
Inflammatory mechanisms underlying cortical injury in progressive multiple sclerosis

Abstract
Multiple sclerosis (MS) is a lifelong inflammatory demyelinating disease of the central nervous system (CNS). While there has been substantial progress in the development of therapeutic strategies for relapsing disease, the field has lagged behind in its understanding and management of progressive stages of the disease, including secondary progressive and primary progressive MS, respectively. It is now thought that distinct but temporally overlapping mechanisms underlie relapsing and progressive aspects of the disease. Relapsing disease is characterized by waves of peripheral immune cell activation and CNS infiltration leading to focal destruction of the white matter, while progressive disease is thought to be driven by chronic, low-grade multifocal inflammation contained within the CNS compartment. Specifically, peripheral B cells, T cells, and myeloid cells take up residence within niches of the inflamed CNS, such as the leptomeninges and the Virchow-Robin spaces, where complex interactions between peripheral and CNS resident cells serve to maintain these cellular aggregates and further propagate CNS injury. In particular, immune infiltrates within the meninges are tightly associated with a specific form of cortical injury, termed subpial cortical demyelination, which is thought to be a key pathologic driver of disease progression. Cortical injury in the MS brain likely occurs via a combination of multiple immune-mediated and degenerative processes, perhaps including the production of diffusible toxic mediators by peripheral immune cells retained within the meninges. A better understanding of the interplay between peripheral immune and CNS resident cells is not only relevant to our concept of the disease process, but also represents a novel target for therapeutic intervention that is more specific to progressive disease biology. This review will focus on the role of CNS-compartmentalized inflammation in the development of cortical injury in MS, with a particular emphasis on the importance of immune-CNS crosstalk in disease progression.
Keywords
Introduction
Multiple sclerosis (MS) is a chronic inflammatory and degenerative demyelinating disease of the central nervous system (CNS) with variable clinical course and rates of progression of neurological disability. Classical teaching has distinguished between relapsing-remitting MS (RRMS) and progressive forms of MS, which in 10%-15% of patients manifests clinically from disease onset [primary progressive MS (PPMS)] or, in most patients, emerges after initial onset with relapsing-remitting disease [secondary progressive MS (SPMS)][1-4]. Mounting evidence from neuropathological, clinical, and imaging studies now support the view that these clinical labels do not define temporally distinct biological entities. Indeed, individuals throughout the MS spectrum can experience both acute focal inflammatory injury (reflecting relapse biology) and the more insidious and distributed progressive injury (reflecting non-relapsing, progressive biology)[4,5]. In addition to their substantial temporal overlap, both of these processes contribute to subclinical CNS injury[6-8]. It follows that MS is now thought of more as a continuum, from radiologically isolated syndrome (RIS), through clinically isolated syndrome (CIS), RRMS, and SPMS, with PPMS differing from SPMS only by the lack of clear-cut clinically evident relapses[4].
While accomplishments of the last several decades are highlighted by the development of increasingly effective disease-modifying therapies (DMTs)[2,9], treating progressive disease remains a major outstanding challenge. Only three DMTs, ocrelizumab, siponimod, and cladribine, have been approved for the treatment of patients with progressive forms of MS, all showing relatively modest benefit on progression[10-14]. The question remains to what extent this benefit indeed reflects targeting non-relapsing progressive disease (i.e., clinical worsening in the absence of clinical or radiographic relapses, sometimes referred to as progression independent of relapse activity, or PIRA)[5], as opposed to progression due to relapses that do not fully recover. In general, even the immunotherapies that are highly effective in targeting the pathophysiological mechanisms underlying relapse biology in MS have a rather limited capacity to impact the temporally overlapping but distinct process(es) that drive(s) non-relapsing disease progression[10-13].
A wealth of neuropathological and imaging data demonstrates the changing features of inflammation and neural tissue degeneration across the spectrum of MS[10-12,14-16]. It is thought that relapses result from waves of peripheral immune cell activation and focal perivascular infiltration into the CNS across a disrupted blood-brain barrier (BBB), resulting in acute inflammatory demyelination and axonal injury[17-19]. Meanwhile, progressive disease biology involves chronic, low-grade inflammation that is thought to foster immune cell retention within the CNS. The resulting CNS-compartmentalized inflammation then propagates ongoing demyelination and neuronal loss without relying on the episodic recruitment of peripheral immune cells into the CNS[18,20-22]. Over time, the reduction in prominent relapse biology and the associated decrease in development of new gadolinium-enhancing lesions on MRI means that the BBB may be less permeable in patients with predominantly progressive disease, which in turn may both foster CNS-compartmentalized inflammation and limit penetration of DMTs[18,22]. Even for the group of small molecule DMTs that do penetrate the CNS[23-25], evidence for a direct effect on CNS protection and/or repair in humans is lacking[26].
With the changing character of inflammation through the disease course, there is also a shift in key pathological findings. While acute, focal, demyelinating plaques predominate early in RRMS, their relative contribution to total white matter lesion (WML) burden wanes in later stages of the disease[10,14,21,27]. The white matter pathology now thought to contribute to progressive MS involves a subset of perivascular lesions with a lower but persistent degree of inflammation, which pathologically have been considered “chronic active” lesions[10,14] and by imaging may be captured as the more recently described slowly evolving lesions (SELs) or “rim” lesions[14,21,28-31]. Other pathological changes also become more prominent with advancing disease, such as demyelination and degeneration of both the superficial and deep gray matter structures as well as more subtle and distributed inflammatory changes in the normal-appearing white and gray matter[10,13,16,32-34]. Though each of these pathological findings is thought to contribute to disease progression in some capacity[32,34-36], a growing body of literature suggests that cortical injury is one of the strongest predictors of progression, including both progression of physical disability and cognitive decline[35-38] as well as evolution of the disease course from initial clinical episodes to SPMS[38-40]. A summary of studies linking cortical pathology and disease progression in MS is provided in Table 1 and builds on the table provided in the detailed review by Geurts et al.[35] in 2012. Though cortical lesions are described as more numerous and more extensive in the brains of progressive MS patients examined at autopsy[16], they are also reported in significant proportions of patients early in their course of relapsing disease[41,42], including CIS[39] and RIS[43]. Because these studies in early-stage MS patients utilized MRI rather than histopathology to detect cortical lesions, the true prevalence of cortical pathology was likely substantially underestimated. This further supports the notion that relapsing and progressive disease biology occurs simultaneously in the MS CNS.
