
Bilirubin and inflammation in neurodegenerative and other neurological diseases

Abstract
Inflammation links neurodegenerative, neuropsychiatric and other neurological diseases (NDs) with acute brain events. It is responsible for the alteration of neurotransmission and circuity, brain architecture, and cell fate, affecting mood and personality (anxiety, depression and schizophrenia) and behavior (decline in cognitive, motor and speech abilities, altered sleep, fatigue, pain sensitivity and dementia). Inflammation is also a key component in systemic chronic diseases (cardiovascular disease, cancer, diabetes, and metabolic syndrome), in which bilirubin has been demonstrated to improve the diseases by acting as a multi-target antiinflammatory molecule, and where the evaluation of pharmacological modulation of the pigment level as a therapeutic approach has already started. While altered serum bilirubin levels have been reported in ND patients, the potential activity of bilirubin in the brain is vague. This review summarizes the available fragmentary information on the interplay of bilirubin with neuroinflammation, aiming to elucidate the pigment’s role in the central nervous system environment.
Keywords
Introduction
Bilirubin (unconjugated - UCB) is the final metabolite of hemoglobin, which is processed in the liver (by conjugation to 1 or 2 molecules of glucuronic acid - CB: conjugated bilirubin) before elimination through urine and feces. Increased level of bilirubin (UCB and CB) in the blood is a well-recognized marker of hepatic damage. Recently, slightly elevated serum bilirubin concentration emerged as a biomarker of resistance versus chronic diseases[1]. Epidemiological data have revealed a reduced prevalence of type 2 diabetes, obesity and metabolic syndrome, certain cancers, and especially cardiovascular diseases and related causes of mortality in Gilbert’s syndrome patients, showing mild hyperbilirubinemia[2,3][Figure 1]. The effect of the pigment has been demonstrated to be due mainly to the unconjugated (or indirect) moiety of bilirubin (UCB). UCB, especially in its free form [free bilirubin (Bf), the portion of UCB exceeding the serum albumin binding capacity], enters tissues from blood, acting as a powerful antioxidant molecule at nanomolar concentrations, where it is able to counteract 10,000 times higher levels of hydrogen peroxide (H2O2)[4]. This capability has been related to the UCB-biliverdin cycle [Figure 2], which is able to regenerate the pigment consumed by oxidants and acts complementary to cellular glutathione (GSH)[4-6]. In the last years, a more complex and fascinating scenario has emerged. First, various cells have been demonstrated to possess the complete enzymatic apparatus necessary for producing UCB themselves [Figure 2][7,8]. A more expanded and intricate view has emerged from the discovery of the interplay of UCB (and the enzymes involved in its production and recycling, altogether called the “yellow players”) with cellular functions, signaling pathways, and defense/adaptation mechanisms (not restricted to redox state). Collectively, these findings suggest a greater role for the yellow players in cellular homeostasis and defense against diseases[2,3]. The interplay of the yellow players with neurological and neurodegenerative diseases is still much less explored. In this article, we review the currently available evidence on the potential roles of bilirubin and other yellow players in neurological disease, with special emphasis on inflammation. We will also address the role of enzyme modulation in bilirubin metabolism, with the goal of increasing the systemic level of bilirubin and the protection it confers.
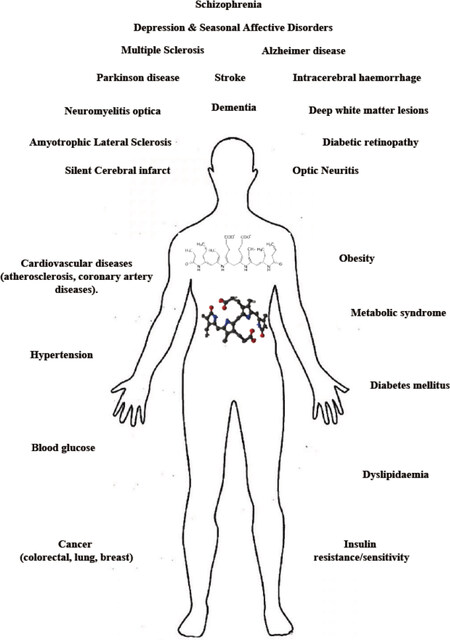
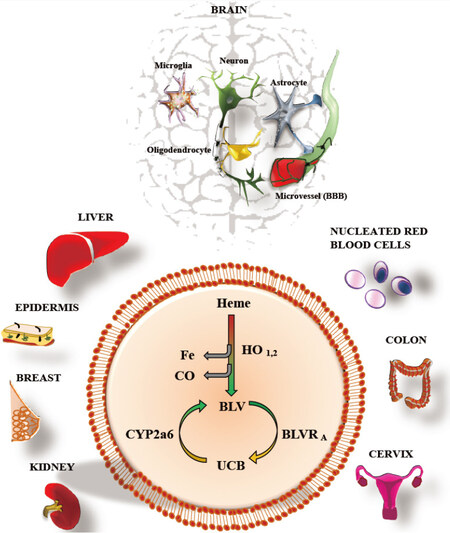
Figure 2. The yellow players. Heme: Haemoglobin; HO: Heme oxygenase; Fe: Iron; Co: Carbon monoxide; BLV: Biliverdin; BLVR: Biliverdin-reductase; UCB: Unconjugated bilirubin (indirect bilirubin); CYP: Cytochrome oxygenase. The picture shows the “yellow players” (see text) and the tissues able to produce de novo bilirubin, based on the cited literature (Takeda et al.[7])
Inflammation and neurological diseases: an overview
Chronic neurodegenerative pathologies are currently the most dominant clinical conditions. They comprise multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), Alzheimer’s disease (AD) and vascular dementia. Consistent data show that the inflammatory process can be triggered by protein misfolding or protein accumulation, which are the initial events of a given pathology (i.e., amyloid for AD, tau protein for frontal dementia, alpha synuclein for PD, etc.)[9].
Aging has been associated with a low-grade sterile inflammatory status of the immune system, in which interleukin-6 (IL6), IL1β and tumor necrosis factor (TNF) are key players, more evident in an unhealthy state. Neuroinflammation can be considered one of the most important etiological factors in age-related neurodegeneration[10], associated with a reduction in neuron number, a decrease in neuronal arborization, and loss of spines[11]. What fits particularly in this theory, is the strong evidence that in the aging brain, both macrophages and microglia react with a prolonged and overactive response to stimuli[12]. This overactivation induces the production of reactive oxygen species and attracts peripheral leukocytes, and both these conditions can activate glial cells[13]. The activation of glial cells promotes telomere shortening, which can be a contributor in different neurological conditions, such as AD[14]. Resulting impaired phagocytosis alters the removal of toxic compounds, such as amyloid-beta (Aβ) and alpha-synuclein (αSyn)[15]. Microglial activation is believed to be involved in the occurrence of deterioration in various neurodegenerative diseases, such as AD, PD, ALS, and MS[15]. Microglia are the main immune cells of the central nervous system (CNS), and as the first line of defense, microglia play an important role in the inflammatory reaction[16]. In AD, microglia are known for their role in cleaning up Aβ. Meanwhile, in PD, a pathological αSyn aggregation can induce microglial activation and dysfunction[17]. Microglia are also directly involved in MS by producing cytokines such as tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ), superoxide (O2-) or nitric oxide (NO), and release of proteolytic and lipolytic enzymes, which can destroy the myelin sheath[15,18].
Neuroinflammation and mitochondrial dysfunction are common features of chronic neurodegenerative diseases. Both conditions can lead to increased oxidative stress, which leads to excess reactive oxygen species and reactive nitrogen species, resulting in a cascade of events, with injury to polarized bilayers, lipid peroxidation, lysosomal intrinsic activity and autophagy; all these events together, permit self-potentiation of an inflammatory cascade[19].
Inflammation also plays a role in cerebral small-vessel disease (cSVD), which usually manifests as stroke, cognitive impairment, dementia, physical disability and depression[20]. Inflammation in cSVD might be explained by the role of endothelial cells in the blood-brain barrier. Endothelial cells communicate with pericytes, astrocytes, microglia, and neural stem cells in the neurovascular unit. Damage to CNS tissues leads to the disruption of tight junctions that link endothelial cells[21]. It is followed by the infiltration of neutrophils and monocytes, the activation of microglia and astrocytes and the invasion of T and B cells. This inflammatory response is mediated by the nuclear factor κ-light-chain-enhancer of activated B cells (NFκB) pathway and strongly associated with neuroinflammation in the acute phase of various vascular injuries such as stroke, diabetic retinopathy, and AD[22,23].
