
Microbiome meets microglia in neuroinflammation and neurological disorders

Abstract
One of the emerging hot topics in biosciences is the intriguing link between gut microbial communities and its influences outside the gastrointestinal tract, such as the central nervous system (CNS), including its cognitive activities and immune responses. Beyond its neuroprotective properties, microglia are also critical for neuronal synaptic pruning and neural remodeling during CNS development. Prolonged microglia activation and neuroinflammation are considered key contributors to neurological disorders. In this regard, it is becoming increasingly important to consider the potential influences underlying the crosstalk between the intestinal microbiota ecosystem and host when determining biomarkers of disease and treatment efficacy. The commensal microbiota is critical for immune development and continuous function through the recognition of bacteria-produced and regulated metabolites. In cases of microbial dysbiosis and microglial dysfunction, chronic neuroinflammation may persist, leading to the propagation of neurological disorders. To address potential mechanisms, this review focuses on the microbiota-gut-brain axis as it relates to communication pathways that have been linked to aberrant CNS immune activity and pathology. We also address anti-inflammatory and neuroprotective mediators which may counteract these detrimental activities. Finally, we explore the potential benefits of current and novel microbiome-targeted approaches to treat neuroinflammation and consequential neurological disease.
Keywords
Introduction
Microglia are the central nervous system (CNS) resident macrophages responsible for initiating innate immune responses to a variety of different stress and damage signals in the brain[1]. For example, when less mobile “resting” microglia recognize these signals with actively surveying processes, they become highly motile and assume an activated phenotype, facilitating the release of pro-inflammatory and cell-recruiting cytokines (e.g., IL-6, IL-12, IL-1β, and TNF-α) at the damage site[2]. After isolating and resolving the problem, it becomes critical for microglia to reestablish homeostatic conditions through the release of anti-inflammatory cytokines (e.g., IL-4, IL-10, and TGF-β) and return to the sentinel deactivated state[3]. On the other hand, prolonged microglial activation has been linked to harmful inflammatory states leading to dysfunctional brain activity and irreversible tissue damage, such as within Alzheimer’s and Parkinson’s diseases[4,5]. Beyond their immune functions, microglia are also important regulators of synaptic pruning and neural patterning during development and throughout adulthood[6]. Despite its evident importance in health and disease, it remains unclear whether signals to modulate microglia activity originate within the CNS only or may also occur externally from other organ/tissue systems. Identification of these additional factors related to microglial function will warrant a better understanding of causes of neuroinflammation and its relationship to CNS disorders.
Recently, one such factor has made a surprisingly strong debut within the scientific community: the vast microflora inhabiting the gastrointestinal (GI) tract has emerged as a critical player connected to multiple host systems, including those outside the GI tract. The gut microbiome (GMB) is able to modulate mucosal immunity and systemic immune activity as well as immune responses within the CNS. For example, the GMB has been demonstrated to affect the development and ongoing activity of microglia[7]. Aberrant changes to the microbiota (“dysbiosis”) and dysregulated microglial activity have both been linked to some of the same neurodevelopmental, neurobehavioral and neurodegenerative disorders, including autism spectrum disorder, anxiety, depression, and Alzheimer’s and Parkinson’s diseases[8-12]. Of note, the GMB and its relationship to immune system maturation and developmental disorders have been extensively described in multiple reviews and are not discussed in detail within this article[13-15]. This review instead focuses on the multidirectional pathways and microbial metabolites within the microbiota-gut-brain axis as they specifically relate to neuroinflammation-induced neuropathologies. Finally, we discuss recent and novel microbiome-targeted strategies as potential treatment options for neurological disorders.
Microbiome, microglia and neurological disorders
A mother’s womb is aseptic, and, as such, we begin to develop our microbiome environments within the first few days after being born, and several factors, including method of birth, institution of breastfeeding or formula diet, and exposure to different environmental elements, determine initial microbiota compositions and continual adaptations[16]. The presence of early microbial colonization is essential for the maturation and healthy function of numerous CNS systems. The innate immune system require microbiota-induced epithelial signaling in order to correctly respond to pathogenic exposure[17]. For example, Erny et al.[7] demonstrated that adult germ-free (GF) mice were found to have major deficits in neuroimmune response compared to conventional controls, such as expressing reduced repertoire of cytokine and chemokine related genetic changes and inactivated microglial morphology (e.g., “failed to display rounded perikarya and small processes”) in response to lipopolysaccharide (LPS) exposure. Furthermore, they were able to demonstrate that reestablishing microbiota diversity and supplementation with microbial-derived short-chain fatty acids (SCFAs), rather than total bacterial abundance load was important for partial recovery of microglial function[7]. In another example, Sudo et al.[18] showed that sterile-bred GF mice, devoid of microbiota, express hyperresponsive irregular hypothalamic-pituitary-adrenal (HPA) activity in response to stress compared to conventionally bred laboratory mice. The HPA axis is considered one of the main relay stations between the GMB and host CNS immune responses, which has been linked to the development of neuropsychiatric disorders later in life[19]. Another study by Thion et al.[20] found that mice born from GF maternal mice expressed changes in genes regulating LPS recognition and processing in utero and continued to exhibit sex-specific alterations in microglial-related gene expressions postnatally. It is important to note that the timeframe when the human GMB begins to stabilize and resemble an adult composition, i.e., around three years of age, also overlaps with critical periods of CNS development, synaptic pruning, and neural remodeling[21-23]. These observations support the assertion that a complex commensal microbiota ecosystem and their metabolites are integral to the early programming of key host physiological systems.
Continuous microbiota-gut-brain axis communication may also play an important role during progression of neurological disorders occurring later in life. Clinical analyses have revealed common comorbidities and correlations between neurological symptoms and GI dysfunction, such as anxiety levels corresponding with irritable bowel syndrome symptoms[24] and GI-related symptoms associated with Parkinson’s disease[25]. Furthermore, “sickness behavior” is a phenomenon descriptive of subjective changes in mood and behavior commonly found in humans and animal models of infection and illness[26]. These relationships highly suggest that GI activity, immunity and microbiome are linked to the CNS function and psychiatric disorders.
There are various methods to modulate the microbiome in order to study its direct impact on health and diseases, ranging from GF, sterile bred rodent models with overt developmental aberrations to antibiotic treatment and fecal matter transplantation (FMT)[27,28]. FMT studies have begun to reveal potential causal and therapeutic roles for the GMB through its ability to endow phenotypes from donor subjects to recipients, such as transferring anxiety-like behaviors and depressive symptoms within rodent models[29,30]. Furthermore, within the past 6 years, FMT has become a standard of care for patients suffering from recurrent C. difficile infections who were unresponsive to antibiotic treatments but responded favorably to the induction of healthy donor microbiota cultures[31]. Colonic samples from healthy donors were also able to improve GI and behavioral symptoms in a small (n = 18) cohort of children diagnosed with autism spectrum disorders[32]. Although the exact mechanisms directing microbiota-gut-brain axis influences in neurological disorders are still being investigated, communication pathways and components have been identified which are related to the systemic immune system, vagus nerve signaling, and neuroendocrine system[33].
The microbiota-gut-brain axis and immune system
The gut microbiota, intestinal immune tolerance and homeostasis
Due to lifelong cohabitation with the intestinal microbiota, mucosal immune tolerance becomes important in differentiating between commensal and pathogenic bacteria[34]. The GMB is not hidden from immune systems but is instead active in maintaining homeostasis through “tolerogenic” signaling[35]. Toll-like receptors (TLRs) on the membrane of epithelial cells and lymphoid cells are responsible for recognizing different broad microbe-associated patterns, including bacterial membrane components, endotoxins such as LPS, and bacterial DNA[36]. TLR stimulation releases nuclear factor kappa-light-chain enhancer of activated B cells (NF-κβ) and involves activation of signaling chemokines, cytokines, and other effector proteins of humoral immune activity[37]. TLR signaling is decreased during the early weeks of development while the GMB ecosystem is being established, and immune-tolerance of bacteria is achieved when recognition of commensal bacteria-produced antigens inhibits inflammatory activation[38]. Specifically, TLR activation on the apical, microbiota-exposed membrane of epithelial cells, rather than on the basolateral membrane, inhibits the inflammatory cascade and limits immune response to microbial antigens found within the GI lumen[39]. Incorrect or incomplete immune-tolerance development can lead to autoimmune diseases, chronic inflammation, and tissue damage. The importance of the GMB in promoting immune homeostasis opens the possibility of microbiota-targeted therapeutics to reduce inflammation in response to GI diseases, such as colon cancer and colitis[40].
Microbiota-derived LPS and neural-immune interactions
The brain and connected CNS have previously been regarded as “privileged” and immunologically isolated from the rest of the body[41]. Consequently, we assumed the peripheral immune system was in place to assure healthy functioning and security for the rest of the body. Despite this separation, we have begun to identify factors outside the CNS which directly impact neurology and behaviors.
The commensal gut microbiota confers colonization protection from pathogens through nutrient and spatial niche competition, in addition to their ability to interact with the mucosal immune system and influence release of soluble IgA antibodies, antimicrobial peptides, and defensins against invaders[42,43]. However, damage to the mucosal wall, for example due to antibiotic-induced microbiota dysbiosis, overgrowth of opportunistic pathogens, and chronic inflammation, can lead to increased susceptibility to infection and permeability of the intestinal epithelial layer, known as “leaky gut”, allowing luminal contents to escape into circulation and induce systemic inflammation[44,45].