Studies investigating the association of cortical lesions with disease progression
| Author | Method | Population(s) studied | Study design | Findings |
|---|---|---|---|---|
| In vivo imaging studies | ||||
| Calabrese et al.[186], 2007 | 1.5T (DIR) | 116 CIS, 163 RRMS, 101 SPMS
40 NC | Cross-sectional | Total number of CLs correlated with EDSS scores in all patient subgroups |
| Calabrese et al.[101], 2009 | 1.5T (DIR) | 70 RRMS | Cross-sectional | Total CL number and volume correlated with cognitive impairment in multiple domains as measured by the Rao’s Brief Repeatable Battery of Neuropsychological Tests. Total CL volume was an independent predictor of cognitive impairment |
| Calabrese et al.[187], 2009 | 1.5T (DIR) | 48 PPMS
22 NC | Longitudinal, 2 years | Total CL volume at baseline was an independent predictor of disability accumulation over the study period |
| Calabrese et al.[90], 2009 | 1.5T (DIR) | 48 Benign MS, 96 RRMS | Longitudinal, 1 year | Low number of CLs at baseline and minimal or absent accrual of both ICLs and LCLs over time were associated with benign MS compared to RRMS |
| Roosendaal et al.[102], 2009 | 1.5T (DIR) | 9 RRMS, 4 SPMS
7 NC | Longitudinal, 3 years | Total CL number at baseline predicted slower processing speed at follow-up. ICL and LCL volume at follow-up was associated with worse performance on tasks of processing speed and visuospatial memory |
| Calabrese et al.[188], 2010 | 1.5T (DIR) | 76 RRMS, 31 SPMS | Longitudinal, 3 years | Increases in CL number and volume were greatest in patients with clinical worsening compared to stable patients. Baseline total CL volume was an independent predictor of EDSS worsening over the study period in both RRMS and SPMS patients |
| Filippi et al.[39], 2010 | 1.5T (DIR) | 80 CIS (training)
39 CIS (validation) | Longitudinal, 4.6 years (training)
2.3 years (validation) | The presence of one or more ICLs on baseline MRI in CIS patients was an independent predictor of subsequent conversion to clinically definite MS |
| Bagnato et al.[189], 2010 | 3T (3D IRSPGR) | 17 RRMS, 4 SPMS
21 NC | Cross-sectional | Patients with CLs demonstrated worse performance on tests of delayed recall, but this relationship was not independent of WML volume |
| Mike et al.[190], 2011 | 3T (3D IRSPGR, 3D FLAIR) | 20 RRMS, 6 SPMS | Cross-sectional | Total CL number and volume correlated with worse performance on working memory as well as worse EDSS scores. Total CL number also correlated with worse performance on verbal learning and memory |
| Nelson et al.[59], 2011 | 3T (DIR, PSIR) | 39 MS, no subtype specified | Cross-sectional | The number of LCLs located predominantly in the cortex correlated with cognitive impairment, as did the number of these LCLs plus ICLs. ICLs alone did not correlate with cognitive impairment, though these lesions tend to be significantly smaller |
| Giorgio et al.[43], 2011 | 1.5T (DIR) | 15 RIS | Cross-sectional | Total CL number and volume correlated with WML volume, presence of OCBs, and DIT criteria on MRI and were present in all subjects classified as having a very high probability of conversion to clinically definite MS |
| Calabrese et al.[37], 2012 | 1.5T (DIR) | 157 RRMS, 35 Pediatric MS, 45 Benign MS, 44 PPMS, 31 SPMS | Longitudinal, 5 years | Patients with relapse independent EDSS progression had a greater number and volume of CLs at baseline, higher rates of CL accumulation, and a greater increase in CL volume. CL volume at baseline was an independent predictor of EDSS and cognitive worsening over the study period in all disease subtypes |
| Nielsen et al.[70], 2013 | 7T ((FLASH)-T2*) | 10 CIS, 8 RRMS, 9 SPMS | Cross-sectional | Numbers of total CLs, LCLs, and SPLs (but not ICLs) were significantly associated with EDSS scores, with SPLs demonstrating the strongest association. Numbers of total CLs and all CL subtypes were negatively correlated with at least one measure of cognitive performance. LCLs correlated with the greatest number of cognitive measures |
| Calabrese et al.[40], 2013 | 1.5T (DIR) | 334 RRMS (training)
88 RRMS (validation) | Longitudinal, 5 years | CL volume at baseline was an independent predictor of progression from RRMS to SPMS |
| Kolber et al.[191], 2015 | 3T (3D DIR, 3D FLAIR) | 29 CIS, 93 RRMS | Cross-sectional | Number of CLs correlated with worse performance on tests of working memory and semantic word fluency. |
| Harrison et al.[69], 2015 | 7T (3D MPRAGE, 3D MPFLAIR) | 30 RRMS & 6 PP/SPMS
15 NC | Cross-sectional | Patients with higher EDSS scores had a higher total CL number and volume as well as higher SPL volume. Total CL volume and LCL volume, in particular, were both independent predictors of cognitive impairment |
| Geisseler et al.[192], 2016 | 1.5T (3D DIR, 3D MPRAGE, 3D FLAIR) | 42 RRMS
43 NC | Cross-sectional | Patients with CLs demonstrated more pronounced cortical thinning and worse performance on memory tasks compared to patients without CLs and controls |
| Curti et al.[193], 2018 | 3T (3D DIR) | 47 RRMS, 11 SPMS, 2 PPMS | Cross-sectional | Total CL number correlated with poor performance in multiple cognitive domains |
| Scalfari et al.[38], 2018 | 1.5T (3D DIR, 3D MPRAGE, 3D FLAIR) | 219 RRMS | Longitudinal, 7.9 years | A greater number of CLs at baseline predicted a higher risk of conversion to SPMS and shorter time to conversion over the study period. Baseline CL volume was an independent predictor of progression to SPMS |
| Treaba et al.[42], 2019 | 7T (T2*-weighted gradient-echo) | 20 RRMS, 13 SPMS
10 NC | Longitudinal, 1.5 years | Total CL volume was an independent predictor of baseline EDSS and EDSS change at follow up |
| Post-mortem clinical-pathological correlation studies | ||||
| Magliozzi et al.[44], 2007 | Post-mortem histology | 29 SPMS, 7 PPMS
3 NC | Clinical-pathological correlation | Patients with follicle-like structures in the meninges had more pronounced cortical (particularly subpial) demyelination and more severe clinical course, characterized by younger age at onset, irreversible disability, and death |
| Magliozzi et al.[45], 2010 | Post-mortem histology | 37 SPMS
14 NC | Clinical-pathological correlation | In a larger SPMS cohort, patients with follicle-like structures in the meninges had more pronounced subpial cortical pathology and more severe clinical course, characterized by younger age at onset, age at death, age at wheelchair dependence, and time to wheelchair use |
| Howell et al.[94], 2011 | Post-mortem histology | 123 SPMS
6 NC | Clinical-pathological correlation | Patients with follicle-like structures in the meninges had a greater extent of meningeal inflammation, cortical demyelination, and activated microglia in the cortex. Cases with follicle-like structures also had a more severe clinical course, characterized by younger age at onset, age at death, age at progression from relapsing to progressive disease, and age at wheelchair dependence |
| Choi et al.[93], 2012 | Post-mortem histology | 26 PPMS
6 NC | Clinical-pathological correlation | More pronounced meningeal inflammation was associated with more severe cortical demyelination and clinical course, characterized by younger age at time of death, shorter disease duration, and shorter time from wheelchair use to death |
| Kooi et al.[56], 2012 | Post-mortem histology | 1 PRMS, 11 SPMS, 23 PPMS, 6 Unclassified MS | Clinical-pathological correlation | A subset of patients with CLs had CLs with a rim of activated microglia. These patients had a younger age at time of death and shorter disease duration than patients with cortical pathology in the absence of rim-associated microglia or patients without extensive CLs |
| Magliozzi et al.[57], 2013 | Post-mortem histology | 44 SPMS | Clinical-pathological correlation | Patients with follicle-like structures in the meninges had more pronounced cortical demyelination, perivascular inflammation in the cortex, and more severe clinical course. No differences in clinical severity were observed between patients with follicle-like meningeal inflammation with cortical perivascular infiltrates and those without perivascular inflammation |
| Bevan et al.[88], 2018 | Post-mortem histology | 12 Acute MS, 18 SPMS, 3 PPMS, 11 NC, 6 OIND | Clinical-pathological correlation | Patients with acute MS (average disease duration of 2 years prior to death) exhibited significant cortical demyelination, the majority in a subpial pattern. Greater myeloid cell activation was observed in cases with more pronounced meningeal inflammation |
| Trapp et al.[91], 2018 | Post-mortem histology | 12 MCMS, 12 TMS | Clinical-pathological correlation | MCMS were characterized by cortical and spinal cord pathology in the absence of cerebral WMLs. Age at death and EDSS did not differ between MCMS and TMS. Cortical demyelinated lesion area was similar between MCMS and TMS. MCMS had decreased cortical neuronal densities compared to controls despite lack of WMLs |
This review will describe the major patterns of cortical injury (leukocortical, intracortical, and subpial lesions) and their relative contribution to disease progression. As will be described in greater detail below, leukocortical/intracortical lesions are thought to reflect mechanisms of perivascular inflammatory injury that are likely shared with classic WMLs. In contrast, the subpial component of cortical injury is thought to reflect progressive, non-relapsing disease biology[44,45], which is the focus of this review. We will therefore emphasize those studies of potential mechanisms underlying subpial lesion formation and their relevance to disease progression. The key word search for this review included variations in the following terms between 1990 and 2020 in the English language: cortical lesions/injury/demyelination, subpial lesions/injury/demyelination, progressive multiple sclerosis, and multiple sclerosis disability progression. The overarching goal of the discussion is to emphasize key pathways that may represent meaningful therapeutic targets in progressive disease, while also recognizing some of the knowledge gaps and technical limitations that must be addressed to ultimately develop and translate such approaches. Given our focus on cellular and molecular mechanisms of cortical injury, and subpial cortical injury in particular, we limit our commentary on imaging to a selection of in vivo studies that provide complementary insights to particular pathophysiological considerations, and we refer the readers to recent comprehensive reviews on imaging of cortical lesions in MS[46-48].
Patterns of cortical injury in multiple sclerosis
Four types of demyelinated lesions have been described in the cortex of MS patients: leukocortical (type I), intracortical (type II), and subpial (type III, sometimes further divided into types III and IV)[49-51]. Leukocortical lesions are thought to originate as a perivascular lesion in the white matter that extends across the gray-white matter junction to involve the cortex. Intracortical lesions reside entirely within the cortex, are typically small, and form in a perivascular distribution, similar to type I lesions and focal lesions in the white matter. In contrast, subpial cortical lesions extend from the pial surface (layer I of the cortex) into the deeper cortical layers. Most lesions involve superficial cortical layer I through layers III or IV (referred to as type III subpial cortical lesions), while some involve the entire thickness of the cortex (type IV subpial cortical lesions), notably with remarkable respect for the gray-white matter junction. The varied topography of these lesions suggests that different mechanisms may drive their development and maintenance [Figure 1], and that different cortical lesion types may contribute to progressive biology and disease progression to varying extents. That being said, certain features distinguish cortical lesions in general from lesions in the white matter, namely that they are typically much less inflammatory, as measured by the number of parenchymal and perivascular infiltrates[49,50], and interestingly, they seem to possess a greater capacity for remyelination, perhaps related to the greater number of oligodendrocyte precursor cells observed in the demyelinated cortex[52-54].