Bilirubin has been reported to act on all the above-mentioned molecular mechanisms. The following paragraphs review the state-of-the-art of what it is known about bilirubin and the brain.
Bilirubin: a marker for neurological diseases
Cumulative clinical evidence demonstrates the alteration of total serum bilirubin (TSB) level in neuroinflammatory diseases, including schizophrenia, MS, PD, AD, ALS, stroke, diabetic retinopathy, etc. (for details, Table 1). For obvious reasons (limited CNS sample availability, especially in the early stages of diseases), few data on the potential molecular role of the pigment in these pathologies are available, and cause-effect studies are possible only by using experimental models.
TSB level in neurological diseases
| Dis. | Study design | # Subjects
Dis/Ctrl | TSB
Dis/Ctrl | CB
Dis/Ctrl | UCB
Dis/Ctrl | P | Ref. |
|---|---|---|---|---|---|---|---|
| AD | CC | 101/101 | 10.26/15.39 | < 0.001 | [119] | ||
| AD | CC | pAD: 12/MC-AD: 12/HS: 12 | 4.62/3.93/12.48 | < 0.05 | [120] | ||
| ALS | CC | Short duration: 19/ long duration: 11 | 12.31/7.70 | 0.011 | [115] | ||
| DR | CC | 67/diabetic no DR: 35 | 10.1/15.1 | < 0.01 | [84] | ||
| DEM | CS/ | Cogn Imp: 31/no Cogn Imp: 33 | 11.3/13.5 | 0.020 | [117] | ||
| CC | Cog. Imp. 31/40 | 11.3/15.0 | 0.003 | ||||
| MS | CC | 133/88 | 11.08/16.47 | 2.20/4.26 | 8.88/2.2 | 0.001 | [87] |
| NMO | CC | 67/98 | 12.25/16.15 | 9.17/13.22 | < 0.001 | [80] | |
| NO | CC | 42/48 | 11.8/15.5 | 2.7/4.1 | 8.7/11.4 | < 0.01 | [79] |
| PD | CC | 420/435 | 9.57/7.70 | < 0.001 | [45] | ||
| PD | CS/CC | drug naïve PD: 75/75 | 12.65/8.72 | < 0.001 | [46] | ||
| PD | CC | LD-PD: 162/untr-PD: 93/ HS: 224 | 12.31/10.94/10.26 | < 0.001 | [48] | ||
| PD | CC | 425/460 | 12.09/12.3 | 3.73/3.15 | 8.36/9.15 | <0.05 | [47] |
| SCI | CC | 343/2522 | 8.3/10.7 | < 0.001 | [88] | ||
| SCZ | CC | 34/114 | 9.74/11.14 | 0.04 | [41] | ||
| SCZ | CC | 72/65 | 6.5/12.2 | 10-13 | [42] | ||
| SCZ vs. BD | CC | 71/BD: 69 | 10.62/8.04* | 7.87/5.47** | *0.02
**0.03 | [26] | |
| SCZ vs. BD | CS | 50/BD: 43 | 8.89/7.01a | 6.84/4.45b | a0.027
b0.004 | [27] | |
| SCZ vs. BD | CS | 44/BD: 56 | 7.01/4.95 | < 0.0001 | [25] | ||
| SCZ vs. SAD | CS | 44/SAD: 99 | 7.01/5.81 | < 0.03 |
Schizoprenia
Multiple contrasting studies exist on the potential correlation between the serum level of UCB and schizophrenia. Studies have reported an increase in UCB level, as in Gilbert’s syndrome in schizophrenia compared to other psychiatric diseases (affective disorder and neuropsychosis) and healthy controls[24]. More interestingly, after antipsychotic treatment, UCB decreases in 80% of subjects. Gama Marques et al.[25] also reported that mean UCB levels are clearly higher in patients with schizophrenia than patients with schizoaffective disorder and bipolar disorder. Additionally, retrospective and recent prospective studies indicate that serum UCB level in patients with schizophrenia is higher than those with bipolar affective disorder (1.40- and 1.53-fold, respectively)[26,27]. Notably, all these studies included only subjects with normal liver enzymes and no pre-existing liver disease or other confounding factors, further emphasizing the pivotal role of UCB increase. UCB can be viewed as a potential biomarker to distinguish schizophrenia from other psychiatric disorders[28].
UCB level also shows a correlation with schizophrenia symptoms. Patients with hyperbilirubinemia have shown significantly higher scores on the positive and general psychiatric subscales of the PANSS (Positive and Negative Syndrome Scale)[24]. In concordance with the previous finding, UCB elevation has been found frequently in psychotic episodes of schizophrenia[25,29]. Radhakrishnan et al.[26] reported that UCB levels are higher in paranoid schizophrenia than non-paranoid schizophrenia. Meanwhile, a recent study by Pradeep et al.[27] found no association between serum UCB levels with the severity of psychopathology in schizophrenia subjects.
Schizophrenic subjects with unconjugated hyperbilirubinemia show a significant enlargement of cerebral ventricles[30] and abnormalities of brain metabolism compared to both normobilirubinemic schizophrenia patients and normal controls[31]. Neuroinflammation in schizophrenia is characterized by increased serum concentrations of proinflammatory cytokines, including IL1β, IL6, and transforming growth factor-β (TGFβ) and microglial activation[32,33]. This inflammatory picture is similar to that observed in kernicterus spectrum disorders, the neurological sequel due to severe neonatal hyperbilirubinemia[34]. Dalman and Cullberg[35] reported that neonates experiencing severe neonatal hyperbilirubinemia (TSB > 15 mg/dL) might later have an increased frequency of mental disorders. This clinical hypothesis is supported by the finding in the Gunn rat, the animal model for severe neonatal hyperbilirubinemia[36], showing severe hyperbilirubinemia in the first weeks of life[37]. The Gunn rat displays a stereotypical behaviour, a supposedly typical symptom of schizophrenia, associated with microglia activation (indicative of inflammation), decreased ribosomal protein synthesis activity in neuronal cells, decreased neurogenesis, and increased apoptosis[38,39].
On the other hand, multiple studies have reported lower TSB levels among schizophrenia patients compared to patients suffering from some other psychiatric disorder and healthy controls[40-42]. To reconcile these contradictory findings, Vítek et al.[42] evaluated the correlation of bilirubin level with variability of the promoter of the gene for UGT1A1 (uridine-diphospho-glucuronosyltransferase 1A1) (responsible for hepatic conjugation of UCB) between schizophrenia patients and controls. They noticed that an increase in bilirubin of 1 μM (0.06 mg/dL) could reduce the odds for schizophrenia status up to 19%.
Notably, UGTs participate in the metabolism of dopamine[26], a critical neurotransmitter, the loss of which is responsible for motor PD, and dopaminergic over activity has recently been suspected responsible for psychosis in schizophrenia[43]. An additional potential link between these two pathologies might be heme oxygenase-1 (HO-1). The overexpression of this enzyme in the astrocytes of glial fibrillary acidic protein-heme oxygenase-1 (GFAP-HMOX1) transgenic mice resulted in increased subcortical oxidative imbalance with the induction of mitochondrial damage and autophagy, augmented dopamine and serotonin levels in the basal ganglia, and reduced dopamine 1 receptor (D1) in the nucleus accumbens, and enzyme overexpression caused degeneration of axons in the hippocampus and hyperkinetic behavior[44]. However, there is still no clear evidence of HO-1 alteration in the clinical setting for schizophrenia.
PD
PD is another neurological disease linked to UCB. A study comparing 420 PD patients and 435 healthy control showed not only a significant increase in bilirubin in PD patients but also a negative correlation between bilirubin level and progression from a less to more severe staging of the disease[45]. Complementary with this study, Moccia et al.[46] also reported higher TSB concentration in drug-naive PD patients compared to controls, and Qin et al.[47] also observed an upregulation of levels of direct bilirubin (conjugated bilirubin, in clinical terminology), accompanied by reduced UCB (indirect bilirubin) levels in PD patients compared to the healthy group. Notably, L-DOPA treatment (the most used therapeutic approach to PD, able to improve symptomatology temporarily), has been found to increase TSB by about 20%[48]. Because L-DOPA may increase the oxidative stress causing dopaminergic neuron loss in PD, and since bilirubin is a well-known antioxidant, an increased bilirubin level has been interpreted as a possible protective response to the disease[48].