The Gram-negative bacterial membrane component LPS is an endotoxin commonly utilized to study the effects of inflammation on behavior in rodent models, including voluntary ethanol intake, anxiety-like behaviors, and blood-brain barrier integrity[46-49]. Studies have demonstrated the direct effects of LPS on microglial activation and subsequent neurological pathologies and behaviors. For example, systemically introduced LPS has been shown to induce depressive-like behaviors in animals, similar to “sickness behavior” commonly comorbid with human infection diseases[50]. Biesmans et al.[50] showed that intraperitoneal injection of LPS increased serum levels of cytokines, including IL-1β, IL-6, TNF-α, IL-10, and MCP-1, peaking at 2 h after administration. This correlated with upregulation of CNS astrocytic immune activity biomarker glial fibrillary acidic protein, decreased locomotion, and reduced sucrose preference which indicates anhedonia associated with sickness behavior[50]. In another example, Hoogland et al.[51] demonstrated increased microglial activation after 48 h of LPS administration and 72 h after live E.coli injection. Furthermore, they found increased inflammatory cytokines within brain homogenates (TNF-α, IL-1β, MCP-1, and M-CSF) at 3 h after LPS stimulation compared to 20 h after E. coli infection. This indicates that E. coli-associated LPS endotoxins induced neuroinflammation before circulatory introduction of LPS-producing bacteria, suggesting the importance of bacterial substrates in triggering immune responses. Furthermore, blocking TLR4-LPS recognition in a rat model prevented sickness behavior following LPS challenge[52]. While unclear of its origin, whether having migrated from the gut microbiome or being derived from a brain microbiome, bacterial LPS has also been identified within the neuronal parenchyma of Alzheimer’s patients[53]. Zhao et al.[54] observed that LPS tended to self-associate and congregate around neurons, indicated by neuronal marker NeuN- and DAPI (nuclear)-staining, within brain tissues from patients with Alzheimer’s Disease compared to age-matched controls that instead expressed more punctate and dispersed LPS. Furthermore, they were able to show that primary co-cultures of human neuronal-glial cells significantly reduced DNA transcription factors when incubated with LPS[54]. Collectively, these observations suggest the critical role of the GMB and microbial endotoxins in influencing systemic and CNS inflammation related to neurological disorders.
The gut and vagus nerve system interactions
Vagal afferents and chemosensing through G-protein coupled receptors
The vagus nerve allows for bidirectional communication between the gut and brain, where afferent signaling conveys sensory information from the gut to a mesh-like system of neurons in the brain. Microglia are sensitive to intestinal microbiome changes and are effective at receiving signals from the vagus nerve to regulate neuroimmune activity and function. Gut endocrine cells (EECs) play important roles in mediating intestinal information to the CNS. They serve as chemosensors that integrate extrinsic and intrinsic signals within the gut. EECs interact with the vagus nerve by responding to different stimuli, such as nutrients, harmful toxins, and bacterial products. Through this cell-mediated sensing mechanism, EECs interact with vagus afferents by releasing serotonin, gut hormones (CCK, PYY, ghrelin, leptin, and GLP-1) and SCFAs that target receptors located on vagus fibers. Long chain fatty acids (LCFAs) also interact with vagus receptors through cholecystokinin-dependent mechanisms.
Direct measurements of EEC activities have been challenging due to their location in the gut wall. Recently, the lab of Reimann et al.[55] has developed a method to directly investigate EEC activity by genetically tagging EECs with a fluorescent protein which expresses under the control of the promoter for a peptide hormone precursor proglucagon, GLP-1. Using this approach, they established the important role of G-protein coupled receptors (GPCRs) in chemosensing and their ability to activate EECs leading to the secretion of peptide hormones. GPCRs are critical for a variety of physiological functions, such as regulation of immune system, autonomic nervous system regulation, sensory (taste and smell) functions, and maintaining energy homeostasis. Recently, some GPCR chemoreceptors were found to be activated by bile acids, SCFAs, and LCFAs, which are also linked to EECs[56]. It was shown that the LCFA receptors GPR40 and GPR120 and the bile acid receptor GPR131 (TGR5) are all expressed on the surface of EECs[55]. In addition, Samuel et al.[57] found that isolated EECs express GPR41, a receptor for SCFAs. It has been shown that these chemosensors are located on the basolateral membrane of of EECs, which interact with the circulatory system[58]. In addition, it is likely that GPCRs co-store and co-release with gut-derived hormones, which indicate that GPCRs may be regulated by associated intestinal hormones. SCFAs and LCFAs, released from gut microbiota or derived nutritionally, can activate release of CKK hormone, which can bind to CCK-A and CCK-B receptors (CCK-r) on vagal afferents that signal the brain[59]. In response, the brain develops immune responses and triggers vagal efferent fibers to release acetylcholine (ACh), which is the principal parasympathetic neurotransmitter[60]. These observations suggest that gut dysbiosis can result in pathological changes in the levels of gut hormones and metabolites, thus influencing GPCR function and dysregulating the vagus nerve and subsequent CNS activities.
The interactions between gamma-aminobutyric acid and vagus nerve
Gut microbiota also produce a number of neurotransmitters similar to mammalian physiological systems, including dopamine, norepinephrine, serotonin, and gamma-aminobutyric acid (GABA). GABA is the major inhibitory neurotransmitter in the CNS; however, GABA receptors are expressed throughout the body, including on the vagus nerve[61]. In human intestines, GABA is produced by the microbiota populations Lactobacillus brevis and Bifidobacterium dentium. GF animals were shown to have reduced levels of GABA, suggesting that the gut microbiota is able to influence GABA levels. Furthermore, altered GABA levels have also been associated with neurological conditions, such as depression, anxiety, autism, and schizophrenia[62,63]. For example, studies into rodents were found to have reduced depressive and anxiety-like behaviors after receiving chronic administration of the probiotic Lactobacillus rhamnosus, which was accompanied by decreases in GABA receptor subunit mRNA expression and corticosterone levels[64]. The GABA-related reductions in behavioral effects did not occur in vagotomized rats and mice[65]. Considering this effect existed only when the vagus nerve was intact, it suggests that intestinal microorganisms regulate GABA signaling through the vagus nerve. In support of this conclusion, animal studies by Takanaga et al.[66] demonstrated that GABA produced by intestinal bacteria are able to cross the blood-brain barrier and influence CNS activities. In addition, the impairment of GABA-mediated neuronal inhibition associated with epilepsy might contribute to the therapeutic efficacy of vagus nerve stimulation, as was demonstrated in patients with drug-resistant partial epilepsy[67].
Vagus nerve pathways in controlling inflammation
The microbiota-gut-brain interaction through the vagus nerve plays a major role in regulating inflammation. The anti-inflammatory properties of vagus nerve function is mediated through several debated pathways, such as the cholinergic anti-inflammatory pathway, the HPA axis and the splenic-sympathetic nerve anti-inflammatory pathway.
Previous studies demonstrated that the cholinergic anti-inflammatory pathway (CAP) plays a pivotal role in controlling neuroinflammation. The CAP modulates inflammation through vagal efferent fibers that synapse onto enteric neurons surrounding the GI tract, which can release acetylcholine[68,69]. Acetylcholine binds to α-7-nicotinic acetylcholine receptors on macrophages, including microglia, and inhibit the release of the pro-inflammatory cytokine TNF-α[70]. Other studies also illustrate the ability of the vagal nerve to regulate neuroinflammation by sensing increased peripheral pro-inflammatory cytokines[71]. As a negative feedback loop, pro-inflammatory cytokine release is prevented if increased levels of inflammation are detected through the acetylcholine-mediated anti-inflammatory signaling system[71]. Wang et al.[72] observed that electrical stimulation of the vagus nerve can inhibit TNF synthesis in wild-type mice but not in α-7-nicotinic acetylcholine receptor-deficient mice. Collectively, these results support the critical role of the vagus nerve in regulating microglia activity and neuroinflammation through CAP signaling.
Studies of vagus nerve stimulation have provided additional evidence for vagus nerve afferent involvement in neuroimmune modulation. For example, non-invasive vagus nerve stimulation is widely used in the treatment of drug resistant depression and has been shown to increase levels of norepinephrine[73,74]. The locus coeruleus is an aminergic brain stem nucleus which represents the main source of norepinephrine in the brain and plays a critical role as a neurotransmitter and neuroimmune modulator, including regulation of microglial activity. By activating β-receptors on the cell surface, norepinephrine affects microglia cell dynamics, which then influence neuronal activity[75,76]. These observations indicate the potential of vagus nerve stimulation to regulate microglial activity.
Recently, the inflammatory reflex was found to be located where vagus afferent fibers activate vagus efferent fibers. Borovikova et al.[77] reported that septic shock was prevented by vagus nerve stimulation of the distal end of the vagus nerve after injection of LPS. This effect is due to CAP activation and the binding of acetylcholine to α-7-nicotinic acetylcholine receptors in order to inhibit macrophages from releasing pro-inflammatory cytokines such as TNF-α[72]. However, the interaction between the vagus nerve and intestinal immune system is indirect because the vagus nerve does not directly interact with resident macrophages in the gut. Therefore, Rosas-Ballina et al.[78] suggested that the vagus nerve tends to activate the splenic sympathetic nerve through a vago-sympathetic co-activation of ventricular contractility and heart rate[78,79]. It is hypothesized that released norepinephrine from the distal end of the spleen can bind to the β2 adrenergic receptor of splenic lymphocytes. Its binding leads to the release of acetylcholine, which in turn binds to α-7-nicotinic acetylcholine receptors on splenic macrophages and inhibits the release of TNF-α[80]. However, this hypothesis is still being debated due to the controversial interaction between the spleen and the vagus nerve[81]. Furthermore, some studies demonstrate the spleen receives not only sympathetic inputs but parasympathetic inputs as well. The sympathetic inputs relay information to the spleen via the arteries while the parasympathetic inputs transfer signals at the tips of the spleen.