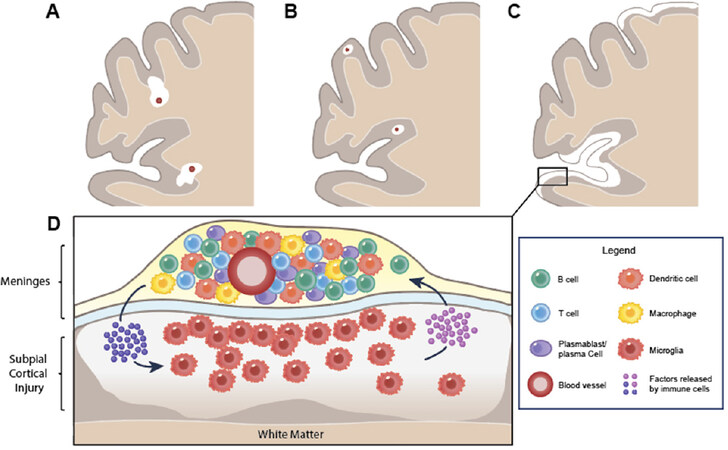
Figure 1. Cortical lesions in multiple sclerosis. Coronal brain sections demonstrating cortical demyelination (white) are shown in panels A-C. (A) Leukocortical (type I) and (B) intracortical (type II) lesions form around post-capillary venules (red) in the subcortical white and cortical gray matter, respectively; (C) subpial (type III) lesions extend from the superficial pial surface into the deeper layers of cortex. Type IV lesions involve all cortical layers and are not shown specifically in this schematic; (D) magnified representation of subpial cortical lesion segment demonstrating proposed inflammatory mechanism underlying subpial injury in MS. Meningeal immune cell aggregates are tightly associated with subpial lesions, in which a gradient of demyelination and microglial activation in the subjacent cortex is represented. Meningeal immune cell aggregates are typically B cell-rich and contain proliferating CD20+ B cells, plasmablasts/plasma cells, and CD4+ and CD8+ T cells contained within a network of follicular dendritic cells (see Legend). Mediators released by infiltrating immune cells in the meninges as well as by CNS-resident cells, such as activated microglia and astrocytes, are thought to contribute to ongoing meningeal inflammation and subpial injury in multiple sclerosis. A range of mechanisms have been implicated as underlying subpial cortical injury, including (1) secretion of diffusible toxic factor(s) (e.g., pro-inflammatory cytokines, extracellular vesicles, complement, etc.) released by immune cells in the meninges and/or perivascular spaces; (2) pro-inflammatory and/or toxic substances (e.g., oxygen radicals; nitrogen species; excitotoxic glutamate) released or insufficiently cleared by activated and injured glial cells; (3) mitochondrial dysfunction leading to bioenergetic failure within neurons and oligodendrocytes; and (4) neural-glial uncoupling (including loss of trophic support). CNS: central nervous system; MS: multiple sclerosis
Prior to discussing the potential mechanisms underlying the development and clinical consequence of these different cortical lesions, there are several challenges to studying cortical lesions in general that should be considered. First, correlating clinical outcomes with pathological findings requires tissue samples that are generally obtained via either post-mortem autopsy or by clinically indicated stereotactic brain biopsy. In either case, the individuals studied may not be representative of the general MS population. Post-mortem tissue typically originates from patients at the latest stages of disease and may fail to capture key mechanisms involved in prior tissue injury. In turn, biopsy specimens are obtained from a group of individuals with enough atypical lesional activity to warrant tissue sampling[16,41,55]. That being said, some post-mortem studies do include patients who have died earlier in their disease course, and cortical lesions with varying degrees of inflammatory activity have been reported even in later stages[44,56-58]. In vivo assessment, including prospective monitoring, of cortical lesions has relied on MRI, though most early studies found that identification of cortical lesions using 3T MRI was largely restricted to the leukocortical lesions. The introduction of ultra-high-field 7T MRI and more sensitive imaging sequences, such as double-inversion recovery[59], phase-sensitive inversion recovery[59,60], magnetization transfer imaging[61], diffusion-weighted imaging[62], and 3D T1-weighted magnetization-prepared rapid gradient echo[63,64], have improved cortical lesion resolution, though capturing all cortical lesions in vivo remains challenging. This is likely due to their small size (particularly the intracortical lesions, which are also less common), the absence of significant surrounding edema, and proximity to the CSF in the case of subpial lesions[35]. For instance, in one post-mortem verification study of cortical lesions, 7T MRI detected 100% of leukocortical lesions identified pathologically, but only 36.2% of intracortical and 13.6% of subpial lesions[65]. Until new or improved imaging modalities enable more complete detection of intracortical and subpial lesions[35,60,63-67], current MRI-based assessment of these lesions may underestimate their association with clinical outcomes. Recent improvement in PET imaging modalities, such as with (11) C-PBR28 MR-PET[34,68], which can detect activated myeloid cells, may provide a window into the relevant sites and pathophysiological mechanisms involved in progressive disease. Further validation is greatly needed to both fully evaluate the respective contributions of cortical lesions to disease progression and to better evaluate treatment effects on distinct mechanisms underlying cortical pathology.
Leukocortical lesions
Leukocortical lesions are demyelinating plaques that involve the subcortical white matter and adjacent cortex. Pathologically, leukocortical lesions appear to originate around a central vein or venule in the subcortical white matter and spread out radially to involve part of the overlying cortex[50,51]. They are generally more inflammatory than the other cortical lesions[49,50] and are strongly associated with features of adjacent WMLs[15,69,70]. Indeed, the inflammatory activity in leukocortical lesions seems to evolve in a similar pattern to that documented in WMLs[55,57], and the degree of oxidative injury and neuronal loss can be explained, at least in part, by retrograde degeneration from injured axons in the white matter[15]. For these reasons, it is thought that leukocortical lesions and WMLs may form via similar mechanisms.
The frequency of leukocortical lesions in MS brains largely depends on the population studied. Leukocortical lesions were shown to represent anywhere from 15% to 34% of the total cortical lesion burden in post-mortem tissue[49,51] or up to 50% in biopsy specimens obtained from early stage MS patients[41]. Because the formation of active WMLs occurs more frequently in early MS[14], this may explain the higher proportion of leukocortical lesions observed in this phase of disease.
A few studies have investigated the relative contribution of different cortical lesions to disease outcomes, and it is interesting to note that several have demonstrated a robust association between leukocortical lesions and cognitive impairment even at early phases of disease[69,70]. The particular effect on cognition may be due to disruption of subcortical U-fibers and, consequently, cortical-cortical signaling, which has previously been implicated in cognitive impairment[69-71].
The presumed white matter origin of leukocortical lesions (as focal perivascular inflammatory plaques) is thought to be driven, like other WMLs, by CNS infiltration of autoreactive T cells, which are subsequently reactivated in the meninges, CSF, or perivascular spaces[9,72,73]. This is thought to trigger a second wave of inflammation and infiltration around a central vein or venule, consisting mainly of CD8+ and CD4+ T cells, B cells, and monocytes[12,74,75]. CD8+ T cells and macrophages may then infiltrate the surrounding parenchyma, and this massive influx of immune cells leads to pronounced demyelination, axonal injury, and further breakdown of the BBB[17-19]. Indeed, contrast-enhancing leukocortical lesions were identified in over one-third of early MS patients, indicating significant BBB disruption is involved in their development[76]. Over time, inflammatory activity within the lesion core subsides, and they may be classified as either mixed active/inactive or inactive lesions depending on the degree of demyelination and inflammation[77]. Some active/inactive WMLs may manifest as SELs at sites of prior perivascular inflammatory demyelinating lesions and are now thought to contribute pathophysiologically to non-relapsing disease progression[14,21,28-31]. The extent to which leukocortical or intracortical lesions may develop into SELs as described for WMLs is not known.
