
Presynaptic mechanisms at prefrontal synapses involved in persistent pain
 0
0
Abstract
The cumulative evidence from animal and human studies revealed that the anterior cingulate cortex (ACC) plays essential roles in pain sensation and persistent pain. It has been evident in the ACC synapses of animals that changes in both the presynaptic and postsynaptic function are caused by peripheral nerve injury. Thus far, postsynaptic changes in the ACC following nerve injury have been primarily studied to understand the mechanisms of chronic pain. In recent years, studies focusing on the presynaptic mechanisms in chronic pain have been progressively increased. In this review, I will discuss molecular mechanisms associated with chronic pain and presynaptic form of long-term potentiation. I will also discuss evidence for presynaptic changes in the ACC caused by disease-related pain.
Keywords
Introduction
The anterior cingulate cortex (ACC) plays critical roles in the processing of pain information in patients and is involved in behavioral responses to tissue damages that cause pain in animals[1-8]. The ACC also contributes to various cognitive processes including decision-making, attention and working memory[9,10]. Humans and animals with persistent pain displayed poor performance in decision-making cognitive tasks[11-13]. Therefore, the understanding of the cellular and molecular mechanisms of chronic pain is very important in ameliorating the cognitive impairments.
Sensory inputs from the periphery are conveyed to the dorsal root ganglion and then to the spinal dorsal horn (SDH). The SDH neurons send ascending projection to the thalamus. Subsequently, the outputs from the thalamus synapse on the neurons in the ACC, amygdala and other cortices including the somatosensory and insular cortex[3,5,14,15][Figure 1].
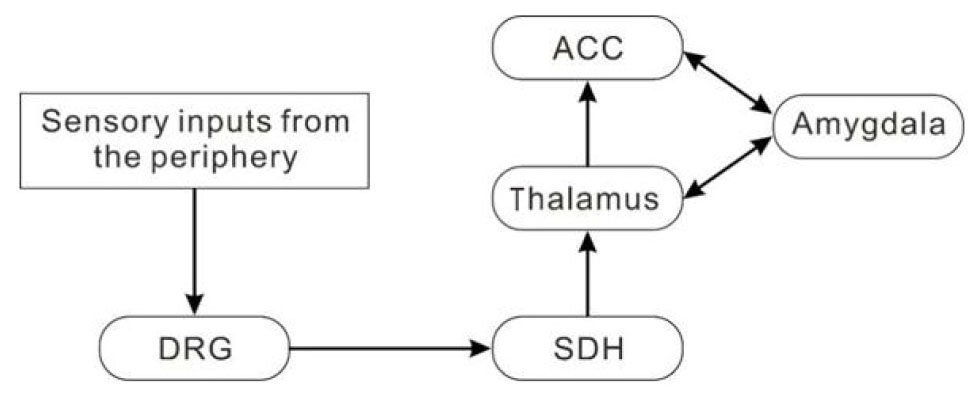
Figure 1. A possible neuronal network for the anterior cingulate cortex (ACC) in pain transmission. Sensory inputs from the periphery are conveyed to the dorsal root ganglion (DRG) and then to the spinal dorsal horn (SDH). Some of the SDH neurons send ascending projection to the neurons in the thalamus. Subsequently, the outputs from the thalamus synapse on the neurons in the amygdala, ACC and other cortical neurons (not shown)
With regard to the synaptic mechanisms of chronic inflammatory and neuropathic pain, it has been proposed that changes in both the presynaptic and postsynaptic function play essential roles[8,16]. To date, a number of studies have shown that tissue injury- or nerve damage-caused central sensitization, a similar phenomenon like long-term potentiation (LTP), in the ACC could contribute to the persistent pain[7,8]. Since it has generally been believed that postsynaptic mechanisms are crucial for the LTP expression, postsynaptic changes following nerve injury in the ACC synapses have been primarily studied to understanding the mechanisms of chronic pain. In behavioral experiments using genetic and pharmacological approaches, inhibiting postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor-mediated synaptic plasticity is shown to be sufficient to produce analgesic effects[17]. These data indicate that postsynaptic LTP in the ACC is involved in chronic pain. Compared to studies focusing on postsynaptic mechanisms, those focusing on presynaptic mechanisms which contribute to persistent pain are relatively few. However, presynaptic changes in the ACC underlying chronic pain have been progressively elucidated in recent years. In this review, I will discuss studies regarding the molecular mechanisms that play pivotal roles in chronic pain and presynaptic form of long-term potentiation (pre-LTP) in the ACC. In addition, I discuss presynaptic changes associated with disease-related pain in the ACC.