As noted in the Introduction, almost every cell in the body possesses the full enzymatic equipment for producing UCB itself[7][Figure 2]. Modulation of the enzymes responsible for UCB production in the brain of subjects with neurological/neurodegenerative diseases has been reported, including PD patients. An increased HO-1 signal has been detected in reactive astrocytes and affected dopaminergic neurons showing Lewy bodies[49]. Although, the authors suggested that the HO-1 induction was uniquely due to its antioxidant properties, it is clear now that the elevation of HO-1 levels also represents an attempt to downregulate inflammation[50].
A direct proof of the protective effect of HO-1 induction has been obtained in experimental models. The injection of adenovirus containing human heme oxygenase-1 gene (Hmox1) into the substantia nigra of methyl-4-phenylpyridinium (MPP+) treated rats (a model for PD) revealed that the overexpression of Hmox1 protects dopaminergic neurons by reducing the expression of TNF-α and IL1β, and increasing brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) expression[51]. Both growth factors, BDNF and GDNF, are able to prevent injury to dopaminergic neurons and improve the behavioural deficits in PD[52-54]. A recent work based on organotypic cultures of substantia nigra (challenged with rotenone to induce PD) demonstrated that inflammation and redox imbalance are early and simultaneous triggers for dopaminergic neuron loss[55]. In this study, Hmox1 upregulation occurs only at the very first phases of neurodegeneration (3 hours after rotenone challenging, a time which represents the pre-diagnosis stage in human PD based on dopaminergic neuron loss). Similarly, Bdnf expression initially increased but then rapidly declined below the control level accompanying dopaminergic neuron demise. The modulation trend of both markers (Hmox1 and Bdnf) was interpreted as a failed tentative reaction to the insult[55], supporting the hypothesis of the interplay between L-DOPA and bilirubin (see above). This interpretation may also agree with a recent finding of Song etal.[56], who reported that the expression levels of HO-1 in the saliva of PD patients with early-stage (stage 1) based on the Hoehn and Yahr scale were significantly higher compared to control subjects or PD patients at stage 3 (advanced PD). The authors proposed an additional value of HO-1 as a potential biomarker in idiopathic PD patients. Notably, hyperactivation of HO-1 may exacerbate oxidative stress by the deposition of iron (Fe), one of its products [Figure 2], a phenomena frequently observed in neurological lesions and known to worsen the disease[49]. This aspect is discussed in detail later on in the review.
Contrary to PD and schizophrenia, other neurological conditions have been associated with a reduced TSB level.
Multiple sclerosis
MS is a chronic inflammatory disease characterized by the destruction of myelin in the brain and spinal cord likely due to loss of immune system tolerance to myelin[57]. TSB levels have been reported to be lower (vs. healthy controls) also in MS subjects with clinically isolated syndrome (predominately by neuroinflammation) and relapsing-remitting multiple sclerosis (predominately by neurodegenerative disorder), where predominate neurodegenerative disorders[58]. Interestingly, bilirubin levels have been shown to be significantly lower in relapsing-remitting multiple sclerosis compared to clinically isolated syndrome patients[59], suggesting a relationship between increased disease severity and decreased TSB. This hypothesis seems to be confirmed by the study of Ljubisavljevic et al.[59] who found a significant correlation between higher TSB and lower disability status, fewer MRI lesions, and shorter disease duration in both study groups. In the brain tissue from MS patients, enhanced nitrotyrosine staining, one of the markers of oxidative damage[60], was found in demyelinated regions, specifically in hypertrophic astrocytes and foamy macrophages on inflammatory lesions[61]. The damaged sites also showed an upregulation of antioxidant enzymes, including HO-1, compared to normal-appearing white matter and white matter tissue from control brains with no neurological disease, and HO-1 immunoreactivity was particularly confined to microglia[61]. These findings emphasize the close interaction between oxidative stress and inflammation in MS.
The effects of bilirubin on MS have also been studied in the animal model for the disease, the so-called experimental autoimmune encephalomyelitis (EAE) model, where the infiltration of lymphocytes, the activation of CD4+ T cells, and death of oligodendrocytes lead to the destruction of myelin sheaths[62,63]. Figure 3 illustrates the most relevant mechanisms of action of bilirubin in EAE, one of the best-known models unravelling the interplay between MS and bilirubin. Bilirubin administration in the EAE model effectively prevented both acute and chronic EAE, even better than did glucocorticoid treatment, the most commonly used therapy for MS[64,65].
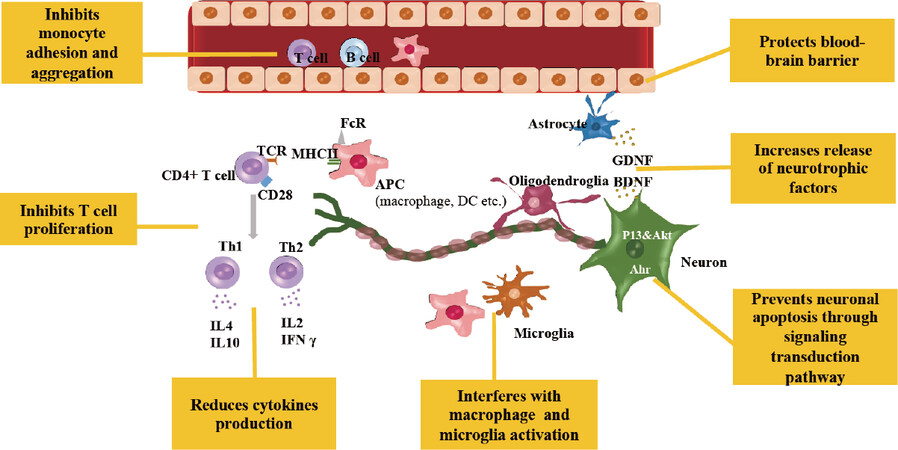
Figure 3. Multiple ways of bilirubin as immunomodulator in neuroinflammation disease. Ahr: aryl hydrocarbron receptor; APC: antigen presenting cell; BDNF: brain-derived neurotrophic factor; DC: dendritic cell; FcR: Fc receptor; GDNF: glial cell line-derived neurotrophic factor; IFNγ: Interferon γ; IL: interleukin; MHC II: major histocompatibility complex; PI3K/Akt: phosphatidylinositol 3-kinase; Th: T helper; TCR: T cell receptor
Histological analyses demonstrated that bilirubin interfered with the infiltration of inflammatory cells into the CNS by protecting the blood-brain barrier from free radical-induced permeability changes[64], stressing the intimal connection between redox stress and inflammation in CNS diseases. Further study supported the antiinflammatory potential of bilirubin in the EAE model[64]. In vitro experiments using spleen-harvested CD4+ T cells showed that bilirubin at non-apoptotic concentrations (20-150 μM) inhibits CD4+ T cell proliferation through various mechanisms. It suppresses the production of T helper-1 (Th1) cytokines (IL2 and IFNγ) and Th2 cytokines (IL4 and IL10), reduces costimulatory molecule activity (CD28 on CD4+ T cell, the co-receptor B7-1 activity in macrophages and dendritic cells), inhibits NFkB activation, which is a key transcription factor involved in T cell receptor-mediated signaling, and downregulates inducible MHC (major histocompatibility complex) class II expression. Bilirubin effectively downregulates EAE in SJL/J mice as confirmed by the reduction of the proliferation capacity of CD4+ T cell. Meanwhile, the reduction of endogenous bilirubin synthesis by zinc-protoporphyrin, a specific inhibitor of the bilirubin producing-enzyme HO-1, dramatically exacerbates this disease[66]. In contrast, induction of HO-1 by cobalt protoporphyrin IX (CoPPIX) inhibits EAE effectively[67].
According to the previous data, it seems beneficial to increase bilirubin synthesis through HO-1 induction. Several clinically used drugs have been reported to induce HO-1, among them are nonsteroidal antiinflammatory drugs (e.g., coxibs, acetylsalicylic acid) and hypolipidemic agents (e.g., niacin, fibrates, statins)[68]. Atorvastatin and rosuvastatin treatment in mice demonstrated protection by increasing not only plasma bilirubin concentrations (up to 70%) but also cardiac tissue bilirubin content (up to 119%)[69]. On the contrary, HO-1 induction in astroglia promotes oxidative mitochondrial membrane damage, iron sequestration, and mitophagy (macroautophagy)[56]. These reasons then increase the doubt of bilirubin synthesis through HO-1 induction as a good strategy to counteract the neuroinflammatory process.