Though the full spectrum of active, mixed active-inactive, and inactive lesions has been described in the cortex of MS patients[57], several studies suggest that the gray matter aspect of leukocortical lesions behaves somewhat differently from its white matter counterpart. For instance, the degree of inflammation (in terms of immune cell infiltrates) is always substantially less in the cortical component of these lesions than in their white matter component[49,50]. This manifests as reduced numbers of parenchymal T cells and activated CD68+ myeloid cells as well as nearly absent perivascular cuffs in post-mortem tissue samples[49], though more significant inflammation can be seen in early MS[41]. The reason for this stark difference is not fully understood. It is possible that different phenotypes of CNS-resident immune cells, such as microglia and astrocytes, exist in these two tissues[49,78-80]. Though these CNS cells may not influence initial lesion formation, distinct glial phenotypes may respond differently to the initial demyelinating injury and, consequently, impact the composition and propagation of ongoing inflammation and damage[49,78-80]. Such potential differences between white and gray matter are worthy of further investigation, as they may ultimately be relevant to the effects of DMTs on various types of cortical pathology.
Intracortical lesions
Intracortical lesions are similar to both leukocortical and typical WMLs in that they also form around post-capillary veins or venules, though they tend to be much smaller and, like the cortical components of leukocortical lesions, less inflammatory[41,51]. Differences in BBB permeability characteristics between the cortex and white matter may explain, at least in part, the reduced inflammatory activity observed in these cortical lesions[18,22]. Meanwhile, their perivascular distribution suggests that they too form via peripheral immune cell activation and infiltration of the brain parenchyma[41,49,51,81] and, indeed, up to 23% of intracortical lesions in biopsy specimens were considered actively demyelinating, with the majority demonstrating both parenchymal and perivascular CD3+ and CD8+ T cells[41]. In autopsy tissue, the extent of intracortical lesions correlated with total WML load and the proportion of mixed active/inactive lesions, as well as with the presence of clusters of activated microglia/macrophages in normal-appearing white and gray matter[36]. Taken together, intracortical lesions have been associated with pathological features that may have elements of both relapse and progressive disease biology, and it remains to be determined whether the development and/or maintenance of these lesions are more closely associated with one or both of these processes. As for the leukocortical lesions noted above, whether and to what extent intracortical lesions may become SELs is unknown.
BBB permeability is an important feature of focal demyelination in the MS CNS. Recently, it has been shown that vascular leakage and deposition of fibrinogen, a serum protein involved in the clotting cascade, may actually precede and trigger neuroinflammation and demyelination in the white matter, and perhaps also the cortex[82-84]. In both rodent and primate models, fibrinogen was shown to activate local microglia in a CD11b-dependent manner, which subsequently triggered recruitment and expansion of myelin-specific Th1 cells[82-84]. Fibrinogen has been detected at the rim of chronic active lesions, further implicating an association with ongoing inflammation and demyelination[83]. With the genesis of both leukocortical and intracortical lesions thought to be surrounding a central vein, it is conceivable that fibrinogen plays a similar pathogenic role in the development of cortical pathology, as well. Indeed, more pronounced fibrinogen deposition has been described in the cortex of progressive MS patients compared to controls and was associated with reduced axonal density, specifically in the deeper layers of the cortex[85]. Additionally, elevated levels of fibrinogen in the CSF were associated with markers of innate immune inflammation and greater cortical lesion burden detected by MRI in recently diagnosed MS patients[86], which together suggests a role for fibrinogen in both cortical demyelination and neurodegeneration[87].
Further mechanistic evidence can be extrapolated from rodent models that recapitulate certain features of inflammatory cortical demyelination. In one recent study using stereotactic injections of IFNg and TNFa into the motor cortex of recombinant MOG1-125-immunized Th/+ mice, it was found that intracortical lesion formation is highly dependent on infiltration of encephalitogenic T cells, NK cells, and CCR2+ monocytes, all of which were also demonstrated in post-mortem demyelinated cortex[55]. While inflammatory macrophages were implicated in the development of both intracortical and subpial lesions in this model, the involvement of T cells and NK cells were only critical for intracortical lesion formation. In fact, the presence of encephalitogenic T cells was necessary to permit NK cell passage into the brain parenchyma of these mice[55]. While murine models are limited in their generalizability, particularly when it comes to progressive MS disease mechanism, these findings support the notion that distinct immune effector mechanisms may be responsible for the formation of different cortical lesion types.
Subpial cortical lesions
Subpial lesions are the most common cortical lesion identified in post-mortem tissue, with varying prevalence and inflammatory activity across the disease spectrum. They are present in almost all cases of progressive disease and represent 50%-90% of the cortical lesion burden[49,51,88,89]. In biopsy material from patients with early RRMS or tissue blocks from RRMS of short duration, subpial lesions can represent anywhere from 34% to 62% of all cortical lesions[41,88]. In some cases, subpial cortical demyelination may even occur in the absence of WMLs[90] or represent the dominant lesion type in the CNS[91]. The large range in lesion prevalence observed across studies may be due to the methods by which the specimens were obtained (post-mortem tissue vs. stereotactic brain biopsy) or sectioned and stained[89], as well as the inherent heterogeneity of disease activity observed across the disease spectrum[56,92].
Pathologically, subpial cortical lesions are distinct from other demyelinating plaques in that they do not form around a central vein and are relatively devoid of parenchymal and perivascular infiltrates[15,50]. Indeed, multiple studies have demonstrated that these lesions form independently of any vascular territory[15,50]. Instead, the demyelination extends from the upper cortical layer (most superficial cortical layer I) to involve several or occasionally all cortical layers, with lesions commonly involving multiple adjacent gyri[15,44,45,49,51,93]. A seminal study by Magliozzi et al.[45] revealed that immune cell collections in the meninges (some with follicle-like features) are associated with areas of adjacent subpial cortical demyelination. The finding that subpial lesions exhibit a surface-in gradient of neuronal injury and microglial activation suggests that one or more toxic soluble factor(s) may be released by immune cells in the meninges and diffuse across the CSF to damage the glia limitans and underlying cortex[44,45,93-95]. Indeed, subpial cortical lesions and adjacent immune-cell meningeal infiltrates have a predilection for the deep invaginations of the cortex, including the cingulate and insular cortices, where CSF stasis may allow more prolonged exposure to such putative toxic molecules[45,94].
It has consistently been shown that patients with a greater degree of meningeal inflammation and more pronounced subpial cortical pathology also have a more rapid and severe clinical course, characterized by younger ages at disease onset, wheelchair dependence, and death[44,57,93,94], with the extent of subpial cortical lesions also correlating with both physical and cognitive disability[70,96]. Taken together, these findings raise several potentially clinically relevant questions: when and how are meningeal immune cell collections formed; how are they maintained in the MS CNS; and what are they releasing that may mediate damage to the subjacent cortex. All these questions are discussed in greater detail below.
An additional intriguing line of investigation pertains to the specificity of this pathological finding (and the presumed associated mechanism of injury) to MS. Subpial lesions were previously considered unique to MS, as they had not observed in a range of other neuroinflammatory or neurodegenerative conditions[21,97,98] until the recent report describing their presence in autopsy and biopsy specimens from patients with myelin oligodendrocyte glycoprotein (MOG) antibody-associated disorders (MOGAD)[99]. Whether the features of cortical injury in MOGAD are identical to those seen in MS remains to be fully elucidated. This initial neuropathological investigation in MOGAD indicated that different cortical lesion subtypes predominate in the two conditions (intracortical in MOGAD vs. leukocortical and subpial in MS), though it should be noted that all but two MOGAD samples in this cohort were obtained via biopsy, which may influence estimates of the relative frequency and activity of cortical lesion subtypes observed[99]. The authors did not comment on whether the subpial lesions in MOGAD exhibited a surface-in gradient of inflammation and injury or whether their presence was associated with overlying meningeal immune-cell collections. Further comparative studies of cortical pathology and meningeal inflammation in MS and MOGAD will likely help elucidate important pathogenic mechanisms, both shared and unique, in these two disorders.
Mechanisms promoting initiation and maintenance of meningeal inflammation
Some degree of meningeal inflammation is now recognized to be a common feature in the MS CNS through all stages of disease, and the more prominent immune cell collections have been associated with greater underlying damage in the cerebral[44,45,88,93,94] and cerebellar[58] cortices as well as in the spinal cord[100]. It should be recognized that methodological differences in tissue preservation, sectioning, and staining as well as differences in disease presentation and phenotype, may all contribute to the differing estimates of the extent and profiles of meningeal immune cell aggregates in MS[13,101,102]. More diffuse (i.e., less organized) meningeal inflammation generally comprises CD20+ B cells and plasma cells/plasmablasts as well as T cells and activated macrophages[44,45,95,103]. A distinction should be made between diffuse meningeal inflammation and B cell-rich follicle-like structures, which are less common, smaller, and more highly structured aggregates. Though the more organized meningeal inflammation in MS can recapitulate some features of lymphoid follicles (hence referred to by some as follicle-like structures), they relatively infrequently recapitulate all features that would qualify them formally as tertiary lymphoid tissue. They are often B cell rich, comprising proliferating CD20+ B cells, plasma cells, and CD4+ and CD8+ T cells in a network of CD35+ follicular dendritic cells[44,94,95]. The more organized meningeal infiltrates have been identified in a subset of patients with both relapsing and progressive clinical phenotypes of MS, who seem to have a more severe clinical course[44,45,57,94,95].
