
Microglial activation and polarization after subarachnoid hemorrhage


Abstract
Subarachnoid hemorrhage (SAH) is a devastating stroke type, with high mortality and morbidity. The neuroinflammatory response evolves over time from early brain injury to delayed cerebral deterioration. Microglia, the resident immune cells of the central nervous system, respond to the acute brain injury through activation and polarization. Microglia are able to polarize along two pathways, classic M1 and alternative M2, towards tissue injury and tissue repair respectively. The modulation of microglial activation has gained appreciation as a means to prevent the detrimental effects. In this review, we describe the progression of microglial polarization after SAH and summarize the key studies on mediators of microglial activation, including M1 and M2 specific microglial markers, transcription factors and key signaling pathways. Interactions between microglia and other cells are critical in modulating microglial activation and function, which are discussed as well. The preclinical application of microglia-dependent treatments is presented, aiming for a better understanding of modulating microglial function and suggesting future investigation for therapeutic approaches.
Keywords
Introduction
Subarachnoid hemorrhage (SAH) is a devastating condition accounting for 5% of the stroke population[1]. The mortality rate is as high as 30%-40% and approximately 50% of the survivors remain permanently disabled[2,3]. Primary brain damage develops within the first few hours to days after SAH as a result of extravasate blood or intracranial circulatory arrest. The increased intracerebral pressure and mass effect result in the sudden herniation and death. Recently, more attention has been focused on early brain injury (EBI) which appear to contribute to subsequent adverse cerebral events[4]. Current treatment for SAH mainly concentrates on aneurysm repair to avoid further bleeding. Also, cerebral blood flow augmentation is needed to reduce vasospasm. Identification of specific events enriches our understanding of the pathophysiological processes of SAH and enhances the investigation of potential therapeutic strategies. The recent prospective studies failing to confirm the association between vasospasm and outcomes in SAH patients attests to the need to develop new therapeutic directions[5,6].
Microglia, the resident immune cells of the central nervous system (CNS), are a pivotal component of neuroinflammation after SAH[7,8]. Microglia respond to brain injury through altering their morphology and polarization to activate in response to pathophysiological brain insults. Microglia are activated and M1-dominated polarization is demonstrated in the early phase of SAH. Microglial activation contributes to the SAH pathogenesis including brain edema, blood-brain barrier permeability, and neuronal apoptosis. The depletion of microglia leads to a significant decrease in neuronal apoptosis in the early phase[9-11]. Microglial activation involves numerous mediators and signaling pathways, especially the toll-like receptor 4 (TLR4). TLR4 plays an important role in inflammatory response and neuronal death after SAH and the majority of TLR4 is expressed by microglia. TLR4 activation in microglia results in secretion of inflammatory factors such as tumor necrosis factor-α (TNF-α) and deletion of TLR4 significantly reduces the neuronal apoptosis in SAH[10,12]. Increasing experimental evidence supports manipulation of microglial polarization as a strategy for preventing disease progression and improving outcomes[13-15].
Microglia are the primary source of cytokines and chemokines, which contribute to the immunomodulatory signaling after SAH[11,16,17]. These molecules initiate secondary brain injury but can also participate in the subsequent brain repair processes. Elevations in neuroinflammatory factors including monocyte chemoattractant protein-1 and TNF-α are regarded as predictors of overall negative outcome, but not necessarily useful predictors of vasospasm[18,19]. The anti-inflammatory factors such as interleukin-4 (IL-4), IL-10 and transforming growth factor-β (TGF-β) are released following the acute pro-inflammatory response in SAH[11].
In this review, we summarize microglial function after SAH, with a specific focus on microglial activation and polarization. Furthermore, we discuss the potential modulators of microglial polarization and function. The interactions of microglia with other cells are discussed as well.
Microglia
Microglia derive from yolk sac primitive progenitors and migrate into the CNS in early embryogenesis. Microglia have the capabilities of proliferation and differentiation, which are basically associated with disease states[8]. Microglia are highly dynamic in the resting state and activate rapidly in response to a set of transcription factors and growth factor receptors in brain injury or degeneration conditions[20]. Microglial cells activate through modulating the phenotype and generating a large number of cytokines and chemokines. Recent studies indicate that microglia have highly motile processes and continually survey the microenvironment, even in the normal brain[21]. Therefore, microglia represent the guardians of the brain in various injuries and diseases.
Microglia are classically considered as CNS-resident macrophage, as they share many macrophage-associated markers, such as CD11b[22]. However, the lineage relationship between microglia and macrophages clearly indicates that they are separate cell types. Microglia proliferate locally and tend to remain viable longer than macrophage. In addition, microglia are not normally supplied by bone marrow-derived cells[23].
The microglial phenotype is modified in response to the brain injury. Experimental evidence indicates that microglia dynamically and temporally polarize into a classically activated state and alternatively activated state, which contributes to tissue damage or repair respectively[24-26]. Microglial polarization is considered a functional means by which they release inflammatory factors that contribute to the neuroinflammation responses in CNS disease.