In another part of inflammation signalling pathways, both UCB and biliverdin are known as activators of aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor that plays critical modulatory roles in various immune cells during innate and adaptive immune responses[2,70,71]. AhR has critical roles in MS by modulating IL17 producing T-helper (Th17) cells, and regulatory T cells[72-74], as well as B cells, macrophages and dendritic cells. In macrophages, AhR regulates IL1β production and also IL6, IL12 and TNFα expression[73,75]. AhR also inhibits the transcriptional activity of NFκB in stimulated macrophages[76]. Meanwhile, in dendritic cells, AhR mediates the generation of T regulatory cells and Th17 cells from naive T cells and also IL10 production[77]. Notably, the injection of low doses of UCB (20 µg/kg body weight, injected into C57Bl/6 mice) increased the population of regulatory T cells by more than 50% compared to controls, prolonging graft survival[78]. Regulatory T cells are also inducible by HO-1 activity, CO, and UCB[78] and all “yellow players” [Figure 2].
Optic neuritis and neuromyelitis optica
Reduced TSB has been noticed in both optic neuritis, an acute inflammatory demyelinating disorder of the optic nerve, and neuromyelitis optica, also known as Devic disease, a severe autoimmune demyelinating disease that selectively affects the optic nerve and spinal cord[79-82]. Although the main focus was on the potential antioxidant role of bilirubin, suggesting that the low TSB is a result of the overconsumption of bilirubin due to oxidative stress, immunohistochemistry of damaged nerves is similar as in MS[83], with an evident inflammatory component. Thus, the interplay between bilirubin and inflammation described in MS also applies to optic neuritis and neuromyelitis optica. Further studies are needed.
Diabetic retinopathy and stroke
TSB levels have been found to be decreased also in patients with diabetes mellitus and diabetic retinopathy vs. subjects with diabetes mellitus without retinopathy, suggesting TSB as a biomarker of diabetic retinopathy[84]. In the pathogenesis of diabetic retinopathy, particularly in the non-proliferative type, hyperglycemia exposure causes the alteration of retinal microvasculature with pericyte loss[85], endothelial apoptosis and endothelial thickening of the basement membrane, which further lead to capillary occlusion and ischemia[86].
Similarly, damage and even destruction of the cerebral vasculature are landmarks of stroke and related vascular events[87]. In a large study conducted by Li et al.[88], 343 subjects with silent cerebral infarct presented with lower TSB levels. In another study, patients with deep white matter lesions (DWMLs) had a decreased TSB level compared to non-DWMLs subjects. Again, the low and intermediate bilirubin groups showed a higher prevalence of severe DWMLs than did the group with higher bilirubin levels[89]. Notably, DWMLs are a recognized predictor for the development of impaired cognitive function and stroke[90,91].
A recent study also explored the association of UCB and intracranial atherosclerosis, which was found in approximately 50% of patients with transient ischemic attack and up to 47% of ischemic stroke patients in Asia[92-94]. In this study, UCB was significantly negatively associated with intracranial atherosclerosis. The odds of intracranial atherosclerosis was 0 0.67-fold lower in participants in the high UCB concentration group (≥ 10.10 mMol/L) when compared with those in the low UCB concentration group[94]. An increased bilirubin level has been proposed as a novel independent predictor for hemorrhagic transformation and symptomatic intracranial hemorrhage after mechanical thrombectomy[95].
The role of bilirubin in all previously mentioned conditions might be explained by a large number of studies suggesting bilirubin as a protector against microvascular complications. Bilirubin exhibits potent antiinflammatory effects [via HO-1, nuclear factor erythroid 2-related factor 2 (Nrf2) and NFκB] by the inhibition of monocyte transmigration [through a decrease in TNFα-induced monocyte chemoattractant protein-1 (MCP-1) secretion], reducing endothelial vascular cell adhesion molecule-1 (VCAM-1) expression, and improving endothelial cell dysfunction and hyperproliferation after damage[96-99]. UCB also appears to affect the immune system by inhibiting the activation of the complement cascade[100], and also by modulating the phagocytic and antigen-presenting function of macrophages (by changing the expression of Fc receptors)[101].
The potential protective role of HO-1 induction has been demonstrated in several studies. HO-1 increases early (24 h) after brain trauma[102], and in intracerebral haemorrhage models, after infiltration of the brain by blood components activating the inflammatory response[103]. HO-1 activity is observed in both endothelial cells and microglia surrounding the haematoma site. The protective effects of HO-1 in neurons was studied by Orozco-Ibarra et al.[104], who revealed that the upregulation of HO-1 prevented the death of cerebellar granule neurons due to mitochondrial toxicity. Further investigation found that the HO-1 products, bilirubin and CORM2 (carbon monoxide releasing molecule), were involved in preventing cell death.
HO-1 has neuroprotective effects by regulating the phosphatidylinositol 3-kinase (PI3K/AKT) signalling pathway and by reducing apoptosis in rats with cerebral haemorrhage[105]. Feng et al.[106] showed that the PI3K/AKT and extracellular signal regulated kinase pathways are involved in oleanolic acid induced HO-1 expression by activating Nrf2 in vascular smooth muscle cells. As one of the signalling pathways for cell survival, the PI3K/AKT signal transduction pathway plays an important role in cell proliferation and differentiation and inhibition of neuronal apoptosis[107]. Therefore, HO-1 may protect the nerves of rats with cerebral haemorrhage by regulating the PI3K/AKT signalling pathway.
A second isoform of HO exists in the brain, called heme oxygenase-2 (HO-2). Considered constitutive, it looks like HO-2 in the brain plays the vital function of maintaining adequate levels of UCB to guarantee normal cellular homeostasis, participating in brain protection[108]. The protective activity of HO-2 has been reported in intracerebral haemorrhage models, where heme oxygenase-2 gene (Hmox2) deletion led to greater brain injury volumes and neurological deficits than in wild-type mice after intracerebral haemorrhage[109]. In brain cultures, Doré et al.[108] expanded our knowledge on the role of HO-2 and bilirubin as neuroprotective factors by showing increased neuronal death in cerebellar granule cultures of Hmox2 knockout mice (Hmox2-/-). On the contrary, Hmox2 transfection in human embryonic kidney 293 (HEK293) cells rescued cells from apoptotic death. In another study, the induction of HO-2 activity by phorbol esters enhanced the production of bilirubin, which protected primary hippocampal and cortical neuronal cultures from the neurotoxicity of H2O2. Of note, HO-2 was immunolocalised in neurons both before and after traumatic brain injury, whereas HO-1 was highly upregulated in glia only after traumatic brain injury. Cell loss was significantly greater in Hmox2-/- mice in areas including the cortex, hippocampus and lateral dorsal thalamus[110].
Biliverdin (BV), another yellow player, also showed neuroprotective effects by ameliorating cerebral reperfusion injury in rats most probably via its antiinflammatory activity[111]. Two pathways are known to be involved in the antiinflammatory mechanism of BV: by activating the nitric oxide-dependent biliverdin reductase, BV reduces the expression of toll-like receptor-4 (TLR-4) in murine macrophages[112], and BV regulates the expression of complement C5a receptor[113]. Furthermore, a study by Zou et al.[111] found the downregulation of miR-204-5p and its target gene, ETS protooncogene 1 (Ets1), in cerebral ischemia-reperfusion injury rats following BV administration. Ets1 is known to be responsible for inducing proinflammatory Th1 type response and causing neuronal death[114]. Therefore, BV may play a crucial role in preventing injury in stroke by interfering in miRNAs levels.
Additional neurological conditions underpinned by reduced TSB levels
ALS
In ALS, the reduction of TSB levels correlates with both the clinical state and disease duration. Patients with long-lasting ALS (where motor neuron degeneration is noticed) have lower TSB levels than do patients with a shorter duration[115].
Seasonal affective disorder
Nocturnal plasma bilirubin levels were evaluated in individuals with unipolar recurrent winter seasonal major depressive disorder and age-/gender-matched controls. Lower bilirubin levels were found in patients with seasonal affective disorder. The reduction in bilirubin level in this study was proposed as a vulnerable risk for depression, although only an associative and not a causative link was provided[116].