Evidence from both MS patients and experimental autoimmune encephalomyelitis (EAE) models has implicated pro-inflammatory cytokines and lymphocyte chemoattractants in the formation and maintenance of immune cell aggregates in the meninges[104]. Specifically, molecules promoting B cell recruitment and survival, such as CXCL13 and B cell activating factor (BAFF), are expressed by dendritic cells in the meninges[44,95] and by infiltrating lymphocytes and astrocytes within active MS[105,106] and EAE lesions[107]. Increased levels of BAFF and CXCL13 have also been reported in the CSF of patients with MS, particularly in SPMS patients with superimposed relapses or in RRMS patients during relapse[108]. Additionally, TNFa and IFNg[55,96,109-112] as well as Th17[111,113,114] signaling have been consistently implicated in the formation and maintenance of immune cell aggregates in EAE and potentially MS. Expanding on these findings, a recent study identified increased expression of the pro-inflammatory cytokines IFNg, TNFa, IL-2, and IL-22, as well as the molecules CXCL13, CXCL10, LTa, IL-6, and IL-10 in the meninges and CSF of SPMS cases with more pronounced meningeal inflammation and cortical pathology at time of death[112]. Similar patterns of inflammation were then demonstrated in the CSF from two independent cohorts of RRMS patients with more significant cortical injury at diagnosis, suggesting that these key inflammatory mediators may be important for the development and propagation of meningeal inflammation and potentially also cortical damage across all phases of disease[112]. The cellular source(s) of the abnormally increased levels of pro-inflammatory cytokines in the CSF of MS, and whether they are produced within the inflamed CNS or in the meningeal compartment, remain unknown.
As with in vivo monitoring of cortical lesions, understanding the natural history and evolution of meningeal inflammation in the MS CNS is greatly limited by the availability of an appropriate biomarker. Identifying foci of leptomeningeal contrast enhancement (LME) on MRI has been studied as a potential proxy for meningeal inflammatory aggregates, albeit with some limitations and conflicting results[115-117]. In one prospective imaging study, two progressive MS patients followed ante-mortem ultimately presented for autopsy, where it was possible to correlate in vivo imaging data with post-mortem 7T MRI and immunohistochemistry[118]. In these patients, MRI foci of LME present antemortem correlated with regions of meningeal T cell, B cell, and macrophage aggregates identified in the post-mortem tissue[118]. Of note, these meningeal aggregates were associated with regions of more pronounced cortical demyelination. Several other studies have since demonstrated an association between foci of LME in vivo and more pronounced cortical atrophy[115,117,119], while others have failed to show an association[116]. It is important to note that the described structures with LME, which are visible to the eye, are of course orders of magnitude larger than microscopic aggregates of meningeal inflammation. The frequency of detection of LME is also relatively low compared to the relatively common meningeal immune cell aggregates, suggesting that if LME indeed represents actual sites of immune cell aggregates, they may be identifying relatively atypical ones, or just “the tip of the iceberg”. Additionally, LME is not specific to MS and has been described in other neuroinflammatory conditions[120]. Further study of LME and/or development of a separate imaging biomarker are greatly needed to better characterize and evaluate treatment effects on meningeal immune cell aggregates, which may play key pathophysiological roles in progressive disease mechanisms throughout the disease course.
Mechanisms by which meningeal immune cell aggregates may drive subpial cortical injury
Absence of direct contact between meningeal immune cells and the subjacent demyelinating cortex has drawn attention to the potential for soluble factors released by cells in the meninges to mediate injury to underlying cortical structures both directly and indirectly. For example, soluble factors released by meningeal immune cells could influence the transcriptional profile of underlying neurons and oligodendrocytes in such a way that increases their susceptibility to injury. A recent study using archival MS tissue specimens demonstrated that meningeal inflammation was associated with selective loss of CUX2-expressing excitatory neurons in the upper layers of the cortex, while other nearby excitatory and inhibitory neurons were relatively spared[80]. These vulnerable neurons upregulated genes for self-antigen presentation, including HLA-C and b-2 microglobulin, which may have been responsible for their increased susceptibility to injury. Similarly, another recent study in post-mortem SPMS tissue demonstrated that cortical neurons and oligodendrocytes in the brains of those with more pronounced meningeal inflammation, compared to those without, were found to exhibit increased expression of genes implicated in pro-apoptotic and pro-necroptotic signaling vs. pro-survival pathways[92]. The finding that many of these differentially expressed genes are regulated by TNF receptor signaling suggests that perhaps the presence of meningeal immune aggregates increases the sensitivity of cortical neurons and oligodendrocytes to TNF-mediated cell death[92]. Indeed, continuous expression of TNFa and IFNg in the meningeal compartment of MOG-immunized DA rats, delivered via lentiviral transfer vectors, recapitulated many aspects of the relationship between meningeal inflammation, subpial demyelination, and neuronal loss (including the upregulation of necroptotic pathways) observed in the post-mortem tissue of MS patients[121]. Further support comes from in vitro experiments in which rat primary neurons exposed to combinations of pro-inflammatory Th1- and monocyte/macrophage-derived cytokines exhibited altered gene expression in key pathways involved in neuronal health and apoptosis[122]. The fact that many rodent models of inflammatory cortical injury often incorporate injection[55,109,110,123] or targeted delivery[121,124] of TNFa and IFNg into the CSF or cortex indicates that these cytokines may be directly responsible for the cortical damage observed in these models, and perhaps also in MS patients, especially if a subset of cells exhibit increased susceptibility to cell death, as described above[80,92]. That being said, the introduction of these cytokines into the CNS compartment is also associated with meningeal inflammation in these models, and the observed pathology may be attributable to indirect effects on other cells[125,126].
Beyond traditionally implicated molecules, a series of studies have demonstrated that products released into the medium by cultured MS patient-derived B cells were selectively cytotoxic in vitro to rat and human neurons and rat oligodendrocytes, independent of measured cytokine, immunoglobulin, or complement activation[127,128]. The observed effect was attributable, at least in part, to B cell-derived extracellular vesicles (EVs)[129]. EVs have been increasingly recognized for their importance in intercellular communication within both the healthy and diseased CNS[130-133], and increased levels of EVs have been demonstrated in the CSF of MS patients[134]. EVs carry protein, lipid, and RNA cargo and could thus mediate toxicity in multiple ways. For instance, another in vitro study demonstrated that CSF from MS patients induced bioenergetic failure in rat neurons, again independent of levels of pro-inflammatory cytokines or other metabolites[135]. In this case, toxicity was mediated by elevated levels of certain ceramide species that are known to be enriched on the membranes of EVs[136]. It is also worth noting that, despite the emphasis above on toxic mediators, MS CSF and/or EVs may actually contain deficient levels of protective factors (i.e., an imbalance between injury and repair signals). Further study is warranted to better characterize the potential role of soluble CSF factors, including EVs, in the development of or protection from cortical damage in MS as well as the molecular pathways involved to identify potential novel therapeutic targets that may slow or prevent disease progression.
Another potential mechanism by which B cells may be involved in cortical injury relates to the description of EBV-infected plasma cells in both the MS meninges and parenchyma and their association with cytotoxic T cells in the perivascular cuffs of actively demyelinating lesions[137]. This suggests that EBV reactivation and the resulting antiviral response by CD8+ T cells within both the perivascular and meningeal compartments may propagate inflammation and mediate damage to the cortical tissue[57,137]. Additional mechanisms by which CD4+ and CD8+ T cells may injure neurons and oligodendrocytes have been described extensively in the context of WMLs, where both antigen-specific and antigen-independent mechanisms have been invoked (Reviewed[138]). It is reasonable to assume that similar mechanisms may take place in the cortex, albeit to a lesser extent given the relative paucity of parenchymal infiltration in cortical lesions.