Microglial polarization
Microglia generally polarize in two directions from a resting state. The classical activation is known as M1, which is the mediator of pro-inflammatory responses. The alternative activation, known as M2, is responsible for resolution and repair. The polarization of microglia has been clarified by measuring the markers both in vitro and in vivo. In vitro treatment mouse microglia with lipopolysaccharide (LPS) and IL-4 results in M1 and M2 phenotypes respectively[27]. The M1 microglial polarization is characterized by an increase in the expression of pro-inflammatory molecules. Alternatively, activated M2 microglia are categorized into four subtypes, M2a, M2b, M2c, and M2d. The subtypes of the M2 phenotype are firstly defined in the macrophage activation. Analogous to macrophage, we hypothesize the similar subtypes of M2 microglia[28-30].
Recently, the mixture of M1 and M2-phenotyped response are reported in experimental studies, which raise a controversy to the traditional concept of M1 vs. M2 microglial polarization. In addition, the classic M1 marker IL-6, which is induced by IL-4, functions as an anti-inflammatory mediator in a mouse model of experimental autoimmune encephalomyelitis[31].
Although the classification of M1/M2 phenotypes is now recognized as an oversimplification, as the pure M1 or M2 polarization is executively observed in in vitro studies, this conception remains useful for understanding the functional role of microglia in CNS diseases. The recent studies on SAH address the functional role of microglia by investigating the M1-like and M2-like markers, which are discussed below.
Microglial M1 polarization
Microglial polarization can be assessed by immunohistochemical analysis of the specific markers. Pro-inflammatory cytokines are considered to be produced predominantly by classically activated M1 microglia, and these pro-inflammatory factors are integral in the activation of downstream pathways. Therefore, the changes in pro-inflammatory cytokine profiles and pathways can be indicators of microglial polarization after SAH.
An intraparenchymal accumulation of KiM1P-positive microglia/macrophage cells is documented in the SAH patients. The microglia accumulation is evident between day 5 and 15[32]. On experimental SAH, the increase of Iba-1-positive microglial cells is observed around day 4 and 28 within the brain parenchyma. The peak occurs on day 14 after SAH induction. The microglia accumulation presents a centrifugal pattern, starting at the base to the cortex of both hemispheres and spreading globally to other regions of the brain[32].
Cell surface markers including CD11b[9], CD68[33], and ED-1[17] are used to distinguish the activated from resting microglia, both of which can be defined by immunostaining of iba1. In culture, microglia treated with oxyhemoglobin leads to M1 polarization, indicated by CD16+/CD11b+ cells [Figure 1][9]. Other M1-associated markers CD86 and inducible nitric oxide synthase (iNOS) increase immediately after SAH, peak at 24-48 h, and remain highly expressed for 72 h[11].
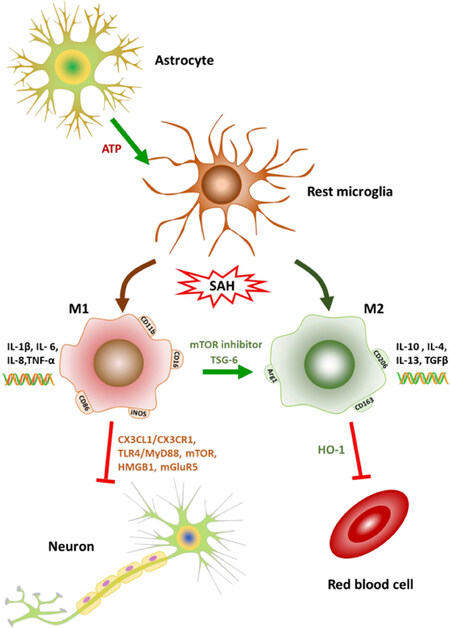
Figure 1. Schematic description of microglial polarization after SAH and the interaction of microglia with other cell types in central nervous system. Microglia are activated and polarized to M1 or M2 direction after SAH. M1 phenotype is characterized by the cell surface marker CD11b, CD16, CD32, CD86. M2 phenotype is characterized by the cell surface markers CD206, CD163, Arg1. M1 microglia exhibit the pro-inflammatory responses with the expression of IL-1β, IL-6, IL-8 and TNF-α. M2 exhibit the pro-inflammatory responses with the expression of IL-10, IL-4, IL-13 and TGF-β. The M1 1 microglial activation has an unfavorable effect on the neurons through the interaction of CX3CL1 and CX3CR1. The neuronal apoptosis is modulated by microglia-dependent TLR4-MyD88, mTOR, HMGB1 and mGluR5. The extracellular ATP released from the surrounding astrocytes trigger the rapid chemotactic response of microglia towards injury. This response can be inhibited by blocking G protein-coupled purinergic receptors and connexin channels, which are highly expressed in astrocytes. The expression of HO-1 in microglia helps to clear the extravasated red blood cells and attenuated neuronal apoptosis. SAH: subarachnoid hemorrhage; IL: interleukin; TNF-α: tumor necrosis factor-α; TGF-β: transforming growth factor-β; CX3CL1: CX3C motif chemokine ligand 1; CX3CR1: CX3C chemokine receptor 1; TLR4: toll-like receptor 4; MyD88: myeloid differentiation factor 88; mTOR: mammalian target of rapamycin; HMGB1: high-mobility group box 1 protein; mGluR5: metabotropic glutamate receptor 5; HO-1: heme oxygenase-1; TSG-6: tumor-specific glycoprotein-6
TNF-α and IL-1β are known to be the most important pro-inflammatory cytokines in human SAH pathology, as well as in other experimental models of SAH. M1-signature pro-inflammatory cytokines are upregulated prominently after SAH. IL-1β, IL- 6, TNF-α increase rapidly within 24 h and last for 48 h on experimental SAH model[9,17,33-35]. The increased pro-inflammatory cytokine expression is associated with poor outcomes in SAH[36].