Dementia
A significant reduction in TSB levels was reported in patients with cognitive impairment compared to those with normal cognitive function, although no significant correlation between bilirubin and disease duration was found[117]. In a further study by Baierle et al.[118], the authors showed that the significant increase in the proinflammatory cytokines IL1β and TNFα correlated with the increase in oxidative stress and inversely associated with low cognitive performance in elderly patients.
AD
As in the majority of neurological conditions, TSB levels are also decreased in AD[119]. The lower concentration of bilirubin is supposed to have a role in the transition from mild cognitive impairment to clear AD[120]. Similarly, reduced Hmox1 mRNA[121] and protein[122] levels were detected in the serum of full-blown AD subjects, and proposed to correlate with cognitive decline[120]. Notably, the diagnosis of AD and cognitive decline is usually a late event and performed after the pathological alteration has already existed for some time. In the early stages, the brain tries to protect itself from the oxidative and inflammatory insult of AD by increasing the endogenous production of UCB. Both HO-1 and biliverdin reductase A (BLVR-A), the two key enzymes in biliverdin and bilirubin production [Figure 2], have been reported to be upregulated in the brain of AD patients[103,123]. However, the upregulation of HO-1 will also cause Fe deposition in the brain, which worsens and accelerates disease progression by enhancing the redox imbalance in ongoing AD lesions[49,124-126]. It should be recalled that the brain possesses very low antioxidant capacity compared to other organs, being much lower in neonatal life[127,128] and in the elderly[129,130]. Thus, the enhanced pro-oxidant milieu due to HO-1 hyperactivation leads to an increased oxidative and nitrosative post-translational modification of cellular enzymes, with their consequent inactivation[131]. One of the targets of this mechanism is BLVRA, and thus, UCB production will be stopped[123].
The chains of events described here, leading to the disruption of UCB-mediated protection and contributing to the progression of neurological damage fits well with what we described in PD.
Is the brain different from the rest of the body?
Differently from extra-CNS diseases involving oxidative imbalance and inflammation, where a positive correlation between higher TSB level (Gilbert syndrome-like) and reduced disease incidence has been frequently found[2,3,68], the most readily evident finding in this review is that brain diseases have lower TSB levels in common. Two major explanation have been given: (1) lower TSB levels in subjects with neurological disease may reflect the consumption of UCB due to oxidative stress, although it seems unlikely that the local redox imbalance occurring in the brain may affect the systemic levels of bilirubin; and (2) individuals with lower TSB, thus a lower systemic antioxidant status, might be more vulnerable to oxidative stress[118] and related diseases (both in the brain and extra-CNS organs) [Figure 1].
Experimental evidence strongly supports the notion that an increased concentration of bilirubin in the CNS exerts antiinflammatory and antioxidant effects. On the other hand, it should be recalled that hyperactivation of HO-1 in the CNS, reported to be a tentative reaction against brain insults, may enhance the damage[68,104,131-148]. This specificity of the CNS has to be taken into account in the growing research aimed at modulating the bilirubin players to increase levels of bilirubin and the protection it confers.
Conclusion
Studies assessing clearly the molecular events correlating bilirubin and neurological damage are still lacking, and many conclusions are based on clinical series. Understanding the role of bilirubin and all the other yellow players in the development and progression of the different neurological disorders may help in deciphering the efficacy of the modulation of bilirubin level to prevent CNS diseases. However, due to the side effects of the drugs inducing HO-1 activity, caution must be taken before their recommendation. More intriguing and promising is the in situ CNS elevation of UCB level achievable by the development of molecules modulating BLVR. The future will hopefully provide the much-needed answer due to the medical and social burden of neurological disorders.
Declarations
Authors’ contributionsMade substantial contributions to the search of the literature, the writing of the manuscript, and the revision and discussion of the contents of the paper: Jayanti S, Moretti R, Tiribelli C, Gazzin S
Availability of data and materialsNot applicable.
Financial support and sponsorshipJayanti S was supported in part by a fellowship from the Lembaga Pengelola Dana Pendidikan of Ministry of Finance of Indonesia and an internal grant from the Italian Liver Foundation. The funders had no role in data analysis and interpretation and also writing of the manuscript.
Conflicts of interestAll authors declare that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Gazzin S, Masutti F, Vitek L, Tiribelli C. The molecular basis of jaundice: an old symptom revisited. Liver Int 2017;37:1094-102.
2. Gazzin S, Vitek L, Watchko J, Shapiro SM, Tiribelli C. A novel perspective on the biology of bilirubin in health and disease. Trends Mol Med 2016;22:758-68.
3. Wagner KH, Wallner M, Mölzer C, Gazzin S, Bulmer AC, et al. Looking to the horizon: the role of bilirubin in the development and prevention of age-related chronic diseases. Clin Sci (Lond) 2015;129:1-25.
4. Baranano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci U S A 2002;99:16093-8.
5. Sedlak TW, Snyder SH. Bilirubin benefits: cellular protection by a biliverdin reductase antioxidant cycle. Pediatrics 2004;113:1776-82.
6. Sedlak TW, Saleh M, Higginson DS, Paul BD, Juluri KR, et al. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc Natl Acad Sci U S A 2009;106:5171-6.
7. Takeda TA, Mu A, Tai TT, Kitajima S, Taketani S. Continuous de novo biosynthesis of haem and its rapid turnover to bilirubin are necessary for cytoprotection against cell damage. Sci Rep 2015;5:10488.
8. Yuan X, Rietzschel N, Kwon H, Walter Nuno AB, Hanna DA, et al. Regulation of intracellular heme trafficking revealed by subcellular reporters. Proc Natl Acad Sci U S A 2016;113:E5144-52.
10. Stephenson J, Nutma E, van der Valk P, Amor S. Inflammation in CNS neurodegenerative diseases. Immunology 2018;154:204-19.
12. Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, et al. Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain 2016;139:653-61.
13. Chinta SJ, Woods G, Rane A, Demaria M, Campisi J, et al. Cellular senescence and the aging brain. Exp Gerontol 2015;68:3-7.
14. Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res 2007;10:61-74.
15. Xu L, He D, Bai Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol Neurobiol 2016;53:6709-15.
17. Zhang W, Wang T, Pei Z, Miller DS, Wu X, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 2005;19:533-42.
18. Bsibsi M, Peferoen LA, Holtman IR, Nacken PJ, Gerritsen WH, et al. Demyelination during multiple sclerosis is associated with combined activation of microglia/macrophages by IFN-γ and alpha B-crystallin. Acta Neuropathol 2014;128:215-29.
19. Fischer R, Maier O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxid Med Cell Longev 2015;2015:610813.
20. Brown R, Benveniste H, Black SE, Charpak S, Dichgans M, et al. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc Res 2018;114:1462-73.
21. Choi YK, Kim KW. Blood-neural barrier: its diversity and coordinated cell-to-cell communication. BMB Rep 2008;41:345-52.
22. Lee H, Choi YK. Regenerative effects of heme oxygenase metabolites on neuroinflammatory diseases. Int J Mol Sci 2018;20:78.
23. Dong J, Jimi E, Zeiss C, Hayden MS, Ghosh S. Constitutively active NF-kappaB triggers systemic TNFalpha-dependent inflammation and localized TNFalpha-independent inflammatory disease. Genes Dev 2010;24:1709-17.
24. Miyaoka T, Seno H, Itoga M, Iijima M, Inagaki T, et al. Schizophrenia-associated idiopathic unconjugated hyperbilirubinemia (Gilbert’s syndrome). J Clin Psychiatry 2000;61:868-71.
25. Gama Marques J, Pedro I, Ouakinin S. Unconjugated bilirubin and acute psychosis: a five years retrospective observational and controlled study in patients with schizophrenia, schizoaffective and bipolar disorders. Int J Psychiatry Clin Pract 2019;23:281-5.
26. Radhakrishnan R, Kanigere M, Menon J, Calvin S, Janish A, et al. Association between unconjugated bilirubin and schizophrenia. Psychiatry Res 2011;189:480-2.
27. Pradeep JR, Acharya MS, Radhakrishnan R, Srinivasan K. Elevated unconjugated bilirubin in schizophrenia compared to bipolar affective disorder. Prim Care Companion CNS Disord 2019;21:19m02448.
28. Pommerening Dornelles E, Gama Marques J, Ouakinin S. Unconjugated bilirubin and schizophrenia: a systematic review. CNS Spectr 2019;24:577-88.
29. Gama Marques J, Ouakinin S. Clinical profile in schizophrenia and schizoaffective spectrum: relation with unconjugated bilirubin in a prospective and controlled study with psychopathological and psychosocial variables. CNS Spectr 2019:1-8.