Pro-inflammatory factors released into the CSF may also indirectly lead to cortical damage by activating CNS-resident cells, such as microglia and astrocytes[88,109,112,139-141]. It was previously shown that particular combinations of cytokines, defined by characteristic Th1, Th2, and monocyte/macrophage responses, greatly altered gene expression of MS relevant pathways in rat glial cultures[139-141]. Specifically, genes involved in antigen presentation, neuroprotection, axon-glial interactions, and metabolism were differentially regulated, suggesting that the activation state of nearby immune cells could impact the phenotype and likely also the function of glia in the MS CNS. Indeed, microglial activation is more robust in the cortex of individuals with more pronounced meningeal inflammation[88,94], and these activated microglia are tightly associated with ongoing demyelination, axonal transections, and neuronal loss[27,28,49]. Microglia are themselves thought to propagate injury either directly by the release of pro-inflammatory cytokines[125] or indirectly by exacerbating oxidative stress[142-144], excitotoxicity[125,145], and synaptic dysfunction[125,145] in the surrounding cortex. Meanwhile, activated astrocytes may also promote CNS injury by influencing the recruitment and activation states of infiltrating and CNS-resident immune cells[126,146,147] and, more directly, by reducing the crucial metabolic support available to surrounding neurons[148]. Injured astrocytes may also compromise the barrier function of the glia limitans and thus increase the susceptibility of the cortex to injury[13,45]. Furthermore, a certain population of pro-inflammatory astrocytes that was recently implicated in EAE-related CNS inflammation was also identified in the cortex of MS patients[149]. To what extent these or other populations of astrocytes drive cortical injury and progressive disease biology in MS patients is currently unknown, though in the context of EAE, it seems that activated astrocytes may be particularly pathogenic in later stages of disease[146].
Overall, multiple mechanisms are likely to drive cortical injury in MS, and the heterogeneity of progression observed in patients both clinically and pathologically suggests that the predominant process(es) at play may differ across patients as well as within any given patient over time.
Demyelination vs. neurodegeneration in the MS cortex
While axonal transections and neuronal loss are associated with demyelination in WMLs[150], the relationship between demyelination and neuronal injury is less consistently described in the cortex of MS patients[33,45,49,91,145,151]. One possible explanation is that cortical neuronal loss results predominantly from the degeneration of axons injured in classic WMLs, rather than injury within the cortex itself. While this phenomenon has been shown to predominate in certain regions of the cortex, including leukocortical lesions[15] and areas with extended axonal projections[152,153], a growing body of literature now indicates that mechanisms driving neuronal injury in other areas of the cortex cannot be explained by retrograde degeneration alone[15,69,154]. In support of this, a recent post-mortem analysis defined a pathological subtype of MS, termed “myelocortical MS”, in which patients have typical spinal cord pathology and cortical injury in the absence of classic WMLs[91]. The substantial degree of cortical neuronal loss present in these patients suggests that cortical injury can occur independent of WMLs and that certain patients may be more or less susceptible to these different mechanisms.
The question then remains as to what additional pathogenic mechanisms are at play that culminate in the profound degree of cortical atrophy observed in MS patients over time. Demyelination itself may contribute to cortical neurodegeneration to a certain extent via loss of trophic support to axons[155,156], reduced insulation from the local inflammation milieu[49,156], and disruption of efficient signal transduction and energy production[156,157], which together can result in axonal degeneration and cell death. However, several studies have failed to demonstrate a robust association between demyelination and neuronal/axonal loss in the cortex of MS patients[33,45,91,151]. While neuronal density was in fact reduced in demyelinated cortical lesions compared to control cortex, significant neuronal loss was also observed in the normal-appearing gray matter (NAGM)[33,45]. Though this finding indicates a dissociation between neurodegeneration and demyelination in the cortex, it also suggests there may exist a shared mechanism that contributes to cortical neurodegeneration more globally. Indeed, more pronounced meningeal inflammation is not only associated with subpial cortical lesions, but also with a gradient of microglial activation and neuronal loss in the NAGM[44,45]. It may be that the mechanisms by which meningeal inflammation drives subpial cortical injury, as described in the preceding sections, are also relevant to more diffuse cortical neuronal loss in MS, especially in the more superficial layers. It is interesting to note that several pathological differences between the superficial and deep layers of the cortex have been described in relation to neurodegeneration. Meningeal inflammation is generally associated with more superficial injury, while consequences of retrograde degeneration are seen in the deeper layers. Additionally, complement deposition is more extensive in cortical layers V and VI and is associated with greater neuronal loss, though notably independent of the extent of underlying WMLs[158,159]. Furthermore, differences in mitochondrial abnormalities can be seen with varying cortical depth[160]. Taken together, there are likely multiple processes that influence cortical neurodegeneration and the relative contribution of these processes may vary by cortical region (i.e., cortical lesions vs. NAGM, superficial vs. deep layers, etc.). It will be critical to disentangle exactly which mechanisms contribute to cortical demyelination and these different aspects of neurodegeneration in the future, as such discoveries may guide the development of distinct or perhaps complementary treatment approaches for progressive disease.
Importance of immune-CNS crosstalk in cortical injury
Several lines of evidence support the concept that CNS-resident cells and infiltrating immune cells, whether contained within the perivascular spaces or meninges, communicate bidirectionally to both propagate CNS-compartmentalized inflammation and incite damage in the cortical parenchyma. Regarding the compartmentalized immune response, meningeal immune cell collections have been associated with more pronounced perivascular inflammation in the cortex, which may reflect greater meningeal inflammation solicited in response to more perivascular inflammation in the subjacent tissue, or alternatively, may indicate that more meningeal inflammation promotes greater immune cell perivascular infiltration in the underlying cortex via upregulation of certain chemokines and/or adhesion molecules on brain or endothelial cells[57].
Next, with regard to cortical damage, B cells and other immune cells in the meninges (or in Virchow-Robin spaces) may produce factors that diffuse into the parenchyma and indirectly or directly induce cortical demyelination and neurodegeneration[45,80,92,127,128,161]. For example, secreted factors from immune cells may result in activated microglia that have been associated with ongoing demyelination and axonal injury in cortical lesions, most commonly at the lesion rim[44,49,94], as well as in the NAGM[44,45]. Though microglial activation in response to a single insult may be generated as a compensatory response to acute CNS injury, it is thought that the homeostatic repair capacity of chronically activated microglia in the MS CNS is impaired[125,162]. Indeed, chronically activated microglia, as well as surrounding astrocytes, produce ROS and other toxic metabolites that lead to a number of metabolic derangements that impact neuronal viability[13,49,125]. For instance, ROS can directly damage neuronal and axonal mitochondria by inducing mitochondrial DNA (mtDNA) deletions and by reducing the expression of key enzymes in the respiratory chain complex[160,163,164]. Impaired and energetically inefficient mitochondria are then primed to produce more ROS, resulting in a deleterious feedforward mechanism[165]. Furthermore, the increased metabolic demand in the demyelinated axon, coupled with the surrounding inflammatory milieu, has been shown to amplify mtDNA deletions[160,166] and further compounds the intracellular bioenergetic strain, resulting in cell death[167]. Activated microglia and astrocytes have also been shown to propagate excitotoxic damage in cortical neurons[125,126,145]. Specifically, in post-mortem tissue from progressive MS patients, activated microglia were associated with decreased expression of excitatory amino acid transporters (EEAT1 and EEAT2), which are critical for maintaining low extracellular glutamate concentration[145]. The loss of the protective mechanism was associated with increased axonal and synaptic damage in the cortex of progressive MS patients[145]. Taken together, microglial and astrocytic activation, which is increased in the brains of those with more pronounced meningeal inflammation, results in key changes in important aspects of neuronal metabolism that can perpetuate or perhaps incite neurodegeneration. It follows then that more pronounced meningeal inflammation is associated with increased microglial/astrocytic activation and more pronounced axonal injury[21]. However, it should be noted that certain metabolic changes, such as those associated with mitochondrial damage, have been observed in the cortex of MS patients without pronounced meningeal inflammation[160], suggesting that other perhaps non-inflammatory mechanisms may contribute to progressive disease biology as well.
Nonetheless, the communication between meningeal immune collections and CNS cells is likely bi-directional in that activated cells within the CNS, including microglia, astrocytes, and perivascular infiltrates, signal back to both immune and stromal cells in the meninges to promote lymphocyte retention within the meningeal compartment[104-106,168,169]. Together, this complex network of cell-cell communication constitutes the dynamic CNS-compartmentalized immune responses that are likely central to disease progression.
Implications for therapeutic intervention and future directions
The literature often refers to relapsing MS disease mechanisms as “inflammatory” and progressive disease mechanisms as “degenerative”. However, the now well-documented association between meningeal inflammation, cortical injury, and clinical progression in MS points to underlying inflammatory mechanisms that may be viable targets for therapeutic intervention in progressive MS. This does not preclude an effort to further study as well as target degenerative disease mechanisms, but suggests that different types of inflammation, playing out in different anatomic compartments, contribute to distinct processes of CNS injury throughout the MS course.
With the recent approval of three drugs for the treatment of progressive forms of MS -ocrelizumab for PPMS and siponimod and cladribine for SPMS - it is of interest to consider the mechanisms by which these therapeutic benefits are mediated and, in particular, whether their ability to limit disease progression reflects, at least in part, targeting of CNS-compartmentalized immune responses relevant to cortical injury. A major challenge to establishing this is that each treatment has a robust effect on relapsing MS biology, which would be expected to limit relapse-related progression of disability. Here, we briefly consider how each agent may impact non-relapsing disease mechanisms.