Molecular mechanisms of presynaptic changes in the ACC following nerve injuries and inflammation
Calcium/calmodulin-stimulated adenylyl cyclase
Cyclic adenosine monophosphate (cAMP) is a nucleotide that acts as a key second messenger in a number of physiological functions including chronic pain, learning and memory, emotional fear and drug abuse[18,19]. The adenylyl cyclase (AC) is the important enzyme that converts ATP to cAMP. The AC family is composed of nine membrane-bound isoforms (AC1-9) and one soluble isoform (sAC). These isoforms are differentially distributed in the body, and each AC isoform has distinct physiological functions[20]. Among AC1-9 and sAC, AC1 and AC8 are the key AC isoforms that respond positively to calcium-calmodulin[20]. AC1 is 4 to 5 times more sensitive to an increase in calcium concentration than AC8. It has been shown that AC1 is abundantly expressed in the mouse ACC neurons[21]. We have previously demonstrated that deletion of AC1 and AC8 genes significantly reduced pain sensitization in mice with chronic inflammatory pain[22] and in those with chronic neuropathic pain[16,21]. In ACC synapses of mice with inflammatory pain, the enhancement of presynaptic transmitter release was suppressed by inhibition of AC1 and/or AC8[22]. Thus, presynaptic AC1 and/or AC8 could be the key molecules that contribute to the enhancement of both the probability of transmitter release and the number of available vesicles in response to inflammatory pain. Subsequently, AC1 and/or AC8 would activate protein kinase A (PKA) and then phosphorylate cAMP response element binding protein[21]. In addition, we have shown that AC1 plays essential roles in both the presynaptic and postsynaptic changes in ACC synapses in the mouse spinal nerve ligation model of neuropathic pain[16].
An aplysia octopamine receptor (Ap oa1) is G protein-coupled and selectively activates cAMP/PKA pathway[23]. By using transgenic mice heterologously expressing Ap oa1, we have previously examined whether and how cAMP in the ACC synapses is involved in the presynaptic modulation of neurotransmitter release. We found that the activation of Ap oa1 by octopamine augmented glutamatergic synaptic transmission in the ACC synapses[24]. Also, behavioral responses to inflammatory pain were apparently facilitated by bilateral microinjection of octopamine into the ACC[24]. These findings provide the evidence that the presynaptic modulation by cAMP contributes to chronic pain caused by peripheral inflammation. Therefore, AC1 may be potential therapeutic targets for treatment of chronic pain. Indeed, intraperitoneal or oral application of NB001, an AC1 inhibitor, reduced pain sensitivity in neuropathic pain mice without no apparent side effects[25,26].
Inflammation-related factors
Tumor necrosis factor alpha (TNF-α) is a potent prototypic inflammatory cytokine and is produced by a wide variety of cells including neuronal and glial cells[27,28]. TNF-α increases glutamatergic synaptic transmission by inhibiting glutamate transporters expressed in glial cells and/or by increasing the surface expression of AMPA and N-methyl-d-aspartate (NMDA) receptors[29]. In addition, TNF-α is known to serve as a trigger for other cytokines in response to inflammation or injury. There is a large number of evidence that TNF-α is responsible for the development and maintenance of chronic inflammatory and neuropathic pain[30]. A previous finding proposed that TNF-α may induce acute mechanical sensitization of peripheral nociceptors by acting on TNF receptor 1[31]. In the mouse ACC, the protein level of TNF-α was significantly increased following hind-paw administration of complete freund’s adjuvant[32]. Together with this finding, the glutamatergic synaptic transmission was significantly enhanced by TNF-α in the ACC synapses. These data suggest that that presynaptic changes caused by peripheral nerve inflammation are partly brought about by the increased TNF-α in the ACC synapses.