30. Miyaoka T, Seno H, Itoga M, Inagaki T, Horiguchi J. Structural brain changes in schizophrenia associated with idiopathic unconjugated hyperbilirubinemia (Gilbert’s syndrome): a planimetric CT study. Schizophr Res 2001;52:291-3.
31. Miyaoka T, Yasukawa R, Mizuno S, Sukegawa T, Inagaki T, et al. Proton magnetic resonance spectroscopy (1H-MRS) of hippocampus, basal ganglia, and vermis of cerebellum in schizophrenia associated with idiopathic unconjugated hyperbilirubinemia (Gilbert’s syndrome). J Psychiatr Res 2005;39:29-34.
32. Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B. Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 2011;70:663-71.
33. Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, et al. Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol 2006;112:305-16.
34. Le Pichon JB, Riordan SM, Watchko J, Shapiro SM. The neurological sequelae of neonatal hyperbilirubinemia: definitions, diagnosis and treatment of the kernicterus spectrum disorders (KSDs). Curr Pediatr Rev 2017;13:199-209.
35. Dalman C, Cullberg J. Neonatal hyperbilirubinaemia--a vulnerability factor for mental disorder? Acta Psychiatr Scand 1999;100:469-71.
36. Chowdhury JR, Kondapalli R, Chowdhury NR. Gunn rat: a model for inherited deficiency of bilirubin glucuronidation. Adv Vet Sci Comp Med 1993;37:149-73.
37. Gazzin S, Zelenka J, Zdrahalova L, Konickova R, Zabetta CC, et al. Bilirubin accumulation and Cyp mRNA expression in selected brain regions of jaundiced Gunn rat pups. Pediatr Res 2012;71:653-60.
38. Liaury K, Miyaoka T, Tsumori T, Furuya M, Wake R, et al. Morphological features of microglial cells in the hippocampal dentate gyrus of Gunn rat: a possible schizophrenia animal model. J Neuroinflammation 2012;9:56.
39. Hayashida M, Miyaoka T, Tsuchie K, Yasuda H, Wake R, et al. Hyperbilirubinemia-related behavioral and neuropathological changes in rats: a possible schizophrenia animal model. Prog Neuropsychopharmacol Biol Psychiatry 2009;33:581-8.
40. Pae CU, Paik IH, Lee C, Lee SJ, Kim JJ, et al. Decreased plasma antioxidants in schizophrenia. Neuropsychobiology 2004;50:54-6.
41. Yin XL, Jia QF, Zhang GY, Zhang JP, Shirao T, et al. Association between decreased serum TBIL concentration and immediate memory impairment in schizophrenia patients. Sci Rep 2019;9:1622.
42. Vítek L, Novotná M, Lenícek M, Novotný L, Eberová J, et al. Serum bilirubin levels and UGT1A1 promoter variations in patients with schizophrenia. Psychiatry Res 2010;178:449-50.
43. Duan J, Göring HHH, Sanders AR, Moy W, Freda J, et al; MGS. Transcriptomic signatures of schizophrenia revealed by dopamine perturbation in an ex vivo model. Transl Psychiatry 2018;8:158.
44. Song W, Zukor H, Lin SH, Hascalovici J, Liberman A, et al. Schizophrenia-like features in transgenic mice overexpressing human HO-1 in the astrocytic compartment. J Neurosci 2012;32:10841-53.
45. Macías-García D, Méndez-Del Barrio C, Jesús S, Labrador MA, Adarmes-Gómez A, et al. Increased bilirubin levels in Parkinson’s disease. Parkinsonism Relat Disord 2019;63:213-6.
46. Moccia M, Picillo M, Erro R, Longo K, Amboni M, et al. Increased bilirubin levels in de novo Parkinson’s disease. Eur J Neurol 2015;22:954-9.
47. Qin XL, Zhang QS, Sun L, Hao MW, Hu ZT. Lower serum bilirubin and uric acid concentrations in patients with Parkinson’s Disease in China. Cell Biochem Biophys 2015;72:49-56.
48. Scigliano G, Girotti F, Soliveri P, Musicco M, Radice D, et al. Increased plasma bilirubin in Parkinson patients on L-dopa: evidence against the free radical hypothesis? Ital J Neurol Sci 1997;18:69-72.
49. Schipper HM, Song W, Zukor H, Hascalovici JR, Zeligman D. Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. J Neurochem 2009;110:469-85.
50. Cuadrado A, Rojo AI. Heme oxygenase-1 as a therapeutic target in neurodegenerative diseases and brain infections. Curr Pharm Des 2008;14:429-42.
51. Hung SY, Liou HC, Kang KH, Wu RM, Wen CC, et al. Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Mol Pharmacol 2008;74:1564-75.
52. Gill SS, Patel NK, Hotton GR, O’Sullivan K, McCarter R, et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med 2003;9:589-95.
53. Love S, Plaha P, Patel NK, Hotton GR, Brooks DJ, et al. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat Med 2005;11:703-4.
54. Patel NK, Bunnage M, Plaha P, Svendsen CN, Heywood P, et al. Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: a two-year outcome study. Ann Neurol 2005;57:298-302.
55. Dal Ben M, Bongiovanni R, Tuniz S, Fioriti E, Tiribelli C, et al. Earliest mechanisms of dopaminergic neurons sufferance in a novel slow progressing ex vivo model of parkinson disease in rat organotypic cultures of substantia nigra. Int J Mol Sci 2019;20:2224.
56. Song W, Kothari V, Velly AM, Cressatti M, Liberman A, et al. Evaluation of salivary heme oxygenase-1 as a potential biomarker of early Parkinson’s disease. Mov Disord 2018;33:583-91.
57. Ortiz GG, Pacheco-Moisés FP, Bitzer-Quintero OK, Ramírez-Anguiano AC, Flores-Alvarado LJ, et al. Immunology and oxidative stress in multiple sclerosis: clinical and basic approach. Clin Dev Immunol 2013;2013:708659.
58. Gonsette RE. Oxidative stress and excitotoxicity: a therapeutic issue in multiple sclerosis? Mult Scler 2008;14:22-34.
59. Ljubisavljevic S, Stojanovic I, Vojinovic S, Milojkovic M, Dunjic O, et al. Association of serum bilirubin and uric acid levels changes during neuroinflammation in patients with initial and relapsed demyelination attacks. Metab Brain Dis 2013;28:629-38.
60. Halliwell B, Zhao K, Whiteman M. Nitric oxide and peroxynitrite. The ugly, the uglier and the not so good: a personal view of recent controversies. Free Radic Res 1999;31:651-69.
61. van Horssen J, Schreibelt G, Drexhage J, Hazes T, Dijkstra CD, et al. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic Biol Med 2008;45:1729-37.
62. Artemiadis AK, Anagnostouli MC. Apoptosis of oligodendrocytes and post-translational modifications of myelin basic protein in multiple sclerosis: possible role for the early stages of multiple sclerosis. Eur Neurol 2010;63:65-72.
63. Kostic MS, Rajkovic JS, Floranovic MSP, Dimov ID, Pavlovic DD. Multiple sclerosis and oxidative stress - a clinical perspective. Neurochem J 2013;7:76-86.
64. Liu Y, Zhu B, Wang X, Luo L, Li P, et al. Bilirubin as a potent antioxidant suppresses experimental autoimmune encephalomyelitis: implications for the role of oxidative stress in the development of multiple sclerosis. J Neuroimmunol 2003;139:27-35.
65. Goodin DS. Chapter 21 - Glucocorticoid treatment of multiple sclerosis. In: Goodin DS, editor. Handbook of Clinical Neurology. Elsevier; 2014. pp. 455-64.
66. Liu Y, Li P, Lu J, Xiong W, Oger J, et al. Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J Immunol 2008;181:1887-97.
67. Chora AA, Fontoura P, Cunha A, Pais TF, Cardoso S, et al. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest 2007;117:438-47.
68. Vitek L, Bellarosa C, Tiribelli C. Induction of mild hyperbilirubinemia: hype or real therapeutic opportunity? Clin Pharmacol Ther 2019;106:568-75.
69. Muchova L, Wong RJ, Hsu M, Morioka I, Vitek L, et al. Statin treatment increases formation of carbon monoxide and bilirubin in mice: a novel mechanism of in vivo antioxidant protection. Can J Physiol Pharmacol 2007;85:800-10.