TLR4 plays an important role in mediating the microglia-dependent neuroinflammation. The activation of TLR4 towards the M1 microglial polarization and increased expression of TLR4 are correlated with the poor outcomes in SAH[37-39]. TLR4 expression in microglia increases 2-6 h after SAH and remains elevated for 12-48 h[40]. High-mobility group box 1 protein (HMGB1) is a nuclear factor and potent pro-inflammatory mediator, and expressed by Iba1-positive microglia[41]. HMGB1 increases in the cerebrospinal fluid (CSF) of SAH patients with a poor functional outcome. The upregulation of HMGB1 correlates significantly with IL-6, IL-8, and TNF-α expression in CSF, developing towards a pro-inflammatory response after SAH[42-44]. While another study suggests no correlation between HMGB1 and IL-6 concentrations in plasma, which may indicate the acute activation of HMGB1 occurs only in the CNS[45].
Microglial M2 polarization
Although M2 microglia play a critical role in tissue repair and toxicity clearance in the other CNS diseases[46,47], M2-directed polarization in SAH has been less studied than M1 phenotype.
All M2a, b and c phenotypes are considered as anti-inflammatory repair M2 microglial cells. The subtypes are generally classified by the different profiles of pro-inflammatory cytokines. M2a activation is considered to be IL4Rα-dependent and follows exposure to IL-4 or IL-13. M2a microglia play a role in the cell repair and regeneration by expressing anti-inflammatory and immune-regulatory molecules. M2b and M2c microglia are largely phagocytic. M2b microglia, in particular, express high levels of IL-10 and low levels of IL-12, whereas, M2c is characterized by high TGF-β expression[28-30,48]. M2d, distinguished from the above subtypes of M2 polarization, results from the classically activated status through the activation of adenosine A2A receptors in activated M1 pro-inflammatory cells[48].
Upregulation of the M2-associated markers Arg1, CD163 and CD206 are observed in experimental SAH. The expression of the markers increases slowly and peaks at 48-72 h[11]. The mRNAs encoding IL-4, IL-10, and TGF-β, show corresponding increments along with M2 polarization in SAH[33]. IL-4 is an important anti-inflammatory cytokine and instrumented in M2-like microglial responses leading to improved functional recovery in ischemic stroke and in intracerebral hemorrhage (ICH) as well[15,30,37,49].
Peroxisome proliferator-activated receptor-γ (PPARγ) is a superfamily of nuclear receptors with the antioxidant and anti-inflammatory properties. Treatment with PPARγ agonists has beneficial effects on SAH, reportedly due in part to reduced microglial activation and reduced pro-inflammatory cytokine expression[50,51]. PPARγ is considered as an important mediator in pro-inflammatory reaction toward M2-like microglial phenotype.
TNF stimulated gene-6 (TSG-6), a multifunctional glycoprotein, acts as a protective regulator against inflammation. The endogenous TSG-6 is mainly expressed in microglia with the peak release in 12-24 h after SAH injury. TSG-6 is considered as an endogenous inhibitor in pro-inflammation progression and induced M2-like polarization. The rh-TSG-6 treated SAH rats show improved neurobehavioral outcomes and reduced brain edema. The decreased M1 polarization and elevated M2 phenotype are observed with the remarkable downregulation of TNF-α and upregulation of IL-10 in SAH rats[11].
The activation of microglia to an M1 phenotype appears mainly in the acute phase after SAH and M2 microglial activation occurs in the subacute and delayed phase. The similar theme of microglial activation is observed in ICH[52]. However, microglia demonstrate the M2-dominated activation in early phase in ischemic stroke and gradually transforms to the M1 phenotype in peri-infarct regions. The ischemic neurons lead microglial polarization toward M1 phenotype[25]. The discrepant responses of microglia to the brain injuries largely depend on the different pathophysiological processes. Hemorrhagic stroke triggers an acute pro-inflammatory response mainly in proximity to the bleeding. The delayed ischemia occurred in one-third of SAH patients[53]. The ischemic neurons may critically contribute to the delayed M2 polarization and the protective M2 phenotype may involve blood clearance and tissue repair in reaction.
Mediator of microglial polarization
Microglia dynamically polarize in response to brain damage or pathogens. The mechanism mediating the microglial polarization remains to be fully elucidated. Recently, several mediators are characterized to help in understanding the underlying process and provide potential targets to suppress the M1-like pro-inflammation [Figure 2].