Ocrelizumab, a humanized monoclonal antibody against CD20, was shown to have a modest but significant beneficial effect on limiting confirmed disability progression in both RRMS and PPMS patients[170,171]. Given the evidence supporting a pathogenic role of meningeal B cells in disease progression, it is interesting to speculate whether anti-CD20 therapy depletes these B cells in the CNS. While anti-CD20 therapy does reduce B cell counts in the CSF of MS patients[172], it is not clear to what extent this reflects peripheral B cell depletion and hence less ongoing trafficking of B cells into the CNS vs. a direct effect of the anti-CD20 antibody on depletion of CNS B cells. There is likely only a small amount of the antibody that accesses the CNS and the extent to which the necessary killing machinery is present in the CNS (i.e., to enable complement-mediated and/or antibody-dependent cellular cytotoxicity of B cells) is unknown. The apparent persistence of CSF oligoclonal bands in the face of anti-CD20 therapy (at least over the initial two years of therapy) suggests that certain B cell populations, particularly plasma cells and plasmablasts, are highly resistant to such B cell depletion therapy[173]. Indeed, intrathecal delivery of anti-CD20 therapy in SPMS patients did not substantially reduce focal leptomeningeal enhancement, which was used as a proxy for the effect on discrete meningeal immune cell collections[174]. The apparent resistance of certain B cell populations to depletion in the CNS may be responsible for ongoing progression even in the absence of relapse in these patients[175].
Siponimod, a sphingosine-1-phosphate receptor modulator, was approved for the treatment of active SPMS[176]. Siponimod, in addition to sequestering circulating immune cells in peripheral lymphoid structures, is expected to readily access the CNS where it may have a direct effect on CNS immune cells that express the S1P receptors, possibly contributing to CNS protection and/or repair, which to date has been demonstrated only in EAE[177]. A recent study using a T cell adoptive transfer model of EAE demonstrated that siponimod substantially reduced meningeal inflammation and subpial cortical injury via modulation of Th17 recruitment to and signaling within the CNS[114]. Notably, efficacy was highly dependent on the timing of drug initiation in that administration later in the disease course failed to prevent cortical damage[114]. This provides mechanistic insight into a plausible mechanism by which siponimod may exert beneficial effects on cortical pathology which, if true, would suggest that a greater effect on limiting progression may be achieved with earlier introduction of the therapy.
As with siponimod, the oral agent cladribine is approved for use in active SPMS patients[178]. Cladribine is a purine nucleoside analog that, when phosphorylated, accumulates in the nuclei of lymphocytes, resulting in DNA damage and eventually cell death[179]. Beyond selective depletion of lymphocytes, cladribine effectively penetrates the CNS where it could theoretically impact infiltrating and CNS-compartmentalized lymphocytes. Cladribine has also been shown to alter the inflammatory responses of microglia in vitro, such that they adopted a less activated phenotype[180]. Separately, in EAE models, intracerebroventricular administration of cladribine improved disease, in part, via a reduction in excitotoxicity[181]. While some studies have indicated that cladribine slows disability accumulation in both progressive and relapsing MS patients[178,182,183], others have failed to show a statistically significant effect. Further studies are needed to better elucidate the ways in which cladribine may slow non-relapsing disease progression.
Efforts are ongoing to better understand how current and emerging therapies may beneficially impact both the CNS-compartmentalized immune response and neurodegeneration, wherein therapies could either interfere with detrimental cell processes or promote remyelination and repair of neural elements[184,185]. It is also possible that a combination of therapies addressing both inflammatory and de- or re-generative pathways may provide the greatest benefit. Of note, therapies directed at CNS-compartmentalized inflammation, including meningeal immune cell aggregates, would provide a critical biological proof-of-principle in progressive disease. The capacity to develop and translate such therapies into clinical practice rests, in part, on our ability to (1) improve upon existing imaging modalities to enhance the detection and monitoring of cortical lesions in vivo, and (2) identify key molecules and cell subsets in the meninges or parenchyma responsible for promoting and protecting against ongoing damage.
Declarations
Authors’ contributionsManuscript and illustration preparation: Zuroff LR
Manuscript review and oversight: Benjamins JA, Bar-Or A, Lisak RP
Approval of the manuscript for publication: Zuroff LR, Benjamins JA, Bar-Or A, Lisak RP
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (TL1TR001880), the Melissa and Paul Anderson President’s Distinguished Professor (University of Pennsylvania), and the Parker Webber Chair in Neurology (DMC Foundation/Wayne State University School of Medicine). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of interestBar-Or A participated as a speaker in meetings sponsored by and received consulting fees and/or grant support from: Janssen/Actelion; Atara Biotherapeutics, Biogen Idec, Celgene/Receptos, Roche/Genentech, Medimmune, Merck/EMD Serono, Novartis, Sanofi-Genzyme. Lisak RP participated as a speaker in meetings sponsored by and received consulting fees and/or grant support from: Alexion, Argenx, Ra Pharmaceuticals, Novartis, Mallinckrodt, Catalyst, Teva Pharmaceuticals, Genentech, Chugai, Medimmune, GLG Consulting, Alpha Sites Consulting, Schlesinger Group Consulting, Slingshot Consulting, Health Sources, Adivo Associates, and Smart Analyst.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
3. Lorscheider J, Buzzard K, Jokubaitis V, Spelman T, Havrdova E, et al; MSBase Study Group. Defining secondary progressive multiple sclerosis. Brain 2016;139:2395-405.
4. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014;83:278-86.
5. Cree BAC, Hollenbach JA, Bove R, Kirkish G, Sacco S, et al; University of California, San Francisco MS-EPIC Team. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol 2019;85:653-66.
6. Stevenson VL, Smith SM, Matthews PM, Miller DH, Thompson AJ. Monitoring disease activity and progression in primary progressive multiple sclerosis using MRI: sub-voxel registration to identify lesion changes and to detect cerebral atrophy. J Neurol 2002;249:171-7.
7. Stevenson VL, Miller DH. Magnetic resonance imaging in the monitoring of disease progression in multiple sclerosis. Mult Scler 1999;5:268-72.
8. Stevenson VL, Miller DH, Leary SM, Rovaris M, Barkhof F, et al. One year follow up study of primary and transitional progressive multiple sclerosis. J Neurol Neurosurg Psychiatry 2000;68:713-8.
9. Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron 2018;97:742-68.
11. Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol 2018;9:3116.
12. Machado-Santos J, Saji E, Tröscher AR, Paunovic M, Liblau R, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 2018;141:2066-82.
13. Calabrese M, Magliozzi R, Ciccarelli O, Geurts JJ, Reynolds R, et al. Exploring the origins of grey matter damage in multiple sclerosis. Nat Rev Neurosci 2015;16:147-58.
14. Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 2015;78:710-21.
15. Haider L, Zrzavy T, Hametner S, Höftberger R, Bagnato F, et al. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 2016;139:807-15.
16. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005;128:2705-12.
17. Steinman L. Immunology of relapse and remission in multiple sclerosis. Annu Rev Immunol 2014;32:257-81.
18. Hochmeister S, Grundtner R, Bauer J, Engelhardt B, Lyck R, et al. Dysferlin is a new marker for leaky brain blood vessels in multiple sclerosis. J Neuropathol Exp Neurol 2006;65:855-65.
19. Kirk J, Plumb J, Mirakhur M, McQuaid S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J Pathol 2003;201:319-27.
20. Meinl E, Krumbholz M, Derfuss T, Junker A, Hohlfeld R. Compartmentalization of inflammation in the CNS: a major mechanism driving progressive multiple sclerosis. J Neurol Sci 2008;274:42-4.
21. Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009;132:1175-89.
22. van Horssen J, Brink BP, de Vries HE, van der Valk P, Bø L. The blood-brain barrier in cortical multiple sclerosis lesions. J Neuropathol Exp Neurol 2007;66:321-8.
23. Brennan MS, Matos MF, Li B, Hronowski X, Gao B, et al. Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro. PLoS One 2015;10:e0120254.
25. Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol 2010;33:91-101.
26. Hunter SF, Bowen JD, Reder AT. The direct effects of fingolimod in the central nervous system: implications for relapsing multiple sclerosis. CNS Drugs 2016;30:135-47.
27. Fischer MT, Wimmer I, Höftberger R, Gerlach S, Haider L, et al. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain 2013;136:1799-815.
28. Prineas JW, Kwon EE, Cho ES, Sharer LR, Barnett MH, et al. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol 2001;50:646-57.