The vagus nerve also plays an important role within the neuroendocrine-immune axis, which can regulate coordinated neural, behavioral, and endocrine responses with the immune system in order to prevent chronic neuroinflammation. The vagus nerve recognizes peripheral pro-inflammatory cytokines released by macrophages, such as IL-1, IL-6, and TNF-α and conveys this information to the neurons within HPA pathway in order to decrease peripheral inflammation[82]. Overall, the vagus nerve has anti-inflammatory properties both through its afferent end (activation of HPA axis) and through its efferent end (activation of CAP).
The gut and the endocrine system
In addition to the vagus nerve, intestinal microbiota are able to communicate with the CNS through hormones secreted by glands within the endocrine system. Steroid hormones take part in many critical physiological processes in our body, such as survival of stress, injury, metabolism, inflammation, salt and water balance, immune functions, and development of sexual characteristics. Studies show that gut microbiota are also able produce and regulate these hormones to affect brain activity, including the state of microglia and neuroinflammation.
Glucocorticoids through HPA axis
The HPA axis is a complex set of direct pathways and feedback interactions which include the hypothalamus, the pituitary gland, and the adrenal glands. The hypothalamus produces and releases corticotropin-releasing hormone (CRH), which can induce the pituitary to release adrenocorticotropic hormone (ACTH). ACTH then stimulates the adrenal cortex, producing glucocorticoid hormones. Each of these hormones can in turn act back on the hypothalamus and pituitary in a negative feedback cycle. Glucocorticoids are corticosteroids which bind to glucocorticoid receptors present in almost every vertebrate animal cell. They can reduce certain aspects of immune activities through a feedback mechanism. Cortisol is the most important human glucocorticoid which has a variety of cardiovascular, metabolic, immunologic, and homeostatic functions. The influence of microbiota on the HPA axis depends on many factors including bacterial strain, host age and sex, and different mouse strains[83-89]. Individual strains of bacteria can regulate the HPA axis and the microbiota as a whole participate in developmentally programming stress responses[90]. Conversely, microglial activity can also affect hormone release through HPA axis. In response to cerebral insults, microglia secrete a variety of inflammatory molecules, such as cytokines, stimulating neuronal activity within the paraventricular nucleus of the hypothalamus to activate the HPA axis anti-inflammatory feedback loop to reduce prolonged neuroinflammation.
Glucocorticoids released by the HPA axis bind to glucocorticoid receptors, which are highly expressed in neurons and microglia to affect cellular responses[85,90]. Glucocorticoids work to suppress both stress and immune responses by binding to specific glucocorticoid receptors and mineralocorticoid receptors in CNS and immune cells[91]. Studies have demonstrated that acute stress induced higher levels of ACTH and corticosterone in the serum of GF mice compared to conventionally-raised control mice[85-87]. Recent targeted microarray analysis found 23 upregulated glucocorticoid receptor pathway genes in the hippocampus of GF mice compared to controls, of which six genes (Slc22a5, Aqp1, Stat5a, Ampd3, Plekhf1, and Cyb561) were confirmed by PCR validation[87]. Among these six genes, two (Stat5a and Ampd3) were upregulated in E. coli-derived LPS-treated mice. The GF mice demonstrated reduced anxiety-like behaviors in response to acute stress, whereas LPS-treated control mice demonstrated anti-depressive but not anti-anxiety behavior and a decrease in the basal serum cortisol levels. LPS-induced abnormal behavior was consistent with previous findings that E. coli colonization in GF mice enhanced the HPA axis response to stress[86]. In another study, plasma ACTH and corticosterone hormones were decreased in mice monocolonized with Bifidobacterium infantis, but were increased in E. coli-monocolonized mice. In addition, after receiving fecal samples from patients diagnosed with severe depression (“depression microbiota”), control mice exhibited anxiety- and depressive-like behaviors with parallel downregulation of Stat5a gene in their hippocampus compared with “healthy microbiota” recipient mice[92]. Stat5a is a member of STAT family encoded transcription factors, mediating signals for a broad spectrum of cytokines. The JAK2-STAT5 signaling pathway plays a critical role in mediating IL-3-induced activation of microglia[93]. Furthermore, STAT5 may play a protective role in damaged nerve cells and has been implicated in cellular functions of proliferation, differentiation, and apoptosis with relevance to processes including hematopoiesis and immunoregulation[92]. Collectively, these observations suggest that microbiota related STAT5 levels may influence neuroinflammation and related disorders.
CRH and glucocorticoids from the HPA axis have been shown to directly affect microglia activity by binding to functional CRH-R1 receptors on microglia and initiate apoptosis of microglia[94]. In that study, Ock et al.[94] demonstrated that CRH-induced apoptosis did not induce nitric oxide production or increase expression of pro-inflammatory genes, which indicates that CRH does not affect inflammatory activation of microglia. This mechanism has been linked to the mitochondrial pathway and induction of reactive oxygen species (ROS) production, which can damage microglia cells and promote apoptosis[95]. In support, the antioxidant N-acetyl cysteine inhibited CRH-induced microglial cell death suggesting that ROS was a main cause of apoptosis.
Glucocorticoid levels are strongly related to the activation of the HPA axis, and distinctively affect macrophage function. Low levels of corticosterone enhanced pro-inflammatory factors, while high corticosterone concentrations suppressed macrophage activation[96]. Steroid hormones directly target mature microglia; glucocorticoids predominantly modulate expression of glucocorticoid receptors to regulate microglial inflammatory activity[97]. Anti-inflammatory effect of glucocorticoids on microglia can reverse the pro-inflammatory function of CRH by attenuating the production of TNF-α, IL-6, and nitric oxide from LPS + IFN-γ-activated murine microglia. Physical or emotional stress may induce microglial activation in the brain as determined by changes in morphology[98,99]. The stress-induced elevation of glucocorticoids can activate microglia in rats, and chronic stress can cause a marked transition from a resting to non-resting state[100]. Temporal treatment of glucocorticoids can exhibit the opposite results[101]. Stress and administration of glucocorticoids prior to peripheral immune stimuli exerted pro-inflammatory effects on microglia, while exposure to glucocorticoids after stimuli had anti-inflammatory effects in a rodent model[101]. Corticosteroids limit microglial activation that occurs during acute stress, serving as an important endogenous suppressive signal limiting neuroinflammation[98,99]. Moreover, glucocorticoid level increases and microglial morphological complexity decreases with aging[88]. Increasing glucocorticoid levels in young mice enhanced microglial ramifications, pointing to their increased neuroprotective function. The opposite, amoeboid state of microglia renders them to move freely in the brain tissue and is indicative of inflammatory activation. Amoeboid microglia occur more frequently with aging. The effects of glucocorticoids or corticosteroids on microglia morphology are dependent on treatment time and concentration of glucocorticoids.
Estrogen through hypothalamic-pituitary-gonadal axis
The hypothalamic-pituitary-gonadal axis (HPG axis) plays an important role in the reproductive and immune systems, and controls development, reproduction, and aging in animal models. The hypothalamus secretes gonadotropin-releasing hormone, the pituitary gland produces luteinizing hormone and follicle-stimulating hormone, and the gonads release estrogen and testosterone. Although the HPG axis has not been as deeply studied as the HPA axis, strong evidence suggests that estrogen has the capacity to inhibit neuroinflammatory processes and can impact immune cells, including microglial functions.
Estradiol (E2) is an estrogen steroid hormone and the major female sex hormone. Studies show that 17β-estradiol (E2) inhibits microglia activation[102] and reduces the expression of inflammatory mediators[102]. For example, E2 was able to inhibit Aβ-induced expression of scavenger receptor-A in microglia cells from an animal model of Alzheimer’s disease[102]. Ovarian hormone deprivation can alter the expression of major components of estrogen and neuronal inhibitory signaling, participating in the control of microglia reactivity[103]. Moreover, aging is related to exaggerated responses to acute inflammatory stimuli, modulated by the duration of hormone deprivation. This deprivation is due to decreased estrogen receptor activity, which, despite the continuous synthesis of the receptors, induces neuroinflammation[89].
Short-chain fatty acids in the gut and neuroinflammation
Microbiota are able to influence brain functions through the production of metabolites such as SCFAs. In addition to being derived from dietary sources, SCFAs are also produced by the microflora in the distal small intestine and colon though the fermentation of dietary fibers. The most abundant SCFAs in the human gut are acetate, propionate, and butyrate. Acetate is used for host synthesis of lipids and cholesterol, and propionate is mostly absorbed by the liver and serves as a substrate during gluconeogenesis. Butyrate functions as the main energy source for colonic enterocytes[104]. SCFAs are mainly absorbed in both the small and large intestine through similar mechanisms, such as diffusion of the dissociated forms and through active transport by SCFA transporters[105].
High doses of systemic or locally injected butyrate has been found to exert neuroprotective effects, such as memory enhancement and cognitive function restoration[106,107]. Physiological levels of butyrate may influence and improve neuroinflammation through different mechanisms. Butyrate is a known inhibitor of histone deacetylases (HDACs)[108,109], which control the innate inflammatory system by regulating the number of microglia cells and astrocytes[110]. Histone acetylation is a post-translational modification through epigenetic process and causes the chromatin structure to loosen by weakening electrostatic attraction between the histone proteins and DNA backbone. Activation of microglia are suppressed by this process. Therefore, increased HDACs have been shown to be involved in neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases[110,111].