Chemokines that belong to proinflammatory cytokines play essential roles in cell growth, immune system, cell development and inflammation. The two chemokine superfamilies, the CC and the CXC chemokines, are thought to be important for these functions[33]. Five receptors for CXC chemokines and eight receptors for CC chemokines have been characterized[34]. Among CXC chemokines, interleukin-8 (IL-8), known as CXCL8, is a critical proinflammatory CXC chemokine that is associated with the neutrophil recruitment and degranulation in inflammatory response[35]. In response to inflammatory responses within the central nervous system, IL-8 is produced by activated microglia and astrocytes[36] and may affect excitability of neurons through modulating intercellular interaction between glial cells and neurons. It has been demonstrated that the expression level of IL-8 was increased in the ACC synapses of mice with persistent inflammatory pain, and the glutamatergic synaptic transmission was enhanced in the ACC synapses[37]. These results indicate that up-regulation of IL-8 in the ACC synapses contributes to the enhanced excitatory synaptic transmission in mice with chronic inflammatory pain.
Synaptic proteins
Neurotransmitters are released from presynaptic nerve terminals by exocytosis of the synaptic vesicles. During this event, many synaptic proteins interact with cytosol and plasma membrane proteins[38]. Cumulative evidence has shown that synaptic proteins such as synaptobrevin, synaptogyrin, synaptophysin, synaptotagmin, syntaxin and synaptosome-associated protein-25 (SNAP-25) are involved in the synaptic plasticity in the central nervous system[39-41]. Increases in synaptic vesicle proteins including synapsin, synaptophysin and synaptotagmin contribute to the enhanced synaptic potentiation in the hippocampal neurons[42,43]. Furthermore, in the spinal cord of chronic neuropathic pain rat models, the number of presynaptic boutons that are synaptophysin-immunoreactive was significantly increased[44]. In synaptosomal fractions obtained from the medial prefrontal cortex of neuropathic pain rat models, expression levels of presynaptic proteins (including synaptotagmin, synaptophysin, syntaxin, synaptobrevin and SNAP-25) were significantly enhanced, compared to those obtained from sham rats[45]. In addition, the electron microscopy revealed that synaptic vesicles in the synaptosomes were significantly increased in the medial prefrontal cortex of neuropathic pain rat models[45]. These observations suggest that the enhancement of both the number and availability of synaptic vesicles play critical roles in the enhancement of excitatory synaptic transmission in the medial prefrontal cortex of neuropathic pain rat models.
SOS3-interacting protein 3 (SIP3), which is known as a SNAP25-interacting protein 30, was first identified in the hair cells of guinea pig cochlea[46]. This novel protein is composed of 266 amino acids is one of the soluble N-ethylmaleimide-sensitive factor attachment protein receptors and plays essential roles for regulated exocytosis of synaptic vesicles in neurotransmission[47]. SIP30 is highly expressed in various brain regions including the cortex[46]. SIP30 was previously shown to be associated with the modulation of pain. For example, the increase in the expression of SIP30 in the spinal cord of chronic constriction injury rats was involved in the formation and maintenance of neuropathic pain[48]. In addition, the same group revealed that chronic constriction injury induced an increase of SIP30 in both sides of the ACC together with the behavioral hypersensitivity[49]. Interestingly, they showed that knockdown of SIP30 by lentiviral vector-mediated short hairpin RNA in the ACC significantly suppressed not only the behavioral hypersensitivity but also the glutamate release within the ACC. These results suggest that SIP30 is critical for the increased excitatory synaptic transmission in the ACC.