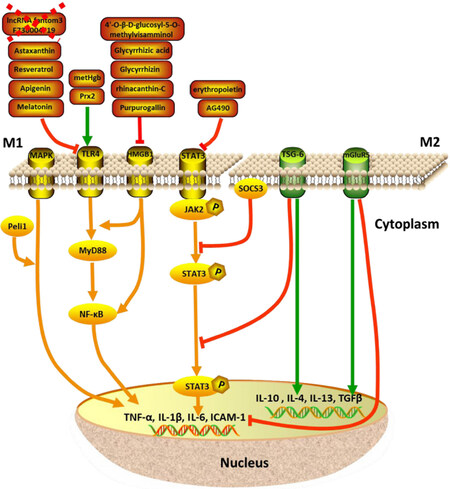
Figure 2. Mediators of microglial polarization after SAH. Activation of TLR4, HMGB1, STAT3 and MAPK signaling pathways promotes microglial M1 polarization with the expression of TNF-α, IL-1β, IL-6 and ICAM-1. The downstream mediators are shown in the figure. Activation of TSG-6 and mGluR5 promotes microglial M2 polarization, therefore reverses the pro-inflammatory responses. SAH: subarachnoid hemorrhage; TLR4: toll-like receptor 4; HMGB1: high-mobility group box 1 protein; STAT3: signal transducer and activator of transcription 3; MAPK: mitogen-activated protein kinase; TNF-α: tumor necrosis factor-α; IL: interleukin; ICAM-1: intercellular adhesion molecule 1; TSG-6: tumor-specific glycoprotein-6; mGluR5: metabotropic glutamate receptor 5; JAK2: Janus kinases 2; MyD88: myeloid differentiation factor 88; SOCS3: suppressor of cytokine signaling 3; TGF-β: transforming growth factor-β; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; Prx2: peroxiredoxin 2; metHgb: methemoglobin
TLR4 is thought the most studied signaling pathway in regulating the microglial activation after SAH. In both SAH patients and animal models, TLR4 is upregulated and associated with cerebral vasospasm, delayed brain ischemia, and neuronal apoptosis in aneurysmal SAH[10,40,54,55]. The majority of TLR4 is expressed in microglia rather than astrocytes or neurons[10]. TLR4 is markedly increased in microglia in a neuron-microglia co-culture system in vitro, with consequent increases in pro-inflammatory cytokines and neuronal apoptosis. The downstream molecules, such as myeloid differentiation factor 88 (MyD88) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) are upregulated as well[37,38]. TLR4/MyD88/NF-κB signaling pathway is more related to the M1 polarization. Based on the high-throughput sequencing and co-expression network analysis of long non-coding RNAs (lncRNAs) and mRNAs, knockdown of lncRNA fantom3_F730004F19 attenuates inflammation in LPS-treated BV-2 microglial cells through the downregulation of TLR4 and CD14, with decreased TNF-α, IL-1β and IL-6 expression[39]. Methemoglobin (metHgb) is considered as an endogenous TLR4 ligand. The application of metHgb into the rat subarachnoid space induces the widespread TLR4-mediated neuroinflammation resulting in the microglial activation and TNF-α upregulation[56].
Substantial treatment strategies have been investigated based on targeting TLR4. Treatment with astaxanthin significantly reduced the TLR4 activation in SAH rats. The subsequent pro-inflammatory releases of IL-1β, TNF-α, and intercellular adhesion molecule 1 are downregulated accordingly at both the protein and mRNA levels in vivo and in vitro[38]. Peroxiredoxin 2 (Prx2) is a member of Prx protein family. Prx2 activates microglia through TLR4/MyD88/NF-κB signaling pathway and the TLR4 knock-out mitigates the Prx2-induced neuronal cytotoxicity after SAH, which suggests the critical role of Prx2 in the inflammatory modulation responding to hemorrhage attack[37]. Post-SAH treatment with resveratrol, a naturally occurring polyphenolic compound with the anti-inflammatory activity, apigenin or melatonin ameliorates EBI after SAH by suppressing the activation of the TLR4 pathway and expression of MyD88 and NF-κB. The reduced microglia activation and inflammatory cytokines results in the mitigation of neural apoptosis, brain edema, and neurological deficits[12,57,58].
HMGB1 has endogenous cytokine-like activity and involves both in the EBI and delayed cerebral ischemia after SAH by modulating the microglia-dependent pro-inflammation[59]. Treatment with anti-HMGB1 antibody significantly reduces the expression of TLR4, IL-6, TNF-α, and iNOS and reverses the basilar artery vasospasm in the SAH model[35]. The application of glycyrrhizin and glycyrrhizin acid, rhinacanthin-C, purpurogallin, and 4’-O-β-D-glucosyl-5-O-methylvisamminol attenuating the expression of HMGB1, ameliorate inflammatory effect by downregulating M1-related cytokines[60-65]. The molecules targeting HMGB1 may be potential candidates for the treatment of inflammatory brain injury after SAH.
Signal transducer and activator of transcription 3 (STAT3) inflammatory signaling mediates microglial activation both in primary microglia and microglial cell lines. Increased STAT3 expression is accompanied by the elevated expression of IL-6 rather than IL-10. The STAT3/Janus kinases (JAK) cascade is a pivotal inflammatory signaling pathway and widely expressed in the brain maintaining[66,67]. Based on our studies and published data, STAT3 responses to the hemorrhage attack immediately through phosphorylation and translocation into the nuclei. STAT3/JAK pathway is activated and upregulated within 24 h after SAH with the increased pro-inflammatory cytokines release. The mediators and inhibitors, such as erythropoietin and AG490, suppress the STAT3/JAK pathway activation and reduce the M1-like inflammatory response and ameliorate brain injury after SAH[68,69]. The neuroprotective effect of TSG-6 on modulating microglial phenotype involves suppression of the STAT3/suppressor of cytokine signaling 3 pathway activation through impeding the translocation of phosphorylated STAT3[11].