Butyrate is one of the most important microbial end-products of the human colon fermentation process which displays several physiological effects via different mechanisms. One function is mentioned above: butyrate is a well-established HDAC inhibitor. In addition to having a significant impact on the transcriptional system, butyrate also serves as the energy substrate. Butyrate is the primary source of energy in the colon and microbiome, which accounts for nearly 70% of ATP produced. It may appear that metabolic events in the colon are disconnected with that of the brain. However, it is impossible to ignore the immense energy demand of the brain. In this regard, energy imbalance in the brain has been noted at early stages of neurodegenerative disease such as Alzheimer’s disease[112]. Another function of butyrate is its ability to activate GPCRs, as described in detail above, within the vagus nerve system section[113]. Butyrate can signal through GPR109a, which is widely expressed in colonocytes, T cells and has also been found in microglia. Butyrate is sensed by FFA2 (previously GPR43) and FFA3 (previously GPR41), which modulate the relationship between SCFAs and gut, as well as the whole body energy use[114,115].
Many studies have shown that butyrate can serve as an anti-inflammatory agent, improving gut barrier function, protecting against colon cancer and neurodegenerative diseases, such as Alzheimer’s disease[116-118]. These studies demonstrate that treatment with butyrate inhibited pro-inflammatory cytokines (IFN-γ, TNF-α, IL-1β, IL-6, and IL-8) and upregulated anti-inflammatory cytokines (IL-10 and TGF-β). This effect may be partly due to the inhibition of transcription factor NF-κB that controls the transcription of DNA, cytokine production, and cell survival. Aguilar et al.[119] demonstrated that butyrate suppressed the NF-κB signaling pathway by rescuing the redox machinery and controlling ROS, which also regulate NF-κB activation. In addition, butyrate is known to enhance and repair barrier function of intestinal epithelial cells. In vitro experiments have illustrated that butyrate plays an important role in the maintenance of gut- barrier integrity in order to block the translocation of LPS, which can cause immune activation[120]. For instance, butyrate leads to the upregulation of mucin 2, the most prominent mucin protein, and enhances the protection of the mucosal layer[121]. These effects of butyrate were demonstrated in Caco-2 cell cultures, which are human epithelial colorectal adenocarcinoma cells, and can form confluent monolayers in vitro that both structurally and functionally resemble the small intestinal epithelium. For instance, butyrate leads to the upregulation of mucin 2, the most prominent mucin protein, and enhances the protection of the mucosal layer[121].
Gut dysbiosis and reduced levels of SCFAs have been observed within neurological disease, including Pelizaeus–Merzbacher disease[122]. Unger et al.[123] found changes in gut microbiota and SCFAs in patients diagnosed with Parkinson’s disease. Fecal SCFA concentrations were significantly reduced in Parkinson’s patients compared to controls. This was associated with reduced microbiota populations of Bacteroidetes and Prevotellaceae[123]. Furthermore, some studies have demonstrated beneficial effects of SCFAs during neuronal pathologies, such as against formation of neurotoxic Aβ aggregation, which occurs during the pathogenesis of Alzheimer’s disease[124]. SCFAs have been reported to increase the expression level of retinotic acid in the GI tract, which inhibits Th17 cell differentiation and promotes Treg proliferation, limiting prolonged neuroinflammation[125]. SCFAs, especially butyrate, are able to modulate immune cells and influence cell proliferation and apoptosis. For example, high concentration of butyrate induces cell apoptosis while low concentration will enhance cell proliferation[126]. Collectively, these observations support the ability of SCFAs to have a therapeutic effect on many neurodegenerative disorders.
Microbiome-targeted therapeutics addressing neurological diseases
The conclusion of the 10-year NIH Human Microbiome Project has been integral in providing resources, methods, and discoveries linking humans and their microbiomes to health and disease[127]. The study utilized a combination of shotgun metagenomics, untargeted metabolomics, and immunoprofiling to determine host-microbiota interactions manifest in largely diverse ways, and sampling large population sizes is critical for accurately determining potential mechanisms of microbiome-linked diseases[127]. They demonstrated that microbiome composition alone was not always an accurate representation of host phenotype, and necessitated the consideration of microbial functions of the microbiota ecosystem as they interacted with host immunity, metabolism, and other interconnected activities[128]. Through this accomplishment, microbiome-targeted strategies have begun to gain interest in both studying mechanistic relationships within animal models and in the treatment of pathologies, including those related to the gut-brain axis.
Antibiotics: non-absorbable “eubiotic” rifaximin
Beyond their bacteriostatic and bactericidal effects in treating GI infections, antibiotics have been shown to negatively affect the intestinal flora, a phenomenon considered “collateral damage”. Antibiotic treatment can have long-lasting negative effects on the GMB, which has been shown to decrease diversity and reduce beneficial bacteria, leading to increased susceptibility to pathogens, such as Salmonella and Clostridium difficile[129,130]. Alternatively, rifaximin, a broad-spectrum, non-absorbable antibiotic, prescribed to treat irritable bowel syndrome and traveler’s diarrhea caused by E. coli, has shown unique qualities related to the GMB and symptoms beyond the GI tract[131]. The mechanism of rifaximin action to reduce pathogens is through binding the β-subunit of microbial RNA polymerase and inhibition of bacterial RNA synthesis[132]. However, unlike other antibiotics which commonly reduce microbiota diversity and promote dysbiosis, rifaximin exerts anti-inflammatory properties and has the “eubiotic” ability to enrich beneficial microbiota populations[133]. For example, Maccaferri et al.[134] found that in vitro treatment with rifaximin increased levels of Bifidobacteria, Atopobium, and Faecalibacterium prausnitzii cultured from colonic samples of patients with Crohn’s disease. These changes were also accompanied by increases in SCFAs, microbial metabolites known to be important in host health, metabolism, and immune homeostasis[135]. In a rodent model of ankylosing spondylitis spinal joint inflammation, rifaximin treatment was able to inhibit TLR-4/NF-κβ signaling and decrease levels of pro-inflammatory cytokines, such as TNF-α, IL-6, IL-17A, and IL-21[136]. Another important commensal GMB population and producer of the SCFA lactate, Lactobacillus, was increased in a rat model of visceral hyperalgesia with rifaximin treatment[137]. Furthermore, hepatic encephalopathy is a common complication of patients with acute or chronic liver disease that is detected through neuropsychological testing and presents as neurocognitive decline: forgetfulness, confusion, irritability, and coma at its most severe forms[138]. These symptoms are mainly a result of elevated levels of ammonia. Rifaximin was able to reduce levels of ammonia-producing intestinal bacteria without decreasing GMB diversity, while also significantly reducing hospital stay, mortality rate, and improving psychometric test performance in patients with mild and severe hepatic encephalopathy compared to other treatments[139]. These observations support alternate uses for rifaximin which may be related to beneficial changes in microbiota and SCFAs, including its indication for CNS-related disorders.
Microbial-derived metabolites: sodium butyrate
Beyond directly targeting and supplementing live bacteria in the GMB, the Human Microbiome Project stressed the importance of microbiota functions in influencing host immunity and pathologies. As a HDAC inhibitor, sodium butyrate can change the balance between two types of enzymes, histone acetylase and HDACs[140]. These two enzymes control acetylation, which is an important process in chromatin structure and gene expression associated with many diseases, such as diabetes, Alzheimer’s disease, and various cancers[141-143]. Physiological doses of sodium butyrate (0.25-4.00 mM) were observed to inhibit glioblastoma cell proliferation and induce cancer cell senescence in vitro[143]. Pharmacological treatment of sodium butyrate was also shown to significantly increase survival rate and delay the neuropathological sequelae in the R6/2 transgenic mouse model of Huntington’s disease[106]. The findings of Arnoldussen et al.[144] demonstrated the beneficial effect of dietary butyrate intervention on the detrimental effects of high fat diet, including relieving high fat diet-induced cognitive impairment and dementia in humans. In addition to serving as a therapeutic agent in some specific diseases, sodium butyrate can have complementary effects when administered with other agents, such as metformin. Metformin is the most prescribed oral anti-diabetic agent, whose potential benefit in many diseases has been investigated. Recent research demonstrates that metformin is able to increase butyrate-producing populations within the gut microbiome[145,146]. Additional data indicate metformin and butyrate have anti-inflammatory effects in relation to physiological functions, including transcription, replication, and repair in the process of tumorigenesis[147]. Other SCFAs also have therapeutic effects. For example, glatiramer acetate serves as immunomodulator to reverse detrimental immune reactivity in two murine models of irritable bowel disorder. Collectively, these findings point to the therapeutic potential of sodium butyrate and other SCFAs in the treatment of various pathologies including neurological disorders.
Targeting the vagus nerve
Lewy body aggregates, constituted mainly by α-synuclein and ubiquitin, and GI dysfunctions are physiopathological characteristics of early development of Parkinson’s disease[148]. Braak et al.[149] hypothesized that these early biomarkers initiate within the gut and then progress to the CNS via the vagus nerve and spinal cord. In support, vagus nerve-mediated brain migration of α-synuclein injected into the intestinal wall has been found in a rodent model[150]. Sander and his colleagues further indicated the correlation between the vagus nerve and cognitive fatigue in multiple sclerosis patients[151]. It is thought to be the result of the vagus nerve stimulation due to the pro-inflammatory cytokines causing changes in neural activity in brainstem and hypothalamus[152]. Furthermore, the stimulation of the vagus nerve is used in the treatment of drug resistant depression, which is the major factor for developing Alzheimer’s disease. Experiments in APP/PS1 (a murine model of Alzheimer’s disease) animals were performed to induce morphological changes in microglia towards a neuroprotective phenotype, which was mediated by vagus nerve activation[153]. Therefore, due to its important role in regulating the gut-brain axis through transferring microbial metabolites and neurotransmitters, such as SCFAs and GABA, manipulation of vagus nerve signaling may play a key role in modulation of some neurological conditions, including Parkinson’s disease, Alzheimer’s disease, and multiple sclerosis.