Presynaptic changes in ACC synapses of animals with disease-related pain
Diabetes mellitus
Diabetes mellitus is known as one of metabolic diseases and is caused by deficiency or diminished effectiveness of endogenous insulin. The high blood sugar levels resulting from the reduced insulin produces several symptoms such as increased hunger, frequent urination and increased thirst. If left untreated, diabetes mellitus causes many acute and chronic complications[50]. Peripheral neuropathy is the most common and debilitating complication of diabetic patients[51]. Thus, a number of studies revealed that diabetes-related plasticity occurs in the spinal nociceptive neurons[52-54]. In addition to the spinal cord, diabetes mellitus causes plasticity in the supraspinal brain regions pertinent to the processing of pain information[55,56]. In the ACC of the streptozotocin-induced diabetic rats, there was a significant reduction in paired-pulse facilitation compared to the control mice[56], suggesting that an enhanced presynaptic glutamatergic synaptic transmission occurs in the ACC neurons of the diabetic mice. Therefore, it is strongly suggested that presynaptic changes in the ACC are critically involved in the diabetes-induced pain.
Hypothyroidism
Two thyroid hormones, triiodothyronine (T3) and thyroxine (T4), are primarily responsible for regulation of metabolism and play essential roles in the development and activity of the central nervous system[57]. The thyroid status of neonates and children has a significant long-term impact on locomotor activity and cognition[57]. Indeed, congenital hypothyroidism, which is characterized by deficiencies in thyroid hormone production in newborn infants, causes mental retardation in children[58]. In addition, adult-onset hypothyroidism which is caused by insufficiency of thyroid hormones in the adulthood decreases cognitive functions[59]. The interaction between pain and thyroid hormones has been progressively elucidated[60]. In athyreotic patients, the pain sensitivity was associated with the thyroid status[61]. It has also been reported that an adequate supply of thyroid hormones from mother rats is necessary to acquire a normal nociceptive function in the offspring into adulthood[62]. Using experimental hypothyroidism mice that were treated with potassium perchlorate and methimazole in the drinking water, it was investigated whether and how synaptic transmission and pain perception in the ACC are modulated by thyroid hormones[63]. In hypothyroid mice, the thermal pain thresholds were decreased. Furthermore, whole-cell patch-clamp recordings showed that the frequency of miniature excitatory postsynaptic currents was increased while that of miniature inhibitory postsynaptic currents was decreased in ACC neurons. Treatment with thyroid hormones (T3 or T4) markedly reduced hypersensitivity to noxious stimuli and reversed the synaptic alterations. Therefore, it is likely that the hypersensitivity to noxious heat observed in the hypothyroid mice is caused by presynaptic changes in the ACC synapses.
Pre-LTP in ACC synapses and its molecular mechanisms
In ACC synapses of adult mice, pre-LTP can be induced by low-frequency stimulation and is proposed to be involved in pain-triggered anxiety[64,65]. The pre-LTP in the ACC synapses is reduced in chronic inflammatory and nerve injury model mice[64]. The induction of pre-LTP in the ACC is dependent on GluK1 containing kainate receptors, calcium-stimulated AC1 and extracellular signal-regulated kinase (ERK), but is independent of NMDA receptors, metabotropic glutamate receptors and protein kinase M zeta[64-66]. Activation of AC1 increases cAMP, which binds to hyperpolarization-activated cyclic nucleotide-gated channels to enhance its sensitivity. Also, AC1 activates PKA to enhance the release of glutamate, presumably through activation of ERK. Recently, it was found that SCRAPPER which is an E3 ubiquitin ligase expressed in presynaptic terminals is required for ACC pre-LTP[67]. The pre-LTP in the ACC synapses may be necessary for the activation of both presynaptic molecules (e.g., RIM1) and the ubiquitin–proteasome system including SCRAPPER protein[67]. Provided that cortical pre-LTP is involved in pain-triggered anxiety and fear, these molecules are potential molecular targets for relief from pain-triggered anxiety and fear. The molecular mechanisms involved in pre-LTP are summarized in Figure 2.