Other mediators trigger the M1 classical activated status in brain in response to pro-inflammatory signals. Activation of metabotropic glutamate receptor 5 (mGluR5) attenuates the M1 microglial activation by downregulating both mRNA and protein expression of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, at 24 h after SAH[17]. Albumin suppresses microglial activation resulting in reduced Iba-1 and CD68 staining in the cortex on 1 day after SAH. Expression of M1 microglial markers including iNOS, IL-1b, CD16, and CD32 are remarkably suppressed as well. The albumin induced microglial modulation is associated with binding of albumin to a C-type lectin microglial receptor (mincle), followed by the reduction of mincle/spleen tyrosine kinase/IL-1b signaling in ipsilateral hemisphere subjected to SAH[33]. Peli1, an E3 ubiquitin ligase, mediates the induction of pro-inflammatory cytokines in microglia via mitogen-activated protein kinase (MAPK) signaling pathway. Peli1 promotes the expression of M1 microglia polarization biomarker CD16/32 and iNOS after SAH. Knockdown of Peli1 suppresses microglial activation by inhibiting MAPK signaling and improves neurological outcomes and reduces cerebral edema after SAH[16].
Transition between M1 and M2
The M2 to M1 shift is observed in models of traumatic brain injury[70] and ischemic stroke[14]. However, whether this transition is a result of phenotypic transformation of a single microglial population, or of the M2 microglial migration and infiltration remains to be determined.
In the SAH model, inhibition of mammalian target of rapamycin (mTOR) can induce a shift of microglia polarizing from M1 to M2 phenotype, as indicated by the reduction of the CD16: CD206 ratio. The shift is along with the decrease in the levels of TNF-α and IL-1β, apoptosis and neuronal degeneration index, brain water content and albumin extravasation in the cerebral cortex after SAH[9].
As described above, in the endovascular punctured SAH model, endogenous TSG-6 transforms the SAH-driven M1 polarization to a skewed M2 polarization and balances the M1/M2 ratio to a beneficial phenotype in the group of CD11b+ CD45low labeled microglia. Deficiency of endogenous TSG-6 results in a conversion to pro-inflammatory microglial activation. TSG-6 is a promising candidate to modulate microglial polarization for the neuroprotective effects[11].
Interaction with other cells
CX3C motif chemokine ligand 1 is expressed on neurons and its receptor CX3C chemokine receptor 1, is highly expressed on microglia[71]. Neuronal apoptosis is mediated in a TLR4-MyD88, mTOR and HMGB1 related microglial manner[9,10,62]. Activation of mGluR5 reduces the terminal deoxynucleotidyl transferase dUTP nick end labeling-positive cells and active caspase-3/NeuN-positive neurons in the cortex at 24 h after SAH[17]. The neuronal apoptosis is initiated from 24 h and increases up to two weeks after SAH[32,72,73].
Adenosine triphosphate (ATP) is a pivotal signaling molecule regulating the interactions among different cell types in the CNS and a high concentration of ATP induces microglial activation. Microglia extend the processes toward the site of injection of ATP without cell body movement. This response can be inhibited by blocking G protein-coupled purinergic receptors and connexin channels, which are highly expressed in astrocytes. The extracellular ATP released from the damaged tissue and surrounding astrocytes trigger the rapid chemotactic response of microglia towards injury. This provides evidence of the interaction between microglia and astrocyte in CNS injury[18].
Erythrocyte extravasation is a potential danger factor to the EBI, as heme, released from injured red blood cells, contributes to the pathogenesis of SAH. Heme is metabolized by heme oxygenase-1 (HO-1), which is minimally expressed in the uncompromised brain, but largely upregulated in microglia following injury. Expression of HO-1 in microglia is necessary to attenuate neuronal cell death, vasospasm, impaired cognitive function, and clearance of cerebral blood burden[74].
Clinical implication
Microglial responses affect neuronal survival and contribute to poor outcome after SAH. Therapeutic intervention or suppression of unfavorable microglial response may accelerate the recovery. Several therapeutic strategies have been implicated to prevent the detrimental effects of microglia and attenuate the neurological impairments in preclinical studies. The application of rosuvastatin markedly inhibits microglia activation and therefore reduces cortical apoptosis, brain edema, and improve the neuronal function after SAH. Heparin reduces the microglial activation and reduces its production of pro-inflammation cytokines[75,76]. As discussed above, administration of resveratrol, apigenin, melatonin, and rh-TSG-6 may reverse the M1-like pro-inflammatory effect after SAH. Clinical trials are essential to provide the advanced evidence for their therapeutic effect. With the expanding investigation of microglial polarization and function, the treatment that targets microglial phenotype switching may be an efficient approach for SAH therapy.
Conclusion
Microglial activation is an important pathological mechanism in the progression of SAH. Microglia undergo polarization into mainly M1 and M2 phenotypes contributing differently to neuroinflammation after SAH. These results indicate the presence of M1-related pro-inflammatory state early after SAH. While microglia polarize to M2 phenotype gradually on delayed phase. Although the dynamics of microglial polarization specifically after SAH remain to be defined, modulation of microglial activation is expected to enhance the tissue repair and functional recovery. The transition from M1 to M2 polarization is thought to be a target concerning the amelioration of the pro-inflammatory response. The investigation on the applications of microglia-targeted treatments is expected to improve our understanding of the pathogenesis of SAH and lead to potential therapeutic strategies for affected patients.
