517/2347-8659.2017.65
Chicago/Turabian Style
Verma, Archana, Alok Kumar. 2018. "Hypoparathyroidism presenting with late onset seizures - a report of two cases from rural India" Neuroimmunology and Neuroinflammation. 5: 20. http://dx.doi.org/10.20517/2347-8659.2017.65
ACS Style
Verma, A.; Kumar A. Hypoparathyroidism presenting with late onset seizures - a report of two cases from rural India. Neurosciences. 2018, 5, 20. http://dx.doi.org/10.20517/2347-8659.2017.65
About This Article
Copyright

Data & Comments
Data


 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 19
likes
Like This Article 19
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.