70. Nguyen NT, Hanieh H, Nakahama T, Kishimoto T. The roles of aryl hydrocarbon receptor in immune responses. Int Immunol 2013;25:335-43.
71. Vítek L. Bilirubin as a signaling molecule. Med Res Rev 2020. Epub ahead of print [PMID: 32017160 DOI: 10.1002/med.21660]
72. Nakahama T, Hanieh H, Nguyen NT, Chinen I, Ripley B, et al. Aryl hydrocarbon receptor-mediated induction of the microRNA-132/212 cluster promotes interleukin-17-producing T-helper cell differentiation. Proc Natl Acad Sci U S A 2013;110:11964-9.
73. Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A 2008;105:9721-6.
74. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008;453:65-71.
75. Sekine H, Mimura J, Oshima M, Okawa H, Kanno J, et al. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol Cell Biol 2009;29:6391-400.
76. Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med 2009;206:2027-35.
77. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A 2010;107:19961-6.
78. Lee SS, Gao W, Mazzola S, Thomas MN, Csizmadia E, et al. Heme oxygenase-1, carbon monoxide, and bilirubin induce tolerance in recipients toward islet allografts by modulating T regulatory cells. FASEB J 2007;21:3450-7.
79. Deng J, Liang XM, Zhang XL, Ling SQ, Yang TT, et al. Relationship between serum bilirubin levels and optic neuritis. Chin Med J (Engl) 2013;126:3307-10.
80. Peng F, Yang Y, Liu J, Jiang Y, Zhu C, et al. Low antioxidant status of serum uric acid, bilirubin and albumin in patients with neuromyelitis optica. Eur J Neurol 2012;19:277-83.
81. Wang X, Jiao W, Lin M, Lu C, Liu C, et al. Resolution of inflammation in neuromyelitis optica spectrum disorders. Mult Scler Relat Disord 2019;27:34-41.
82. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485-9.
83. Tsoi VL, Hill KE, Carlson NG, Warner JE, Rose JW. Immunohistochemical evidence of inducible nitric oxide synthase and nitrotyrosine in a case of clinically isolated optic neuritis. J Neuroophthalmol 2006;26:87-94.
84. Cho HC. The Relationship among Homocysteine, Bilirubin, and Diabetic Retinopathy. Diabetes Metab J 2011;35:595-601.
85. Naruse K, Nakamura J, Hamada Y, Nakayama M, Chaya S, et al. Aldose reductase inhibition prevents glucose-induced apoptosis in cultured bovine retinal microvascular pericytes. Exp Eye Res 2000;71:309-15.
86. Beltramo E, Porta M. Pericyte loss in diabetic retinopathy: mechanisms and consequences. Curr Med Chem 2013;20:3218-25.
87. Peng F, Deng X, Yu Y, Chen X, Shen L, et al. Serum bilirubin concentrations and multiple sclerosis. J Clin Neurosci 2011;18:1355-9.
88. Li RY, Cao ZG, Zhang JR, Li Y, Wang RT. Decreased serum bilirubin is associated with silent cerebral infarction. Arterioscler Thromb Vasc Biol 2014;34:946-51.
89. Higuchi S, Kabeya Y, Uchida J, Kato K, Tsukada N. Low bilirubin levels indicate a high risk of cerebral deep white matter lesions in apparently healthy subjects. Sci Rep 2018;8:6473.
90. Garde E, Mortensen EL, Krabbe K, Rostrup E, Larsson HB. Relation between age-related decline in intelligence and cerebral white-matter hyperintensities in healthy octogenarians: a longitudinal study. Lancet 2000;356:628-34.
91. Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ 2010;341:c3666.
92. Suwanwela NC, Chutinetr A. Risk factors for atherosclerosis of cervicocerebral arteries: intracranial versus extracranial. Neuroepidemiology 2003;22:37-40.
93. Wong KS, Li H, Chan YL, Ahuja A, Lam WW, et al. Use of transcranial Doppler ultrasound to predict outcome in patients with intracranial large-artery occlusive disease. Stroke 2000;31:2641-7.
94. Zhong K, Wang X, Ma X, Ji X, Sang S, et al. Association between serum bilirubin and asymptomatic intracranial atherosclerosis: results from a population-based study. Neurol Sci 2020. Epub ahead of print. doi: 10.1007/s10072-020-04268-x
95. Jian Y, Zhao L, Wang H, Li T, Zhang L, et al. Bilirubin: a novel predictor of hemorrhagic transformation and symptomatic intracranial hemorrhage after mechanical thrombectomy. Neurol Sci 2019. Epub ahead of print. doi:10.1007/s10072-019-04182-x
96. Ishikawa K, Navab M, Leitinger N, Fogelman AM, Lusis AJ. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. J Clin Invest 1997;100:1209-16.
97. Kawamura K, Ishikawa K, Wada Y, Kimura S, Matsumoto H, et al. Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler Thromb Vasc Biol 2005;25:155-60.
98. Ollinger R, Bilban M, Erat A, Froio A, McDaid J, et al. Bilirubin: a natural inhibitor of vascular smooth muscle cell proliferation. Circulation 2005;112:1030-9.
99. Pae HO, Oh GS, Lee BS, Rim JS, Kim YM, et al. 3-Hydroxyanthranilic acid, one of L-tryptophan metabolites, inhibits monocyte chemoattractant protein-1 secretion and vascular cell adhesion molecule-1 expression via heme oxygenase-1 induction in human umbilical vein endothelial cells. Atherosclerosis 2006;187:274-84.
100. Basiglio CL, Arriaga SM, Pelusa HF, Almará AM, Roma MG, et al. Protective role of unconjugated bilirubin on complement-mediated hepatocytolysis. Biochim Biophys Acta 2007;1770:1003-10.
101. Vĕtvicka V, Miler I, Síma P, Táborský L, Fornůsek L. The effect of bilirubin on the Fc receptor expression and phagocytic activity of mouse peritoneal macrophages. Folia Microbiol (Praha) 1985;30:373-80.
102. Dohi K, Satoh K, Ohtaki H, Shioda S, Miyake Y, et al. Elevated plasma levels of bilirubin in patients with neurotrauma reflect its pathophysiological role in free radical scavenging. In Vivo 2005;19:855-60.
103. Wang J, Doré S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain 2007;130:1643-52.
104. Orozco-Ibarra M, Estrada-Sánchez AM, Massieu L, Pedraza-Chaverrí J. Heme oxygenase-1 induction prevents neuronal damage triggered during mitochondrial inhibition: role of CO and bilirubin. Int J Biochem Cell Biol 2009;41:1304-14.
105. Zhao Q, Qu R, Teng L, Yin C, Yuan Y. HO-1 protects the nerves of rats with cerebral hemorrhage by regulating the PI3K/AKT signaling pathway. Neuropsychiatr Dis Treat 2019;15:1459-68.
106. Feng J, Zhang P, Chen X, He G. PI3K and ERK/Nrf2 pathways are involved in oleanolic acid-induced heme oxygenase-1 expression in rat vascular smooth muscle cells. J Cell Biochem 2011;112:1524-31.
107. Li Q, Huai L, Zhang C, Wang C, Jia Y, et al. Icaritin induces AML cell apoptosis via the MAPK/ERK and PI3K/AKT signal pathways. Int J Hematol 2013;97:617-23.
108. Doré S, Goto S, Sampei K, Blackshaw S, Hester LD, et al. Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience 2000;99:587-92.
109. Wang J, Zhuang H, Doré S. Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage. Neurobiol Dis 2006;22:473-6.
110. Chang EF, Wong RJ, Vreman HJ, Igarashi T, Galo E, et al. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J Neurosci 2003;23:3689-96.
111. Zou ZY, Liu J, Chang C, Li JJ, Luo J, et al. Biliverdin administration regulates the microRNA-mRNA expressional network associated with neuroprotection in cerebral ischemia reperfusion injury in rats. Int J Mol Med 2019;43:1356-72.
112. Wegiel B, Gallo D, Csizmadia E, Roger T, Kaczmarek E, et al. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc Natl Acad Sci U S A 2011;108:18849-54.
113. Holst B, Raby AC, Hall JE, Labéta MO. Complement takes its Toll: an inflammatory crosstalk between Toll-like receptors and the receptors for the complement anaphylatoxin C5a. Anaesthesia 2012;67:60-4.
114. Pulliam JV, Xu Z, Ford GD, Liu C, Li Y, et al. Computational identification of conserved transcription factor binding sites upstream of genes induced in rat brain by transient focal ischemic stroke. Brain Res 2013;1495:76-85.