Declarations
Authors’ contributionsFormulation of the key concepts and manuscript framework, literature research, manuscript draft and editing: Zheng ZV
Formulation of the key concepts and manuscript framework, manuscript revision: Wong GKC
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
2. Wong GK, Lam S, Ngai K, Wong A, Mok V, et al. Evaluation of cognitive impairment by the Montreal cognitive assessment in patients with aneurysmal subarachnoid haemorrhage: prevalence, risk factors and correlations with 3 month outcomes. J Neurol Neurosurg Psychiatry 2012;83:1112-7.
3. Longstreth WT, Nelson LM, Koepsell TD, van Belle G. Clinical course of spontaneous subarachnoid hemorrhage: a population-based study in King County, Washington. Neurology 1993;43:712-8.
4. Chen S, Wu H, Tang J, Zhang J, Zhang JH. Neurovascular events after subarachnoid hemorrhage: focusing on subcellular organelles. Acta Neurochir Suppl 2015;120:39-46.
5. Vergouwen MD, Ilodigwe D, Macdonald RL. Cerebral Infarction after subarachnoid hemorrhage contributes to poor outcome by vasospasm-dependent and -independent effects. Stroke 2011;42:924-9.
6. Dankbaar JW, Rijsdijk M, van der Schaaf IC, Velthuis BK, Wermer MJ, et al. Relationship between vasospasm, cerebral perfusion, and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neuroradiology 2009;51:813-9.
7. Zheng VZ, Wong GKC. Neuroinflammation responses after subarachnoid hemorrhage: a review. J Clin Neurosci 2017;42:7-11.
8. Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 2011;11:775-87.
9. You W, Wang Z, Li H, Shen H, Xu X, et al. Inhibition of mammalian target of rapamycin attenuates early brain injury through modulating microglial polarization after experimental subarachnoid hemorrhage in rats. J Neurol Sci 2016;367:224-31.
10. Hanafy KA. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J Neuroinflammation 2013;10:83.
11. Li R, Liu W, Yin J, Chen Y, Guo S, et al. TSG-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway. J Neuroinflammation 2018;15:231.
12. Zhang T, Su J, Guo B, Wang K, Li X, et al. Apigenin protects blood-brain barrier and ameliorates early brain injury by inhibiting TLR4-mediated inflammatory pathway in subarachnoid hemorrhage rats. Int Immunopharmacol 2015;28:79-87.
13. Yao X, Liu S, Ding W, Yue P, Jiang Q, et al. TLR4 signal ablation attenuated neurological deficits by regulating microglial M1/M2 phenotype after traumatic brain injury in mice. J Neuroimmunol 2017;310:38-45.
14. Pan J, Jin JL, Ge HM, Yin KL, Chen X, et al. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARγ-dependent manner. J Neuroinflammation 2015;12:51.
15. Liu X, Liu J, Zhao S, Zhang H, Cai W, et al. Interleukin-4 is essential for microglia/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke 2016;47:498-504.
16. Huang XP, Peng JH, Pang JW, Tian XC, Li XS, et al. Peli1 contributions in microglial activation, neuroinflammatory responses and neurological deficits following experimental subarachnoid hemorrhage. Front Mol Neurosci 2017;10:398.
17. Zhang ZY, Sun BL, Liu JK, Yang MF, Li DW, et al. Activation of mGluR5 attenuates microglial activation and neuronal apoptosis in early brain injury after experimental subarachnoid hemorrhage in rats. Neurochem Res 2015;40:1121-32.
18. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005;8:752-8.
19. Kim GH, Kellner CP, Hahn DK, Desantis BM, Musabbir M, et al. Monocyte chemoattractant protein-1 predicts outcome and vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg 2008;109:38-43.
20. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005;308:1314-8.
21. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of Microglia. Physiol Rev 2011;91:461-553.
22. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010;330:841-5.
23. de Haas AH, Boddeke HW, Biber K. Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 2008;56:888-94.
24. Xu F, Huang J, He Z, Chen J, Tang X, et al. Microglial polarization dynamics in dorsal spinal cord in the early stages following chronic sciatic nerve damage. Neurosci Lett 2016;617:6-13.
25. Wang G, Zhang J, Hu X, Zhang L, Mao L, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab 2013;33:1864-74.
26. Perego C, Fumagalli S, De Simoni MG. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation 2011;8:174.
27. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 2016;173:649-65.
28. Anderson CF, Mosser DM. A novel phenotype for an activated macrophage: the type 2 activated macrophage. J Leukoc Biol 2002;72:101-6.
29. Franco R, Fernández-Suárez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol 2015;131:65-86.
30. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med 1992;176:287-92.
31. Casella G, Garzetti L, Gatta AT, Finardi A, Maiorino C, et al. IL4 induces IL6-producing M2 macrophages associated to inhibition of neuroinflammation in vitro and in vivo. J Neuroinflammation 2016;13:139.
32. Schneider UC, Davids AM, Brandenburg S, Müller A, Elke A, et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol 2015;130:215-31.
33. Xie Y, Guo H, Wang L, Xu L, Zhang X, et al. Human albumin attenuates excessive innate immunity via inhibition of microglial Mincle/Syk signaling in subarachnoid hemorrhage. Brain Behav Immun 2017;60:346-60.
34. Kooijman E, Nijboer CH, van Velthoven CT, Mol W, Dijkhuizen RM, et al. Long-term functional consequences and ongoing cerebral inflammation after subarachnoid hemorrhage in the rat. PLoS One 2014;9:e90584.