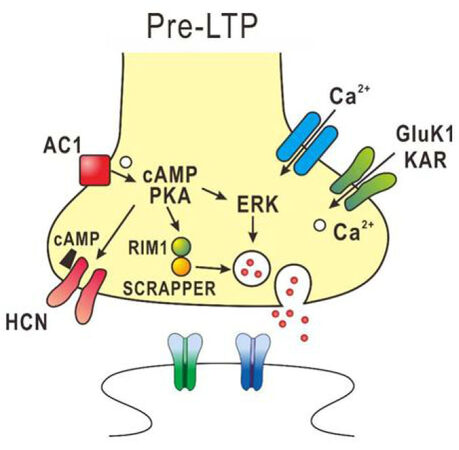
Figure 2. Diagram of the potential mechanisms in presynaptic form of long-term potentiation (pre-LTP) in the anterior cingulate cortex (ACC). The presynaptic Ca2+ influx via Gluk1 KAR s leads to activation of the adenylyl cyclase 1 (AC1)-PKA pathway. Then cAMP binds to the HCN channel to increase its sensitivity and activates PKA to enhance the release of glutamate, presumably through activation of extracellular signal-regulated kinase (ERK). The enhanced PKA activity is likely to activate SCRAPPER and RIM1 to regulate vesicle recycling, which is required for pre-LTP in the ACC synapses. KAR: kainate receptor; PKA: protein kinase A; cAMP: cyclic adenosine monophosphate; HCN: hyperpolarization-activated cyclic nucleotide-gated
Conclusion
Peripheral nerve injuries and peripheral inflammation can cause extensive changes in pain processing in the central nervous system. Many in vitro and in vivo studies have proved compelling evidence that changes in both presynaptic and postsynaptic function are responsible for chronic pain. It is believed that the key presynaptic and postsynaptic events in chronic pain in the central nervous system are synaptic plasticity triggered by peripheral nerve injury and ongoing unusual sensory inputs afterwards. Therefore, understanding of molecular and cellular mechanisms underlying the pain-induced synaptic plasticity will provide new therapeutic targets for treating persistent pain in patients. At present, little is understood about molecular mechanisms underlying presynaptic changes in chronic pain state, compared to those underlying postsynaptic changes. Thus, it is necessary to further elucidate the cellular and molecular mechanisms underlying presynaptic changes in the ACC of chronic pain mice. Treatment of chronic pain is difficult because current available drugs for chronic pain induce unwanted side effects or supply insufficient analgesia. Thus, studies focusing on the molecular mechanisms of presynaptic changes in the ACC of chronic pain mice could lead to the development of effective novel analgesics on treating chronic pain. Although it has been reported that pre-LTP in the ACC synapses is involved in pain-triggered anxiety and fear[64,65], the molecular and cellular mechanisms for the pre-LTP are not well understood. Understanding of these mechanisms for pre-LTP in the ACC synapses will help to develop new therapeutic strategies for pain-triggered anxiety and fear.
Declarations
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by Japan Society for the Promotion of Science KAKENHI Grant Number 17K08538.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Davis KD, Taylor SJ, Crawley AP, Wood ML, Mikulis DJ. Functional MRI of pain- and attention-related activations in the human cingulate cortex. J Neurophysiol 1997;77:3370-80.
2. Singer W. Development and plasticity of cortical processing architectures. Science 1995;270:758-64.
3. Vogt BA. Pain and emotion interactions in subregions of the cingulate gyrus. Nat Rev Neurosci 2005;6:533-44.
5. Zhuo M. Canadian association of neuroscience review: cellular and synaptic insights into physiological and pathological pain. EJLB-CIHR michael smith chair in neurosciences and mental health lecture. Can J Neurol Sci 2005;32:27-36.
7. Zhuo M. A synaptic model for pain: long-term potentiation in the anterior cingulate cortex. Mol Cells 2007;23:259-71.
8. Zhuo M. Long-term potentiation in the anterior cingulate cortex and chronic pain. Philos Trans R Soc Lond B Biol Sci 2013;369:20130146.
9. Devinsky O, Morrell MJ, Vogt BA. Contributions of anterior cingulate cortex to behaviour. Brain 1995;118:279-306.
10. Han CJ, O'Tuathaigh CM, van Trigt L, Quinn JJ, Fanselow MS, Mongeau R, Koch C, Anderson DJ. Trace but not delay fear conditioning requires attention and the anterior cingulate cortex. Proc Natl Acad Sci U S A 2003;100:13087-92.