Life-style interventions
Lifestyle interventions can affect gut microbiome composition, which influence brain activity and immune responses. Since neuroinflammation is strongly linked to neurodegenerative diseases, lifestyle alterations, such as dietary supplement and exercise, are able to play an important role in improving disease states.
Pre-/probiotic supplementation
Probiotics are living beneficial microorganisms (bacteria and yeasts), and prebiotics are the indigestible fibers which feed them[154]. Probiotics have been widely marketed and consumed as dietary supplements or as functional foods, such as “live” yogurts[154]. Probiotic treatments with Lactobacillus acidophilus, L. casei, and L. rhamnosus were able to affect transcription of host genes related to mucosal immunity in healthy human volunteers, supporting the ability of live bacterial cultures to affect host activities[155]. D’Mello et al.[26] demonstrated a probiotic mixture, VSL#3, was able to reduce “sickness behavior” by increasing novel social investigation in a liver inflammation rodent model, which was related to an increase in circulating G-CSF, reduction in TNF-α, and a decrease in activated microglia. In an in vitro study, peripheral blood mononuclear cells isolated from patients with Parkinson’s disease were co-cultured with probiotic bacteria, Lactobacillus and Bifidobacterium, to investigate changes in innate immune cell release of inflammatory signaling markers. Probiotic strains were able to significantly reduce pro-inflammatory (TNF-α, IL-6, and IL-17A) and increase anti-inflammatory cytokines (IL-4 and IL-10)[156]. In another study of a randomized, double-blind trial, patients diagnosed with Alzheimer’s disease given a probiotic mixture for 12 weeks, exhibited a significant score improvement on the mini-mental state examination compared to controls[157].
High fiber prebiotic with or without probiotic supplementation can be a non-invasive strategy to treat neurological conditions. High fiber diets can affect gut microbiota abundance. For example, inulin is a prebiotic fiber and inulin-type fructan supplementation on the fecal microbiota is able to selectively change abundance of specific colon bacteria strains, such as Anaerostipes, Bilophila, and Bifidobacterium[158]. As such, high fiber supplementation has been shown to counter age-related microbiota dysbiosis[159]. Feeding mice with inulin has been shown to beneficially alter gut microbiome resulting in improved neurological outcomes through affecting gut microbiota-produced SCFAs. In support, high fiber diets, in which SCFAs can be derived, have numerous reported health benefits in reducing risk of type 2 diabetes, obesity, stroke, and cardiovascular disease. High fiber diets have been shown to increase circulating levels of butyrate, which may affect CNS function directly[160]. Collectively, these studies provide exciting evidence and demonstrate the need for further investigations into the ability of live bacteria with or without prebiotic supplementation to treat inflammation and neurological pathologies.
Diet
The microbiota composition and diversity are sensitive to host dietary habits[23,161]. Dietary factors may have pro-inflammatory or anti-inflammatory effects[162], which can indirectly affect gut microbiota by providing multiple nutrients and specific compounds. For example, data suggest that the modified Mediterranean-ketogenic diet can modulate the gut microbiome and metabolites in association with improved Alzheimer’s disease biomarkers in cerebrospinal fluid[163]. The abundance of Enterobacteriaceae, Akkermansia, Slackia, Christensenellaceae, and Erysipelotriaceae increases while that of Bifidobacterium and Lachnobacterium reduces after modified Mediterranean-ketogenic diet treatment in subjects. A bad dietary habit, such as chronic alcohol intake, can induce neuroinflammation and neurodegeneration. Reduction of intestinal bacterial load was able to attenuate alcohol-associated CNS and gut inflammation[164]. Alcohol activated microglia and modified its cell morphology, taking on an amoeboid shape with enlarged soma and shortened peripheral processes[164].
Exercise
Exercise is considered as a protective treatment for neurodegenerative diseases[165]. Both voluntary and controlled exercise can alter the gut microbiota[166]. The microbiota composition of exercised rats was notably different from the sedentary rats with a significantly higher butyrate concentration[167]. Voluntary running has neuroprotective effects in an α-synuclein rat model of Parkinson’s disease[168]. It can protect rats against neuronal loss, increase enteric glial expression, and modify gut microbiome composition in the Parkinson’s disease model[169]. Exercise is also considered to enhance immune system. The vagus nerve regulates gastrointestinal inflammatory tone. Parasympathetic neuroimmune reflex depends on vagal afferent neurons for the local release of intestinal inflammatory mediators in response to pathogenic gut bacteria. For this reason, elevated vagal tone and parasympathetic influence in the resting state of athletes foster a preferential anti-inflammatory milieu through conditionally influencing microbial composition[170].
Conclusion and future directions
Recent discoveries link the GBM and neurological disorders through the microbiota-gut-brain axis. It is also increasingly recognized that disruptions in the GBM ecosystem and its function may directly or indirectly impact CNS disease states, implicating the involvement of microglial-induced neuroinflammation and neurodegeneration. In this respect, there is bidirectional communication between the GBM and the brain, which is achieved through several pathways [Figure 1]. This communication involves the immune system, which not only supports the tolerance towards the microbiome ecosystem residing in the GI tract but also can react to dysbiosis and “leaky” gut, thus relaying this information to the CNS. On the other hand, there is the involvement of the vagus nerve in the microbiota-gut-brain interactions, which have several afferent and efferent pathways involving a variety of factors such as gut endocrine cells, neurotransmitters, and receptors. Importantly, the vagus nerve plays an important function in controlling inflammation through cholinergic and splenic-sympathetic anti-inflammatory pathways and the HPA axis. The role of hormones in the microbiota-brain bidirectional communication is also deemed important through regulation of the HPA and HPG axes. In addition, microbiota-derived metabolites, such as SCFAs and LCFAs, are integral in maintaining intestinal health and have been shown to also impact neurological health.
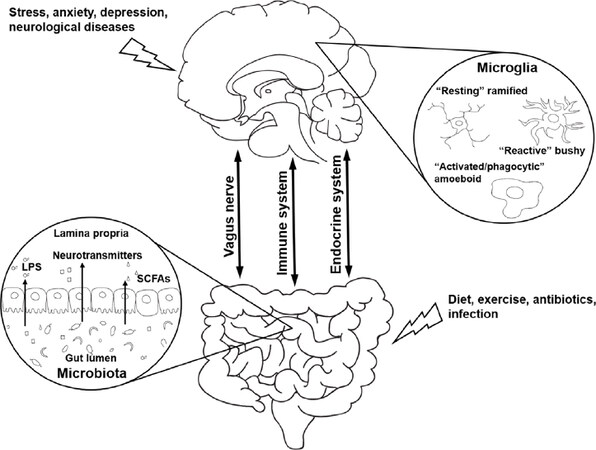
Figure 1. A schematic summarizing the pathways of gut–brain bidirectional communication with the emphasis on the immune system, the vagus nerve, and the endocrine system. The importance of the gut bacterial metabolites in this communication as well as lifestyle changes that affect microglia functioning in normal physiology and during neurological diseases with neuroinflammatory component are indicated. LPS: lipopolysaccharide; SCFAs: short-chain fatty acids
The GMB’s critical influence on host development, immune homeostasis, and metabolism as well as involvement in the development of the CNS disorders, makes it an ideal candidate for novel preventative therapies and treatments. These strategies include the use of beneficial “eubiotic” antibiotics or other means such as lifestyle interventions (diet and exercise) aimed at reversing microbiota “dysbiosis” by targeting microbiota and their metabolites. Although in its infancy, studies into the efficacy of the microbiome-targeted manipulation and FMT to treat diseases, including those beyond the GI tract, promise interesting insights into the importance and impact of the vast and diverse microcosm residing within us every day of our lives from birth to old age and death.
Certainly, we are still in the beginning of the research trying to reveal the causative links between the GMB and brain function as it relates to neurological disorders. There is a huge untapped potential in this area of microbiome in human health and disease, which will be more appreciated with the improvement of new technologies and methods of GMB research.
Declarations
Authors’ contributionsDesigned the focus and scope of the review and substantially contributed to the bulk of the manuscript: Reyes REN, Asatryan L
Contributed to different sections of the review and all participated in editing and finalizing the manuscript: Reyes REN, Zhang Z, Gao L, Asatryan L
Availability of data and materialsNot applicable.
Financial support and sponsorshipRose Hills Foundation Innovator Grant, USC School of Pharmacy and USC Good Neighbors.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
2. Lively S, Schlichter LC. Microglia responses to pro-inflammatory stimuli (LPS, IFNγ + TNFα) and reprogramming by resolving cytokines (IL-4, IL-10). Front Cell Neurosci 2018;12:215.
3. Franco R, Fernandez-Suarez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol 2015;131:65-86.
4. Krause DL, Muller N. Neuroinflammation, microglia and implications for anti-inflammatory treatment in Alzheimer’s disease. Int J Alzheimers Dis 2010;2010:732806.
5. Hunot S, Hirsch EC. Neuroinflammatory processes in Parkinson’s disease. Ann Neurol 2003;53:S58-60.
7. Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci 2015;18:965-77.
8. Golubeva AV, Joyce SA, Moloney G, Burokas A, Sherwin E, et al. Microbiota-related changes in bile acid & tryptophan metabolism are associated with gastrointestinal dysfunction in a mouse model of autism. EBioMedicine 2017;24:166-78.
9. Foster JA, Rinaman L, Cryan JF. Stress & the gut-brain axis: regulation by the microbiome. Neurobiol Stress 2017;7:124-36.
10. Caspani G, Kennedy S, Foster JA, Swann J. Gut microbial metabolites in depression: understanding the biochemical mechanisms. Microb Cell 2019;6:454-81.