115. Iłzecka J, Stelmasiak Z. Serum bilirubin concentration in patients with amyotrophic lateral sclerosis. Clin Neurol Neurosurg 2003;105:237-40.
116. Oren DA, Desan PH, Boutros N, Anand A, Charney DS. Effects of light on low nocturnal bilirubin in winter depression: a preliminary report. Biol Psychiatry 2002;51:422-5.
117. Raszewski G, Gustaw K, Chwedorowicz R. Endogenous antioxidant status in dementia patients with cognitive impairment and normal cognitive function. Clin Endocrinol 2011;1:13-23.
118. Baierle M, Nascimento SN, Moro AM, Brucker N, Freitas F, et al. Relationship between inflammation and oxidative stress and cognitive decline in the institutionalized elderly. Oxid Med Cell Longev 2015;2015:804198.
119. Kim TS, Pae CU, Yoon SJ, Jang WY, Lee NJ, et al. Decreased plasma antioxidants in patients with Alzheimer’s disease. Int J Geriatr Psychiatry 2006;21:344-8.
120. Di Domenico F, Barone E, Mancuso C, Perluigi M, Cocciolo A, et al. HO-1/BVR-a system analysis in plasma from probable Alzheimer’s disease and mild cognitive impairment subjects: a potential biochemical marker for the prediction of the disease. J Alzheimers Dis 2012;32:277-89.
121. Ishizuka K, Kimura T, Yoshitake J, Akaike T, Shono M, et al. Possible assessment for antioxidant capacity in Alzheimer’s disease by measuring lymphocyte heme oxygenase-1 expression with real-time RT-PCR. Ann N Y Acad Sci 2002;977:173-8.
122. Barone E, Di Domenico F, Sultana R, Coccia R, Mancuso C, et al. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic Biol Med 2012;52:2292-301.
123. Barone E, Di Domenico F, Mancuso C, Butterfield DA. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: it’s time for reconciliation. Neurobiol Dis 2014;62:144-59.
124. Liu B, Moloney A, Meehan S, Morris K, Thomas SE, et al. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J Biol Chem 2011;286:4248-56.
125. Schipper HM. Heme oxygenase expression in human central nervous system disorders. Free Radic Biol Med 2004;37:1995-2011.
126. Schipper HM. Heme oxygenase-1: transducer of pathological brain iron sequestration under oxidative stress. Ann N Y Acad Sci 2004;1012:84-93.
127. Buonocore G, Perrone S, Bracci R. Free radicals and brain damage in the newborn. Biol Neonate 2001;79:180-6.
128. Matyas M, Zaharie G. Antioxidants at newborns. Antioxidants 2019. Available from: https:/doi.org/10.5772/intechopen.85175 [Last accessed on 27 Mar 2020].
129. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature 2000;408:239-47.
130. Gemma C, Vila J, Bachstetter A, Bickford PC. Oxidative stress and the aging brain: from theory to prevention. In: Riddle DR, editor. Brain aging: models, methods, and mechanisms. FL: CRC Press/Taylor & Francis; 2007.
131. Nitti M, Piras S, Marinari UM, Moretta L, Pronzato MA, et al. HO-1 induction in cancer progression: a matter of cell adaptation. Antioxidants (Basel) 2017;6:29.
132. Attucks OC, Jasmer KJ, Hannink M, Kassis J, Zhong Z, et al. Induction of heme oxygenase I (HMOX1) by HPP-4382: a novel modulator of Bach1 activity. PLoS One 2014;9:e101044.
133. Campbell NK, Fitzgerald HK, Malara A, Hambly R, Sweeney CM, et al. Naturally derived Heme-Oxygenase 1 inducers attenuate inflammatory responses in human dendritic cells and T cells: relevance for psoriasis treatment. Sci Rep 2018;8:10287.
134. Croft KD, Zhang D, Jiang R, Ayer A, Shengule S, et al. Structural requirements of flavonoids to induce heme oxygenase-1 expression. Free Radic Biol Med 2017;113:165-75.
135. George EM, Stout JM, Stec DE, Granger JP. Heme oxygenase induction attenuates TNF-α-induced hypertension in pregnant rodents. Front Pharmacol 2015;6:165.
136. Gerbitz A, Ewing P, Wilke A, Schubert T, Eissner G, et al. Induction of heme oxygenase-1 before conditioning results in improved survival and reduced graft-versus-host disease after experimental allogeneic bone marrow transplantation. Biol Blood Marrow Transplant 2004;10:461-72.
137. Hamamura RS, Ohyashiki JH, Kurashina R, Kobayashi C, Zhang Y, et al. Induction of heme oxygenase-1 by cobalt protoporphyrin enhances the antitumour effect of bortezomib in adult T-cell leukaemia cells. Br J Cancer 2007;97:1099-105.
138. Kirby RJ, Divlianska DB, Whig K, Bryan N, Morfa CJ, et al. Discovery of novel small-molecule inducers of heme oxygenase-1 that protect human iPSC-derived cardiomyocytes from oxidative stress. J Pharmacol Exp Ther 2018;364:87-96.
139. Maamoun H, Zachariah M, McVey JH, Green FR, Agouni A. Heme oxygenase (HO)-1 induction prevents Endoplasmic Reticulum stress-mediated endothelial cell death and impaired angiogenic capacity. Biochem Pharmacol 2017;127:46-59.
140. Mikkelsen RB, Rabender CS, Graves P, Anscher MS. Induction of HO-1 and CO synthesis radiosensitizes tumor epithelial cells with CO acting as a NO mimetic. Int J Radiat Oncol Biol Phys 2014;90:S105.
141. Muchova L, Vanova K, Suk J, Micuda S, Dolezelova E, et al. Protective effect of heme oxygenase induction in ethinylestradiol-induced cholestasis. J Cell Mol Med 2015;19:924-33.
142. Pittala V, Vanella L, Salerno L, Di Giacomo C, Acquaviva R, et al. Novel caffeic acid phenethyl ester (Cape) analogues as inducers of heme oxygenase-1. Curr Pharm Des 2017;23:2657-64.
143. Ramma W, Ahmed A. Therapeutic potential of statins and the induction of heme oxygenase-1 in preeclampsia. J Reprod Immunol 2014;101-2:153-60.
144. Schipper HM. Is glial heme oxygenase-1 suppression in neurodegenerative disorders permissive for neural repair? Neural Regen Res 2015;10:208-10.
145. Simon T, Pogu J, Rémy S, Brau F, Pogu S, et al. Inhibition of effector antigen-specific T cells by intradermal administration of heme oxygenase-1 inducers. J Autoimmun 2017;81:44-55.
146. Skrzypek K, Tertil M, Golda S, Ciesla M, Weglarczyk K, et al. Interplay between heme oxygenase-1 and miR-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis. Antioxid Redox Signal 2013;19:644-60.
147. Strasky Z, Zemankova L, Nemeckova I, Rathouska J, Wong RJ, et al. Spirulina platensis and phycocyanobilin activate atheroprotective heme oxygenase-1: a possible implication for atherogenesis. Food Funct 2013;4:1586-94.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Jayanti S, Moretti R, Tiribelli C, Gazzin S. Bilirubin and inflammation in neurodegenerative and other neurological diseases. Neurosciences 2020;7:92-108. http://dx.doi.org/10.20517/2347-8659.2019.14
AMA Style
Jayanti S, Moretti R, Tiribelli C, Gazzin S. Bilirubin and inflammation in neurodegenerative and other neurological diseases. Neuroimmunology and Neuroinflammation. 2020; 7(2): 92-108. http://dx.doi.org/10.20517/2347-8659.2019.14
Chicago/Turabian Style
Jayanti, Sri, Rita Moretti, Claudio Tiribelli, Silvia Gazzin. 2020. "Bilirubin and inflammation in neurodegenerative and other neurological diseases" Neuroimmunology and Neuroinflammation. 7, no.2: 92-108. http://dx.doi.org/10.20517/2347-8659.2019.14
ACS Style
Jayanti, S.; Moretti R.; Tiribelli C.; Gazzin S. Bilirubin and inflammation in neurodegenerative and other neurological diseases. Neurosciences. 2020, 7, 92-108. http://dx.doi.org/10.20517/2347-8659.2019.14
About This Article
Copyright

Data & Comments
Data


 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 25
likes
Like This Article 25
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.