11. Dick BD, Verrier MJ, Harker KT, Rashiq S. Disruption of cognitive function in fibromyalgia syndrome. Pain 2008;139:610-6.
12. Park DC, Glass JM, Minear M, Crofford LJ. Cognitive function in fibromyalgia patients. Arthritis Rheum 2001;44:2125-33.
13. Ramírez-Maestre C, Esteve R, López AE. Cognitive appraisal and coping in chronic pain patients. Eur J Pain 2008;12:749-56.
14. Koechlin E, Ody C, Kouneiher F. The architecture of cognitive control in the human prefrontal cortex. Science 2003;302:1181-5.
15. Likhtik E, Pelletier JG, Paz R, Paré D. Prefrontal control of the amygdala. J Neurosci 2005;25:7429-37.
16. Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW, Zhuo M. Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J Neurosci 2008;28:7445-53.
17. Li XY, Ko HG, Chen T, Descalzi G, Koga K, Wang H, Kim SS, Shang Y, Kwak C, Park SW, Shim J, Lee K, Collingridge GL, Kaang BK, Zhuo M. Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science 2010;330:1400-4.
18. Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci 2005;6:267-76.
20. Xia Z, Storm DR. Calmodulin-regulated adenylyl cyclases and neuromodulation. Curr Opin Neurobiol 1997;7:391-6.
21. Wei F, Qiu CS, Kim SJ, Muglia L, Maas JW, Pineda VV, Xu HM, Chen ZF, Storm DR, Muglia LJ, Zhuo M. Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron 2002;36:713-26.
22. Zhao MG, Ko SW, Wu LJ, Toyoda H, Xu H, Quan J, Li J, Jia Y, Ren M, Xu ZC, Zhuo M. Enhanced presynaptic neurotransmitter release in the anterior cingulate cortex of mice with chronic pain. J Neurosci 2006;26:8923-30.
23. Chang DJ, Li XC, Lee YS, Kim HK, Kim US, Cho NJ, Lo X, Weiss KR, Kandel ER, Kaang BK. Activation of a heterologously expressed octopamine receptor coupled only to adenylyl cyclase produces all the features of presynaptic facilitation in aplysia sensory neurons. Proc Natl Acad Sci U S A 2000;97:1829-34.
24. Wu LJ, Steenland HW, Kim SS, Isiegas C, Abel T, Kaang BK, Zhuo M. Enhancement of presynaptic glutamate release and persistent inflammatory pain by increasing neuronal cAMP in the anterior cingulate cortex. Mol Pain 2008;4:40.
25. Wang H, Xu H, Wu LJ, Kim SS, Chen T, Koga K, Descalzi G, Gong B, Vadakkan KI, Zhang X, Kaang BK, Zhuo M. Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci Transl Med 2011;3:65ra3.
26. Zhang MM, Liu SB, Chen T, Koga K, Zhang T, Li YQ, Zhuo M. Effects of NB001 and gabapentin on irritable bowel syndrome-induced behavioral anxiety and spontaneous pain. Mol Brain 2014;7:47.
27. Li Y, Ji A, Weihe E, Schäfer MK. Cell-specific expression and lipopolysaccharide-induced regulation of tumor necrosis factor alpha (TNFa) and TNF receptors in rat dorsal root ganglion. J Neurosci 2004;24:9623-31.
28. Tchelingerian JL, Quinonero J, Booss J, Jacque C. Localization of TNF alpha and IL-1 alpha immunoreactivities in striatal neurons after surgical injury to the hippocampus. Neuron 1993;10:213-24.
29. Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science 2002;295:2282-5.
31. Jin X, Gereau RW, 4th. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci 2006;26:246-55.
32. Jia D, Gao GD, Liu Y, He SM, Zhang XN, Zhang YF, Zhao MG. TNF-alpha involves in altered prefrontal synaptic transmission in mice with persistent inflammatory pain. Neurosci Lett 2007;415:1-5.