11. Minter MR, Zhang C, Leone V, Ringus DL, Zhang X, et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci Rep 2016;6:30028.
12. Parashar A, Udayabanu M. Gut microbiota: implications in Parkinson’s disease. Parkinsonism Relat Disord 2017;38:1-7.
13. Lebovitz Y, Ringel-Scaia VM, Allen IC, Theus MH. Emerging developments in microbiome and microglia research: implications for neurodevelopmental disorders. Front Immunol 2018;9:1993.
14. Verdu EF, Hayes CL, O’ Mahony SM. Chapter 9 - Importance of the microbiota in early life and influence on future health. The Gut-Brain Axis 2016:159-84.
15. Kelly JR, Minuto C, Cryan JF, Clarke G, Dinan TG. Cross talk: the microbiota and neurodevelopmental disorders. Front Neurosci 2017;11:490.
16. Wampach L, Heintz-Buschart A, Hogan A, Muller EEL, Narayanasamy S, et al. Colonization and succession within the human gut microbiome by archaea, bacteria, and microeukaryotes during the first year of life. Front Microbiol 2017;8:738.
17. Dabbagh K, Dahl ME, Stepick-Biek P, Lewis DB. Toll-like receptor 4 is required for optimal development of Th2 immune responses: role of dendritic cells. J Immunol 2002;168:4524.
18. Sudo N. Chapter 13 - The hypothalamic-pituitary-adrenal axis and gut microbiota: a target for dietary intervention? The Gut-Brain Axis 2016:293-304.
19. Kato TA, Hayakawa K, Monji A, Kanba S. Missing and possible link between neuroendocrine factors, neuropsychiatric disorders, and microglia. Front Integr Neurosci 2013;7:53.
20. Thion MS, Low D, Silvin A, Chen J, Grisel P, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell 2018;172:500-16.e16.
22. Knudsen EI. Sensitive periods in the development of the brain and behavior. J Cogn Neurosci 2004;16:1412-25.
23. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, et al. Human gut microbiome viewed across age and geography. Nature 2012;486:222-7.
24. Blanchard EB, Scharff L, Schwarz SP, Suls JM, Barlow DH. The role of anxiety and depression in the irritable bowel syndrome. Behav Res Ther 1990;28:401-5.
25. Yan F, Chen Y, Li M, Wang Y, Zhang W, et al. Gastrointestinal nervous system alpha-synuclein as a potential biomarker of Parkinson disease. Medicine (Baltimore) 2018;97:e11337.
26. D’Mello C, Ronaghan N, Zaheer R, Dicay M, Le T, et al. Probiotics improve inflammation-associated sickness behavior by altering communication between the peripheral immune system and the brain. J Neurosci 2015;35:10821-30.
27. Luczynski P, McVey Neufeld KA, Oriach CS, Clarke G, Dinan TG, et al. Growing up in a bubble: using germ-free animals to assess the influence of the gut microbiota on brain and behavior. Int J Neuropsychopharmacol 2016;19.
28. Lundberg R, Toft MF, August B, Hansen AK, Hansen CHF. Antibiotic-treated versus germ-free rodents for microbiota transplantation studies. Gut Microbes 2016;7:68-74.
29. Li N, Wang Q, Wang Y, Sun A, Lin Y, et al. Fecal microbiota transplantation from chronic unpredictable mild stress mice donors affects anxiety-like and depression-like behavior in recipient mice via the gut microbiota-inflammation-brain axis. Stress 2019;22:592-602.
30. Kelly JR, Borre Y, O’Brien C, Patterson E, El Aidy S, et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J Psychiatr Res 2016;82:109-18.
31. Hui W, Li T, Liu W, Zhou C, Gao F. Fecal microbiota transplantation for treatment of recurrent C. Difficile infection: an updated randomized controlled trial meta-analysis. PloS One 2019;14:e0210016.
32. Kang DW, Adams JB, Gregory AC, Borody T, Chittick L, et al. Microbiota transfer therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome 2017;5:10.
33. Berthoud HR. Vagal and hormonal gut-brain communication: from satiation to satisfaction. Neurogastroenterol Motil 2008;20:64-72.
34. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature 2016;535:65-74.
36. McClure R, Massari P. TLR-dependent human mucosal epithelial cell responses to microbial pathogens. Front Immunol 2014;5:386.
37. Spiljar M, Merkler D, Trajkovski M. The immune system bridges the gut microbiota with systemic energy homeostasis: focus on TLRs, mucosal barrier, and SCFAs. Front Immunol 2017;8:1353.
38. Rakoff-Nahoum S, Pglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of comensal microflora by toll-like receptors in required for intestinal homeostasis. Cell 2004;118:229-41.
39. Lee J, Gonzales-Navajas JM, Raz E. The “polarizing-tolerizing” mechanism of intestinal epithelium: its relevance to colonic homeostasis. Semin Immunopathol 2008;30:3-9.
40. Lazar V, Ditu LM, Pircalabioru GG, Gheorghe I, Curutiu C, et al. Aspects of gut microbiota and immune system interactions in infectious diseases, immunopathology, and cancer. Front Immunol 2018;9:1830.
41. Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev 2006;213:48-65.
43. Taur Y, Pamer EG. The intestinal microbiota and susceptibility to infection in immunocompromised patients. Curr Opin Infect Dis 2013;26:332-7.
44. Croswell A, Amir E, Teggatz P, Barman M, Salzman NH. Prolonged impact of antibiotics on intestinal microbial ecology and susceptibility to enteric Salmonella infection. Infect Immun 2009;77:2741-53.
45. Baraona E, Julkunen R, Tannenbaum L, Lieber CS. Role of intestinal bacterial overgrowth in ethanol production and metabolism in rats. Gastroenterology 1986;90:103-10.
46. Wieczorek M, Swiergiel AH, Pournajafi-Nazarloo H, Dunn AJ. Physiological and behavioral responses to interleukin-1β and LPS in vagotomized mice. Physiol Behav 2005;85:500-11.
47. Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, et al. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Beh Immun 2011;25:S92-105.
48. Fields CT, Chassaing B, Castillo-Ruiz A, Osan R, Gewirtz AT, et al. Effects of gut-derived endotoxin on anxiety-like and repetitive behaviors in male and female mice. Biol Sex Differ 2018;9:7.
49. Chastre A, Belanger M, Nguyen BN, Butterworth RF. Lipopolysaccharide precipitates hepatic encephalopathy and increases blood-brain barrier permeability in mice with acute liver failure. Liver Int 2014;34:353-61.
50. Biesmans S, Meert TF, Bouwknecht JA, Acton PD, Davoodi N, et al. Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediators Inflamm 2013;2013:271359.
51. Hoogland ICM, Westhoff D, Engelen-Lee JY, Melief J, Valls Serón M, et al. Microglial activation after systemic stimulation with lipopolysaccharide and Escherichia coli. Front Cell Neurosci 2018;12:110.
52. Hines DJ, Choi HB, Hines RM, Phillips AG, MacVicar BA. Prevention of LPS-induced microglia activation, cytokine production and sickness behavior with TLR4 receptor interfering peptides. PloS One 2013;8:e60388.
53. Zhao Y, Cong L, Jaber V, Lukiw WJ. Microbiome-derived lipopolysaccharide enriched in the perinuclear region of Alzheimer’s disease brain. Front Immunol 2017;8:1064.
54. Zhao Y, Cong L, Lukiw WJ. Lipopolysaccharide (LPS) accumulates in neocortical neurons of Alzheimer’s disease (AD) brain and impairs transcription in human neuronal-glial primary co-cultures. Front Aging Neurosci 2017;9:407.
55. Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, et al. Glucose sensing in L cells: a primary cell study. Cell Metab 2008;8:532-9.
56. White KL, Eddy MT, Gao ZG, Han GW, Lian T, et al. Structural connection between activation microswitch and allosteric sodium site in GPCR signaling. Structure 2018;26:259-69.e5.
57. Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A 2008;105:16767-72.
58. Karaki S, Mitsui R, Hayashi H, Kato I, Sugiya H, et al. Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell Tissue Res 2006;324:353-60.
59. Whited KL, Thao D, Lloyd KC, Kopin AS, Raybould HE. Targeted disruption of the murine CCK1 receptor gene reduces intestinal lipid-induced feedback inhibition of gastric function. Am J Physiol Gastrointest Liver Physiol 2006;291:G156-62.
60. Yuan M, Han B, Xia Y, Liu Y, Wang C, et al. Augmentation of peripheral lymphocyte-derived cholinergic activity in patients with acute ischemic stroke. BMC Neurol 2019;19:236.
61. Bravo J, Forsythe P, Chew M, Escaravage E, Savignac H, et al. From the cover: ingestion of lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A 2011;108:16050-5.
62. Panzanelli P, Gunn BG, Schlatter MC, Benke D, Tyagarajan SK, et al. Distinct mechanisms regulate GABAA receptor and gephyrin clustering at perisomatic and axo-axonic synapses on CA1 pyramidal cells. J Physiol 2011;589:4959-80.
63. Glykys J, Mody I. Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABA A receptor alpha5 subunit-deficient mice. J Neurophysiol 2006;95:2796-807.
64. Liu GX, Cai GQ, Cai YQ, Sheng ZJ, Jiang J, et al. Reduced anxiety and depression-like behaviors in mice lacking GABA transporter subtype 1. Neuropsychopharmacology 2007;32:1531-9.
65. Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A 2011;108:16050-5.
66. Takanaga H, Ohtsuki S, Hosoya K, Terasaki T. GAT2/BGT-1 as a system responsible for the transport of gamma-aminobutyric acid at the mouse blood-brain barrier. J Cereb Blood Flow Metab 2001;21:1232-9.