35. Haruma J, Teshigawara K, Hishikawa T, Wang D, Liu K, et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci Rep 2016; doi: 10.1038/srep37755.
36. Chou SH, Feske SK, Atherton J, Konigsberg RG, De Jager PL, et al. Early elevation of serum tumor necrosis factor-α is associated with poor outcome in subarachnoid hemorrhage. J Investig Med 2012;60:1054-8.
37. Lu Y, Zhang XS, Zhang ZH, Zhou XM, Gao YY, et al. Peroxiredoxin 2 activates microglia by interacting with toll-like receptor 4 after subarachnoid hemorrhage. J Neuroinflammation 2018;15:87.
38. Zhang X, Lu Y, Wu Q, Dai H, Li W, et al. Astaxanthin mitigates subarachnoid hemorrhage injury primarily by increasing sirtuin 1 and inhibiting the toll-like receptor 4 signaling pathway. FASEB J 2018; doi: 10.1096/fj.201800642RR.
39. Peng J, Wu Y, Tian X, Pang J, Kuai L, et al. High-throughput sequencing and co-expression network analysis of IncRNAs and mRNAs in early brain injury following experimental subarachnoid haemorrhage. Sci Rep 2017;7:46577.
40. Ma CX, Yin WN, Cai BW, Wu J, Wang JY, et al. Toll-like receptor 4/nuclear factor-kappa B signaling detected in brain after early subarachnoid hemorrhage. Chin Med J (Engl) 2009;122:1575-81.
41. Murakami K, Koide M, Dumont TM, Russell SR, Tranmer BI, et al. Subarachnoid hemorrhage induces gliosis and increased expression of the pro-inflammatory cytokine high mobility group box 1 protein. Transl Stroke Res 2011;2:72-9.
42. Nakahara T, Tsuruta R, Kaneko T, Yamashita S, Fujita M, et al. High-mobility group box 1 protein in CSF of patients with subarachnoid hemorrhage. Neurocrit Care 2009;11:362-8.
43. Wang KC, Tang SC, Lee JE, Li YI, Huang YS, et al. Cerebrospinal fluid high mobility group box 1 is associated with neuronal death in subarachnoid hemorrhage. J Cereb Blood Flow Metab 2017;37:435-43.
44. Sokół B, Woźniak A, Jankowski R, Jurga S, Wąsik N, et al. HMGB1 level in cerebrospinal fluid as a marker of treatment outcome in patients with acute hydrocephalus following aneurysmal subarachnoid hemorrhage. J Stroke Cerebrovasc Dis 2015;24:1897-904.
45. Kiiski H, Långsjö J, Tenhunen J, Ala-Peijari M, Huhtala H, et al. Time-courses of plasma IL-6 and HMGB-1 reflect initial severity of clinical presentation but do not predict poor neurologic outcome following subarachnoid hemorrhage. eNeurologicalSci 2017;6:55-62.
46. Tian DS, Li CY, Qin C, Murugan M, Wu LJ, et al. Deficiency in the voltage-gated proton channel Hv1 increases M2 polarization of microglia and attenuates brain damage from photothrombotic ischemic stroke. J Neurochem 2016;139:96-105.
47. Wang G, Shi Y, Jiang X, Leak RK, Hu X, et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc Natl Acad Sci U S A 2015;112:2853-8.
48. Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation 2013;36:921-31.
49. Yang J, Ding S, Huang W, Hu J, Huang S, et al. Interleukin-4 ameliorates the functional recovery of intracerebral hemorrhage through the alternative activation of microglia/macrophage. Front Neurosci 2016;10:61.
50. Xie Z, Huang L, Enkhjargal B, Reis C, Wan W, et al. Recombinant Netrin-1 binding UNC5B receptor attenuates neuroinflammation and brain injury via PPARγ/NFκB signaling pathway after subarachnoid hemorrhage in rats. Brain Behav Immun 2018;69:190-202.
51. Tu L, Yang XL, Zhang Q, Wang Q, Tian T, et al. Bexarotene attenuates early brain injury via inhibiting micoglia activation through PPARγ after experimental subarachnoid hemorrhage. Neurol Res 2018;40:702-8.
52. Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol 2017;13:420-33.
53. Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol 2014;10:44-58.
54. Ma C, Zhou W, Yan Z, Qu M, Bu X. Toll-like receptor 4 (TLR4) is correlated with delayed cerebral ischemia (DCI) and poor prognosis in aneurysmal subarachnoid hemorrhage. J Neurol Sci 2015;359:67-71.
55. Kurki MI, Häkkinen SK, Frösen J, Tulamo R, von und zu Fraunberg M, et al. Upregulated signaling pathways in ruptured human saccular intracranial aneurysm wall: an emerging regulative role of toll-like receptor signaling and nuclear factor-κB, hypoxia-inducible factor-1A, and ETS transcription factors. Neurosurgery 2011;68:1667-75.
56. Kwon MS, Woo SK, Kurland DB, Yoon SH, Palmer AF, et al. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int J Mol Sci 2015;16:5028-46.
57. Zhang XS, Li W, Wu Q, Wu LY, Ye ZN, et al. Resveratrol attenuates acute inflammatory injury in experimental subarachnoid hemorrhage in rats via inhibition of TLR4 pathway. Int J Mol Sci 2016; doi: 10.3390/ijms17081331.