33. Luster AD. Chemokines--chemotactic cytokines that mediate inflammation. N Engl J Med 1998;338:436-45.
34. Murphy PM. Chemokine receptors: structure, function and role in microbial pathogenesis. Cytokine Growth Factor Rev 1996;7:47-64.
36. Ehrlich LC, Hu S, Sheng WS, Sutton RL, Rockswold GL, Peterson PK, Chao CC. Cytokine regulation of human microglial cell IL-8 production. J Immunol 1998;160:1944-8.
37. Cui GB, An JZ, Zhang N, Zhao MG, Liu SB, Yi J. Elevated interleukin-8 enhances prefrontal synaptic transmission in mice with persistent inflammatory pain. Mol Pain 2012;8:11.
38. Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 1995;375:645-53.
39. Isao T, Akiyama K. Effect of acute and chronic treatment with methamphetamine on mRNA expression of synaptotagmin IV and 25 KDa-synaptic-associated protein in the rat brain. Psychiatry Clin Neurosci 2004;58:410-9.
40. Janz R, Südhof TC, Hammer RE, Unni V, Siegelbaum SA, Bolshakov VY. Essential roles in synaptic plasticity for synaptogyrin I and synaptophysin I. Neuron 1999;24:687-700.
41. Zhang Z, Wang D, Sun T, Xu J, Chiang HC, Shin W, Wu LG. The SNARE proteins SNAP25 and synaptobrevin are involved in endocytosis at hippocampal synapses. J Neurosci 2013;33:9169-75.
42. Antonova I, Arancio O, Trillat AC, Wang HG, Zablow L, Udo H, Kandel ER, Hawkins RD. Rapid increase in clusters of presynaptic proteins at onset of long-lasting potentiation. Science 2001;294:1547-50.
43. Lynch MA, Voss KL, Rodriguez J, Bliss TV. Increase in synaptic vesicle proteins accompanies long-term potentiation in the dentate gyrus. Neuroscience 1994;60:1-5.
44. Jaken RJ, Joosten EA, Knüwer M, Miller R, van der Meulen I, Marcus MA, Deumens R. Synaptic plasticity in the substantia gelatinosa in a model of chronic neuropathic pain. Neurosci Lett 2010;469:30-3.
45. Hung KL, Wang SJ, Wang YC, Chiang TR, Wang CC. Upregulation of presynaptic proteins and protein kinases associated with enhanced glutamate release from axonal terminals (synaptosomes) of the medial prefrontal cortex in rats with neuropathic pain. Pain 2014;155:377-87.
46. Lee HK, Safieddine S, Petralia RS, Wenthold RJ. Identification of a novel SNAP25 interacting protein (SIP30). J Neurochem 2002;81:1338-47.
47. Bennett MK, Calakos N, Scheller RH. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 1992;257:255-9.
48. Peng G, Han M, Du Y, Lin A, Yu L, Zhang Y, Jing N. SIP30 is regulated by ERK in peripheral nerve injury-induced neuropathic pain. J Biol Chem 2009;284:30138-47.
49. Han M, Xiao X, Yang Y, Huang RY, Cao H, Zhao ZQ, Zhang YQ. SIP30 is required for neuropathic pain-evoked aversion in rats. J Neurosci 2014;34:346-55.
50. Kim C, Newton KM, Knopp RH. Gestational diabetes and the incidence of type 2 diabetes: a systematic review. Diabetes Care 2002;25:1862-8.
51. Edwards JL, Vincent AM, Cheng HT, Feldman EL. Diabetic neuropathy: mechanisms to management. Pharmacol Ther 2008;120:1-34.
52. Boric M, Jelicic Kadic A, Ferhatovic L, Sapunar D, Puljak L. Calcium/calmodulin-dependent protein kinase II in dorsal horn neurons in long-term diabetes. Neuroreport 2013;24:992-6.
53. Kinoshita J, Takahashi Y, Watabe AM, Utsunomiya K, Kato F. Impaired noradrenaline homeostasis in rats with painful diabetic neuropathy as a target of duloxetine analgesia. Mol Pain 2013;9:59.