67. Marrosu F, Serra A, Maleci A, Puligheddu M, Biggio G, et al. Correlation between GABA(A) receptor density and vagus nerve stimulation in individuals with drug-resistant partial epilepsy. Epilepsy Res 2003;55:59-70.
68. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384-8.
69. Báez-Pagán CA, Delgado-Vélez M, Lasalde-Dominicci JA. Activation of the macrophage α7 nicotinic acetylcholine receptor and control of inflammation. J Neuroimmune Pharmacol 2015;10:468-76.
70. Frasch MG, Szynkaruk M, Prout AP, Nygard K, Cao M, et al. Decreased neuroinflammation correlates to higher vagus nerve activity fluctuations in near-term ovine fetuses: a case for the afferent cholinergic anti-inflammatory pathway? J Neuroinflammation 2016;13:103.
71. Goehler LE, Gaykema RP, Nguyen KT, Lee JE, Tilders FJ, et al. Interleukin-1beta in immune cells of the abdominal vagus nerve: a link between the immune and nervous systems? J Neurosci 1999;19:2799-806.
72. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384-8.
73. Hassert DL, Miyashita T, Williams CL. The effects of peripheral vagal nerve stimulation at a memory-modulating intensity on norepinephrine output in the basolateral amygdala. Behav Neurosci 2004;118:79-88.
74. Follesa P, Biggio F, Gorini G, Caria S, Talani G, et al. Vagus nerve stimulation increases norepinephrine concentration and the gene expression of BDNF and bFGF in the rat brain. Brain Res 2007;1179:28-34.
75. Stowell RD, Sipe GO, Dawes RP, Batchelor HN, Lordy KA, et al. Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat Neurosci 2019;22:1782-92.
76. Liu YU, Ying Y, Li Y, Eyo UB, Chen T, et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat Neurosci 2019;22:1771-81.
77. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458-62.
78. Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, et al. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A 2008;105:11008-13.
79. Paton JFR, Boscan P, Pickering AE, Nalivaiko E. The yin and yang of cardiac autonomic control: vago-sympathetic interactions revisited. Brain Res Brain Res Rev 2005;49:555-65.
80. Olofsson PS, Katz DA, Rosas-Ballina M, Levine YA, Ochani M, et al. alpha7 nicotinic acetylcholine receptor (alpha7nAChR) expression in bone marrow-derived non-T cells is required for the inflammatory reflex. Mol Med 2012;18:539-43.
81. Bratton BO, Martelli D, McKinley MJ, Trevaks D, Anderson CR, et al. Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp Physiol 2012;97:1180-5.
82. Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, Verdu EF, Collins SM. The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology 2006;131:1122-30.
83. Cox LM, Weiner HL. Microbiota Signaling Pathways that Influence Neurologic Disease. Neurotherapeutics 2018;15:135-45.
84. Amini-Khoei H, Haghani-Samani E, Beigi M, Soltani A, Balali-Dehkordi S, et al. On the role of corticosterone in behavioral disorders, microbiota composition alteration and neuroimmune response in adult male mice subjected to maternal separation stress. Int Immunopharmacol 2019;66:242-50.
85. Clarke G, Grenham S, Scully P, Fitzgerald P, Moloney RD, et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry 2013;18:666-73.
86. Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, et al. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol 2004;558:263-75.
87. Luo Y, Zeng B, Zeng L, Du X, Li B, et al. Gut microbiota regulates mouse behaviors through glucocorticoid receptor pathway genes in the hippocampus. Transl Psychiatry 2018;8:187.
88. van Olst L, Bielefeld P, Fitzsimons CP, de Vries HE, Schouten M. Glucocorticoid-mediated modulation of morphological changes associated with aging in microglia. Aging Cell 2018;17:e12790.
89. Benedusi V, Meda C, Della Torre S, Monteleone G, Vegeto E, et al. A lack of ovarian function increases neuroinflammation in aged mice. Endocrinology 2012;153:2777-88.
90. Walker DJ, Spencer KA. Glucocorticoid programming of neuroimmune function. Gen Comp Endocrinol 2018;256:80-8.
91. Miller AH, Spencer RL, Pearce BD, Pisell TL, Azrieli Y, et al. Glucocorticoid receptors are differentially expressed in the cells and tissues of the immune system. Cell Immunol 1998;186:45-54.
92. Sun SL, Li TJ, Yang PY, Qiu Y, Rui YC. Modulation of signal transducers and activators of transcription (STAT) factor pathways during focal cerebral ischaemia: a gene expression array study in rat hippocampus after middle cerebral artery occlusion. Clin Exp Pharmacol Physiol 2007;34:1097-101.
93. Natarajan C, Sriram S, Muthian G, Bright JJ. Signaling through JAK2-STAT5 pathway is essential for IL-3-induced activation of microglia. Glia 2004;45:188-96.
94. Ock J, Lee H, Kim S, Lee WH, Choi DK, et al. Induction of microglial apoptosis by corticotropin-releasing hormone. J Neurochem 2006;98:962-72.
95. Yang Y, Hahm E, Kim Y, Kang J, Lee W, et al. Regulation of IL-18 expression by CRH in mouse microglial cells. Immunol Lett 2005;98:291-6.
96. Lim HY, Muller N, Herold MJ, van den Brandt J, Reichardt HM. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology 2007;122:47-53.
97. Sierra A, Gottfried-Blackmore A, Milner TA, McEwen BS, Bulloch K. Steroid hormone receptor expression and function in microglia. Glia 2008;56:659-74.
98. Sugama S, Fujita M, Hashimoto M, Conti B. Stress induced morphological microglial activation in the rodent brain: involvement of interleukin-18. Neuroscience 2007;146:1388-99.
99. Sugama S, Takenouchi T, Fujita M, Kitani H, Conti B, et al. Corticosteroids limit microglial activation occurring during acute stress. Neuroscience 2013;232:13-20.
100. Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, et al. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun 2010;24:1058-68.
101. Frank MG, Miguel ZD, Watkins LR, Maier SF. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun 2010;24:19-30.
102. Vegeto E, Belcredito S, Ghisletti S, Meda C, Etteri S, et al. The endogenous estrogen status regulates microglia reactivity in animal models of neuroinflammation. Endocrinology 2006;147:2263-72.
103. Sarvari M, Hrabovszky E, Kallo I, Solymosi N, Liko I, et al. Menopause leads to elevated expression of macrophage-associated genes in the aging frontal cortex: rat and human studies identify strikingly similar changes. J Neuroinflammation 2012;9:264.
104. Orgaard A, Jepsen SL, Holst JJ. Short-chain fatty acids and regulation of pancreatic endocrine secretion in mice. Islets 2019;11:103-11.
105. Kumar A, Alrefai WA, Borthakur A, Dudeja PK. Lactobacillus acidophilus counteracts enteropathogenic E. coli-induced inhibition of butyrate uptake in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 2015;309:G602-7.
106. Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci 2003;23:9418-27.
107. Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A. Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J Alzheimers Dis 2011;26:187-97.
108. Silva LG, Ferguson BS, Avila AS, Faciola AP. Sodium propionate and sodium butyrate effects on histone deacetylase (HDAC) activity, histone acetylation, and inflammatory gene expression in bovine mammary epithelial cells. J Anim Sci 2018;96:5244-52.
109. Walsh ME, Bhattacharya A, Sataranatarajan K, Qaisar R, Sloane L, et al. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell 2015;14:957-70.
110. Kannan V, Brouwer N, Hanisch UK, Regen T, Eggen BJL, et al. Histone deacetylase inhibitors suppress immune activation in primary mouse microglia. J Neurosci Res 2013;91:1133-42.
111. Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, et al. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci U S A 2003;100:4281-6.
112. Yin F, Sancheti H, Patil I, Cadenas E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic Biol Med 2016;100:108-22.
113. Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, et al. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci U S A 2011;108:8030-5.
114. Canfora EE, Jocken JW, Blaak EE. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol 2015;11:577-91.
115. Layden BT, Angueira AR, Brodsky M, Durai V, Lowe WL Jr. Short chain fatty acids and their receptors: new metabolic targets. Transl Res 2013;161:131-40.
116. Guilloteau P, Martin L, Eeckhaut V, Ducatelle R, Zabielski R, et al. From the gut to the peripheral tissues: the multiple effects of butyrate. Nutr Res Rev 2010;23:366-84.
117. Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, et al. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 2008;27:104-19.
118. Sun J, Xu J, Yang B, Chen K, Kong Y, et al. Effect of clostridium butyricum against microglia-mediated neuroinflammation in Alzheimer’s disease via regulating gut microbiota and metabolites butyrate. Mol Nutr Food Res 2019;64:e1900636.
119. Aguilar EC, Leonel AJ, Teixeira LG, Silva AR, Silva JF, et al. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFkappaB activation. Nutr Metab Cardiovasc Dis 2014;24:606-13.
120. de la Cuesta-Zuluaga J, Mueller NT, Alvarez-Quintero R, Velasquez-Mejia EP, Sierra JA, et al. Higher fecal short-chain fatty acid levels are associated with gut microbiome dysbiosis, obesity, hypertension and cardiometabolic disease risk factors. Nutrients 2018;11.
121. Elamin EE, Masclee AA, Dekker J, Pieters HJ, Jonkers DM. Short-chain fatty acids activate AMP-activated protein kinase and ameliorate ethanol-induced intestinal barrier dysfunction in Caco-2 cell monolayers. J Nutr 2013;143:1872-81.
122. Bourre JM, Bornhofen JH, Araoz CA, Daudu O, Baumann NA. Pelizaeus--Merzbacher disease: brain lipid and fatty acid composition. J Neurochem 1978;30:719-27.