58. Wang Z, Wu L, You W, Ji C, Chen G. Melatonin alleviates secondary brain damage and neurobehavioral dysfunction after experimental subarachnoid hemorrhage: possible involvement of TLR4-mediated inflammatory pathway. J Pineal Res 2013;55:399-408.
59. Hendrix P, Foreman PM, Harrigan MR, Fisher WS Rd, Vyas NA, et al. Impact of High-Mobility Group Box 1 Polymorphism on Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. World Neurosurg 2017;101:325-30.
60. Sun Q, Wang F, Li W, Li W, Hu YC, et al. Glycyrrhizic acid confers neuroprotection after subarachnoid hemorrhage via inhibition of high mobility group box-1 protein: a hypothesis for novel therapy of subarachnoid hemorrhage. Med Hypotheses 2013;81:681-5.
61. Li Y, Sun F, Jing Z, Wang X, Hua X, et al. Glycyrrhizic acid exerts anti-inflammatory effect to improve cerebral vasospasm secondary to subarachnoid hemorrhage in a rat model. Neurol Res 2017;39:727-32.
62. Ieong C, Sun H, Wang Q, Ma J. Glycyrrhizin suppresses the expressions of HMGB1 and ameliorates inflammative effect after acute subarachnoid hemorrhage in rat model. J Clin Neurosci 2018;47:278-84.
63. Chang CZ, Lin CL, Wu SC, Kwan AL. Purpurogallin, a natural phenol, attenuates high-mobility group box 1 in subarachnoid hemorrhage induced vasospasm in a rat model. Int J Vasc Med 2014;2014:254270.
64. Chang CZ, Wu SC, Kwan AL, Lin CL. Rhinacanthin-C, a fat-soluble extract from rhinacanthus nasutus, modulates high-mobility group box 1-related neuro-inflammation and subarachnoid hemorrhage-induced brain apoptosis in a rat model. World Neurosurg 2016;86:349-60.
65. Chang CZ, Wu SC, Kwan AL, Lin CL. 4’-O-β-d-glucosyl-5-O-methylvisamminol, an active ingredient of saposhnikovia divaricata, attenuates high-mobility group box 1 and subarachnoid hemorrhage-induced vasospasm in a rat model. Behav Brain Funct 2015;11:28.
66. Kim OS, Park EJ, Joe EH, Jou I. JAK-STAT signaling mediates gangliosides-induced inflammatory responses in brain microglial cells. J Biol Chem 2002;277:40594-601.
67. De-Fraja C, Conti L, Govoni S, Battaini F, Cattaneo E. STAT signalling in the mature and aging brain. Int J Dev Neurosci 2000;18:439-46.
68. Wei S, Luo C, Yu S, Gao J, Liu C, et al. Erythropoietin ameliorates early brain injury after subarachnoid haemorrhage by modulating microglia polarization via the EPOR/JAK2-STAT3 pathway. Exp Cell Res 2017;361:342-52.
69. An JY, Pang HG, Huang TQ, Song JN, Li DD, et al. AG490 ameliorates early brain injury via inhibition of JAK2/STAT3-mediated regulation of HMGB1 in subarachnoid hemorrhage. Exp Ther Med 2018;15:1330-8.
70. Hu X, Li P, Guo Y, Wang H, Leak RK, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012;43:3063-70.
71. Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A 2000;97:8075-80.
72. Wu Y, Pang J, Peng J, Cao F, Vitek MP, et al. An apoE-derived mimic peptide, COG1410, alleviates early brain injury via reducing apoptosis and neuroinflammation in a mouse model of subarachnoid hemorrhage. Neurosci Lett 2016;627:92-9.
73. Provencio JJ, Swank V, Lu H, Brunet S, Baltan S, et al. Neutrophil depletion after subarachnoid hemorrhage improves memory via NMDA receptors. Brain Behav Immun 2016;54:233-42.
74. Schallner N, Pandit R, LeBlanc R 3rd, Thomas AJ, Ogilvy CS, et al. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J Clin Invest 2015;125:2609-25.
75. Uekawa K, Hasegawa Y, Ma M, Nakagawa T, Katayama T, et al. Rosuvastatin ameliorates early brain injury after subarachnoid hemorrhage via suppression of superoxide formation and nuclear factor-kappa b activation in rats. J Stroke Cerebrovasc Dis 2014;23:1429-39.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Zheng ZV, Wong KCG. Microglial activation and polarization after subarachnoid hemorrhage. Neurosciences 2019;6:1. http://dx.doi.org/10.20517/2347-8659.2018.52
AMA Style
Zheng ZV, Wong KCG. Microglial activation and polarization after subarachnoid hemorrhage. Neuroimmunology and Neuroinflammation. 2019; 6: 1. http://dx.doi.org/10.20517/2347-8659.2018.52
Chicago/Turabian Style
Zheng, Zhiyuan Vera, Kwok Chu George Wong. 2019. "Microglial activation and polarization after subarachnoid hemorrhage" Neuroimmunology and Neuroinflammation. 6: 1. http://dx.doi.org/10.20517/2347-8659.2018.52
ACS Style
Zheng, ZV.; Wong KCG. Microglial activation and polarization after subarachnoid hemorrhage. Neurosciences. 2019, 6, 1. http://dx.doi.org/10.20517/2347-8659.2018.52
About This Article
Copyright

Data & Comments
Data


 Cite This Article 67 clicks
Cite This Article 67 clicks

 Like This Article 32
likes
Like This Article 32
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.