54. Tan AM, Samad OA, Fischer TZ, Zhao P, Persson AK, Waxman SG. Maladaptive dendritic spine remodeling contributes to diabetic neuropathic pain. J Neurosci 2012;32:6795-807.
55. Joghataie MT, Roghani M, Jalali MR, Baluchnejadmojarad T, Sharayeli M. Dendritic spine changes in medial prefrontal cortex of male diabetic rats using golgi-impregnation method. Arch Iran Med 2007;10:54-8.
56. Li W, Wang P, Li H. Upregulation of glutamatergic transmission in anterior cingulate cortex in the diabetic rats with neuropathic pain. Neurosci Lett 2014;568:29-34.
57. Chan S, Kilby MD. Thyroid hormone and central nervous system development. J Endocrinol 2000;165:1-8.
59. Baldini IM, Vita A, Mauri MC, Amodei V, Carrisi M, Bravin S, Cantalamessa L. Psychopathological and cognitive features in subclinical hypothyroidism. Prog Neuropsychopharmacol Biol Psychiatry 1997;21:925-35.
61. Guasti L, Marino F, Cosentino M, Cimpanelli M, Rasini E, Piantanida E, Vanoli P, De Palma D, Crespi C, Klersy C, Maroni L, Loraschi A, Colombo C, Simoni C, Bartalena L, Lecchini S, Grandi AM, Venco A. Pain perception, blood pressure levels, and peripheral benzodiazepine receptors in patients followed for differentiated thyroid carcinoma: a longitudinal study in hypothyroidism and during hormone treatment. Clin J Pain 2007;23:518-23.
62. Alves IG, da Cruz KM, Mota CM, de Santana DS, Gaujac DP, de Carvalho VC, Reis LC, Sluka KA, Quintans-Junior LJ, Antoniolli AR, Desantana JM, Badauê-Passos D Jr, de Santana-Filho VJ. Experimental hypothyroidism during pregnancy affects nociception and locomotor performance of offspring in rats. Eur J Pain 2013;17:1291-8.
63. Yi J, Zheng JY, Zhang W, Wang S, Yang ZF, Dou KF. Decreased pain threshold and enhanced synaptic transmission in the anterior cingulate cortex of experimental hypothyroidism mice. Mol Pain 2014;10:38.
64. Koga K, Descalzi G, Chen T, Ko HG, Lu J, Li S, Son J, Kim T, Kwak C, Huganir RL, Zhao MG, Kaang BK, Collingridge GL, Zhuo M. Coexistence of two forms of LTP in ACC provides a synaptic mechanism for the interactions between anxiety and chronic pain. Neuron 2015;85:377-89.
65. Zhuo M. Neural mechanisms underlying anxiety-chronic pain interactions. Trends Neurosci 2016;39:136-45.
66. Yamanaka M, Tian Z, Darvish-Ghane S, Zhuo M. Pre-LTP requires extracellular signal-regulated kinase in the ACC. Mol Pain 2016; doi: 10.1177/1744806916647373.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Toyoda H. Presynaptic mechanisms at prefrontal synapses involved in persistent pain. Neurosciences 2018;5:40. http://dx.doi.org/10.20517/2347-8659.2018.48
AMA Style
Toyoda H. Presynaptic mechanisms at prefrontal synapses involved in persistent pain. Neuroimmunology and Neuroinflammation. 2018; 5: 40. http://dx.doi.org/10.20517/2347-8659.2018.48
Chicago/Turabian Style
Toyoda, Hiroki. 2018. "Presynaptic mechanisms at prefrontal synapses involved in persistent pain" Neuroimmunology and Neuroinflammation. 5: 40. http://dx.doi.org/10.20517/2347-8659.2018.48
ACS Style
Toyoda, H. Presynaptic mechanisms at prefrontal synapses involved in persistent pain. Neurosciences. 2018, 5, 40. http://dx.doi.org/10.20517/2347-8659.2018.48
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 0 clicks
Cite This Article 0 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.