123. Unger MM, Spiegel J, Dillmann KU, Grundmann D, Philippeit H, et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 2016;32:66-72.
124. Ho L, Ono K, Tsuji M, Mazzola P, Singh R, et al. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev Neurother 2018;18:83-90.
125. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007;317:256-60.
126. Kurita-Ochiai T, Hashizume T, Yonezawa H, Ochiai K, Yamamoto M. Characterization of the effects of butyric acid on cell proliferation, cell cycle distribution and apoptosis. FEMS Immunol Med Microbiol 2006;47:67-74.
127. Proctor LM, Creasy HH, Fettweis JM, Lloyd-Price J, Mahurkar A, et al. The integrative human microbiome project. Nature 2019;569:641-8.
128. Human Microbiome Project (HMP) Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012;486:207-14.
129. Theriot CM, Koenigsknecht MJ, Carlson PE Jr, Hatton GE, Nelson AM, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 2014;5:3114.
130. Sekirov I, Tam NM, Jogova M, Robertson ML, Li Y, et al. Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect Immun 2008;76:4726-36.
131. Hong KS, Kim JS. Rifaximin for the treatment of acute infectious diarrhea. Therap Adv Gastroenterol 2011;4:227-35.
132. Iorio N, Malik Z, Schey R. Profile of rifaximin and its potential in the treatment of irritable bowel syndrome. Clin Exp Gastroenterol 2015;8:159-67.
133. Ponziani FR, Zocco MA, D’Aversa F, Pompili M, Gasbarrini A. Eubiotic properties of rifaximin: disruption of the traditional concepts in gut microbiota modulation. World J Gastroenterol 2017;23:4491-9.
134. Maccaferri S, Vitali B, Klinder A, Kolida S, Ndagijimana M, et al. Rifaximin modulates the colonic microbiota of patients with Crohn’s disease: an in vitro approach using a continuous culture colonic model system. J Antimicrob Chemother 2010;65:2556-65.
135. Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, et al. The role of short-chain fatty acids in health and disease. Adv Immunol 2014;121:91-119.
136. Yang L, Liu B, Zheng J, Huang J, Zhao Q, et al. Rifaximin alters intestinal microbiota and prevents progression of ankylosing spondylitis in mice. Front Cell Infect Microbiol 2019;9:44.
137. Xu D, Gao J, Gillilland M, Wu X, Song I, et al. Rifaximin alters intestinal bacteria and prevents stress-induced gut inflammation and visceral hyperalgesia in rats. Gastroenterology 2014;146:484-96.e4.
138. Cash WJ, McConville P, McDermott E, McCormick PA, Callender ME, et al. Current concepts in the assessment and treatment of hepatic encephalopathy. QJM 2010;103:9-16.
139. Coronel-Castillo CE, Contreras-Carmona J, Frati-Munari AC, Uribe M, Mendez-Sanchez N. Efficacy of rifaximin in the different clinical scenarios of hepatic encephalopathy. Rev Gastroenterol Mex 2020;85:56-68.
140. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011;21:381-95.
141. Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell 2006;23:289-96.
142. Ghiaghi M, Forouzesh F, Rahimi H. Effect of sodium butyrate on LHX1 mRNA expression as a transcription factor of HDAC8 in human colorectal cancer cell lines. Avicenna J Med Biotechnol 2019;11:317-24.
143. Nakagawa H, Sasagawa S, Itoh K. Sodium butyrate induces senescence and inhibits the invasiveness of glioblastoma cells. Oncol Lett 2018;15:1495-502.
144. Arnoldussen IAC, Wiesmann M, Pelgrim CE, Wielemaker EM, van Duyvenvoorde W, et al. Butyrate restores HFD-induced adaptations in brain function and metabolism in mid-adult obese mice. Int J Obes (Lond) 2017;41:935-44.
145. Mannucci E, Ognibene A, Cremasco F, Bardini G, Mencucci A, et al. Effect of metformin on glucagon-like peptide 1 (GLP-1) and leptin levels in obese nondiabetic subjects. Diabetes Care 2001;24:489-94.
146. Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004;44:595-600.
147. van Engeland M, Derks S, Smits KM, Meijer GA, Herman JG. Colorectal cancer epigenetics: complex simplicity. J Clin Oncol 2011;29:1382-91.
148. Halliday GM, Holton JL, Revesz T, Dickson DW. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol 2011;122:187-204.
149. Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 2006;396:67-72.
150. Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 2014;128:805-20.
151. Sander C, Modes F, Schlake HP, Eling P, Hildebrandt H. Capturing fatigue parameters: the impact of vagal processing in multiple sclerosis related cognitive fatigue. Mult Scler Relat Disord 2019;32:13-8.
152. Hanken K, Eling P, Hildebrandt H. The representation of inflammatory signals in the brain - a model for subjective fatigue in multiple sclerosis. Front Neurol 2014;5:264.
153. Kaczmarczyk R, Tejera D, Simon BJ, Heneka MT. Microglia modulation through external vagus nerve stimulation in a murine model of Alzheimer’s disease. J Neurochem 2017; doi: 10.3389/fneur.2014.00264.
154. Sarao LK, Arora M. Probiotics, prebiotics, and microencapsulation: a review. Crit Rev Food Sci Nutr 2017;57:344-71.
155. van Baarlen P, Troost F, van der Meer C, Hooiveld G, Boekschoten M, et al. Human mucosal in vivo transcriptome responses to three lactobacilli indicate how probiotics may modulate human cellular pathways. Proc Natl Acad Sci U S A 2011;108:4562-9.
156. Magistrelli L, Amoruso A, Mogna L, Graziano T, Cantello R, et al. Probiotics may have beneficial effects in Parkinson’s disease: in vitro evidence. Front Immunol 2019;10:969.
157. Akbari E, Asemi Z, Daneshvar Kakhaki R, Bahmani F, Kouchaki E, et al. Effect of probiotic supplementation on cognitive function and metabolic status in Alzheimer’s disease: a randomized, double-blind and controlled trial. Front Aging Neurosci 2016;8:256.
158. Vandeputte D, Falony G, Vieira-Silva S, Wang J, Sailer M, et al. Prebiotic inulin-type fructans induce specific changes in the human gut microbiota. Gut 2017;66:1968-74.
159. Bourassa MW, Alim I, Bultman SJ, Ratan RR. Butyrate, neuroepigenetics and the gut microbiome: can a high fiber diet improve brain health? Neurosci Lett 2016;625:56-63.
160. Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res 2009;69:2826-32.
161. Katagiri S, Shiba T, Tohara H, Yamaguchi K, Hara K, et al. Re-initiation of oral food intake following enteral nutrition alters oral and gut microbiota communities. Front Cell Infect Microbiol 2019;9:434.
162. Riccio P, Rossano R. Diet, gut microbiota, and vitamins D + A in multiple sclerosis. Neurotherapeutics 2018;15:75-91.
163. Nagpal R, Neth BJ, Wang S, Craft S, Yadav H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019;47:529-42.
164. Lowe PP, Gyongyosi B, Satishchandran A, Iracheta-Vellve A, Cho Y, et al. Reduced gut microbiome protects from alcohol-induced neuroinflammation and alters intestinal and brain inflammasome expression. J Neuroinflammation 2018;15:298.
165. Gubert C, Kong G, Renoir T, Hannan AJ. Exercise, diet and stress as modulators of gut microbiota: implications for neurodegenerative diseases. Neurobiol Dis 2020;134:104621.
166. Bermon S, Petriz B, Kajeniene A, Prestes J, Castell L, et al. The microbiota: an exercise immunology perspective. Exerc Immunol Rev 2015;21:70-9.
167. Matsumoto M, Inoue R, Tsukahara T, Ushida K, Chiji H, et al. Voluntary running exercise alters microbiota composition and increases n-butyrate concentration in the rat cecum. Biosci Biotechnol Biochem 2008;72:572-6.
168. Crowley EK, Nolan YM, Sullivan AM. Neuroprotective effects of voluntary running on cognitive dysfunction in an alpha-synuclein rat model of Parkinson’s disease. Neurobiol Aging 2018;65:60-8.
169. O’Donovan SM, Crowley EK, Brown JR, O’Sullivan O, O’Leary OF, et al. Nigral overexpression of alpha-synuclein in a rat Parkinson’s disease model indicates alterations in the enteric nervous system and the gut microbiome. Neurogastroenterol Motil 2020;32:e13726.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Reyes REN, Zhang Z, Gao L, Asatryan L. Microbiome meets microglia in neuroinflammation and neurological disorders. Neurosciences 2020;7:215-33. http://dx.doi.org/10.20517/2347-8659.2020.13
AMA Style
Reyes REN, Zhang Z, Gao L, Asatryan L. Microbiome meets microglia in neuroinflammation and neurological disorders. Neuroimmunology and Neuroinflammation. 2020; 7(3): 215-33. http://dx.doi.org/10.20517/2347-8659.2020.13
Chicago/Turabian Style
Reyes, Rachel E. N., Zeyu Zhang, Lei Gao, Liana Asatryan. 2020. "Microbiome meets microglia in neuroinflammation and neurological disorders" Neuroimmunology and Neuroinflammation. 7, no.3: 215-33. http://dx.doi.org/10.20517/2347-8659.2020.13
ACS Style
Reyes, REN.; Zhang Z.; Gao L.; Asatryan L. Microbiome meets microglia in neuroinflammation and neurological disorders. Neurosciences. 2020, 7, 215-33. http://dx.doi.org/10.20517/2347-8659.2020.13
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 33 clicks
Cite This Article 33 clicks

 Like This Article 10
likes
Like This Article 10
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.