29. Elliott C, Belachew S, Wolinsky JS, Hauser SL, Kappos L, et al. Chronic white matter lesion activity predicts clinical progression in primary progressive multiple sclerosis. Brain 2019;142:2787-99.
30. Elliott C, Wolinsky JS, Hauser SL, Kappos L, Barkhof F, et al. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult Scler 2019;25:1915-25.
31. Absinta M, Sati P, Fechner A, Schindler MK, Nair G, et al. Identification of chronic active multiple sclerosis lesions on 3T MRI. AJNR Am J Neuroradiol 2018;39:1233-8.
32. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 2012;8:647-56.
33. Klaver R, Popescu V, Voorn P, Galis-de Graaf Y, van der Valk P, et al. Neuronal and axonal loss in normal-appearing gray matter and subpial lesions in multiple sclerosis. J Neuropathol Exp Neurol 2015;74:453-8.
34. Herranz E, Giannì C, Louapre C, Treaba CA, Govindarajan ST, et al. Neuroinflammatory component of gray matter pathology in multiple sclerosis. Ann Neurol 2016;80:776-90.
35. Geurts JJ, Calabrese M, Fisher E, Rudick RA. Measurement and clinical effect of grey matter pathology in multiple sclerosis. Lancet Neurol 2012;11:1082-92.
36. Luchetti S, Fransen NL, van Eden CG, Ramaglia V, Mason M, et al. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol 2018;135:511-28.
37. Calabrese M, Poretto V, Favaretto A, Alessio S, Bernardi V, et al. Cortical lesion load associates with progression of disability in multiple sclerosis. Brain 2012;135:2952-61.
38. Scalfari A, Romualdi C, Nicholas RS, Mattoscio M, Magliozzi R, et al. The cortical damage, early relapses, and onset of the progressive phase in multiple sclerosis. Neurology 2018;90:e2107-18.
39. Filippi M, Rocca MA, Calabrese M, Sormani MP, Rinaldi F, et al. Intracortical lesions: relevance for new MRI diagnostic criteria for multiple sclerosis. Neurology 2010;75:1988-94.
40. Calabrese M, Romualdi C, Poretto V, Favaretto A, Morra A, et al. The changing clinical course of multiple sclerosis: a matter of gray matter. Ann Neurol 2013;74:76-83.
41. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med 2011;365:2188-97.
42. Treaba CA, Granberg TE, Sormani MP, Herranz E, Ouellette RA, et al. Longitudinal characterization of cortical lesion development and evolution in multiple sclerosis with 7.0-T MRI. Radiology 2019;291:740-9.
43. Giorgio A, Stromillo ML, Rossi F, Battaglini M, Hakiki B, et al. Cortical lesions in radiologically isolated syndrome. Neurology 2011;77:1896-9.
44. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007;130:1089-104.
45. Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, et al. A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 2010;68:477-93.
46. Filippi M, Rocca MA. Cortical lesions on 7-T MRI in multiple sclerosis: a window into pathogenetic mechanisms? Radiology 2019;291:750-1.
47. Filippi M, Preziosa P, Banwell BL, Barkhof F, Ciccarelli O, et al. Assessment of lesions on magnetic resonance imaging in multiple sclerosis: practical guidelines. Brain 2019;142:1858-75.
48. Schindler MK, Sati P, Reich DS. Insights from ultrahigh field imaging in multiple sclerosis. Neuroimaging Clin N Am 2017;27:357-66.
49. Peterson JW, Bö L, Mörk S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 2001;50:389-400.
50. Bø L, Vedeler CA, Nyland H, Trapp BD, Mørk SJ. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler 2003;9:323-31.
51. Bø L, Vedeler CA, Nyland HI, Trapp BD, Mørk SJ. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol 2003;62:723-32.
52. Strijbis EMM, Kooi EJ, van der Valk P, Geurts JJG. Cortical remyelination is heterogeneous in multiple sclerosis. J Neuropathol Exp Neurol 2017;76:390-401.
53. Chang A, Staugaitis SM, Dutta R, Batt CE, Easley KE, et al. Cortical remyelination: a new target for repair therapies in multiple sclerosis. Ann Neurol 2012;72:918-26.
54. Albert M, Antel J, Brück W, Stadelmann C. Extensive cortical remyelination in patients with chronic multiple sclerosis. Brain Pathol 2007;17:129-38.
55. Lagumersindez-Denis N, Wrzos C, Mack M, Winkler A, van der Meer F, et al. Differential contribution of immune effector mechanisms to cortical demyelination in multiple sclerosis. Acta Neuropathol 2017;134:15-34.
56. Kooi EJ, Strijbis EM, van der Valk P, Geurts JJ. Heterogeneity of cortical lesions in multiple sclerosis: clinical and pathologic implications. Neurology 2012;79:1369-76.
57. Magliozzi R, Serafini B, Rosicarelli B, Chiappetta G, Veroni C, et al. B-cell enrichment and Epstein-Barr virus infection in inflammatory cortical lesions in secondary progressive multiple sclerosis. J Neuropathol Exp Neurol 2013;72:29-41.
58. Howell OW, Schulz-Trieglaff EK, Carassiti D, Gentleman SM, Nicholas R, et al. Extensive grey matter pathology in the cerebellum in multiple sclerosis is linked to inflammation in the subarachnoid space. Neuropathol Appl Neurobiol 2015;41:798-813.
59. Nelson F, Datta S, Garcia N, Rozario NL, Perez F, et al. Intracortical lesions by 3T magnetic resonance imaging and correlation with cognitive impairment in multiple sclerosis. Mult Scler 2011;17:1122-9.
60. Sethi V, Yousry TA, Muhlert N, Ron M, Golay X, et al. Improved detection of cortical MS lesions with phase-sensitive inversion recovery MRI. J Neurol Neurosurg Psychiatry 2012;83:877-82.
61. Crespy L, Zaaraoui W, Lemaire M, Rico A, Faivre A, et al. Prevalence of grey matter pathology in early multiple sclerosis assessed by magnetization transfer ratio imaging. PLoS One 2011;6:e24969.
62. Rovaris M, Judica E, Ceccarelli A, Ghezzi A, Martinelli V, et al. A 3-year diffusion tensor MRI study of grey matter damage progression during the earliest clinical stage of MS. J Neurol 2008;255:1209-14.
63. Beck ES, Sati P, Sethi V, Kober T, Dewey B, et al. Improved visualization of cortical lesions in multiple sclerosis using 7T MP2RAGE. AJNR Am J Neuroradiol 2018;39:459-66.
64. Nelson F, Poonawalla A, Hou P, Wolinsky JS, Narayana PA. 3D MPRAGE improves classification of cortical lesions in multiple sclerosis. Mult Scler 2008;14:1214-9.
65. Kilsdonk ID, Jonkman LE, Klaver R, van Veluw SJ, Zwanenburg JJ, et al. Increased cortical grey matter lesion detection in multiple sclerosis with 7 T MRI: a post-mortem verification study. Brain 2016;139:1472-81.
66. Mainero C, Benner T, Radding A, van der Kouwe A, Jensen R, et al. In vivo imaging of cortical pathology in multiple sclerosis using ultra-high field MRI. Neurology 2009;73:941-8.
67. Geurts JJ, Roosendaal SD, Calabrese M, Ciccarelli O, Agosta F, et al; MAGNIMS Study Group. Consensus recommendations for MS cortical lesion scoring using double inversion recovery MRI. Neurology 2011;76:418-24.
68. Herranz E, Louapre C, Treaba CA, Govindarajan ST, Ouellette R, et al. Profiles of cortical inflammation in multiple sclerosis by 11C-PBR28 MR-PET and 7 Tesla imaging. Mult Scler 2019;1352458519867320.
69. Harrison DM, Roy S, Oh J, Izbudak I, Pham D, et al. Association of cortical lesion burden on 7-T magnetic resonance imaging with cognition and disability in multiple sclerosis. JAMA Neurol 2015;72:1004-12.
70. Nielsen AS, Kinkel RP, Madigan N, Tinelli E, Benner T, et al. Contribution of cortical lesion subtypes at 7T MRI to physical and cognitive performance in MS. Neurology 2013;81:641-9.
71. Walsh M, Montojo CA, Sheu YS, Marchette SA, Harrison DM, et al. Object working memory performance depends on microstructure of the frontal-occipital fasciculus. Brain Connect 2011;1:317-29.
72. Kivisäkk P, Imitola J, Rasmussen S, Elyaman W, Zhu B, et al. Localizing central nervous system immune surveillance: meningeal antigen-presenting cells activate T cells during experimental autoimmune encephalomyelitis. Ann Neurol 2009;65:457-69.
73. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 2009;10:514-23.
74. Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain 2007;130:2800-15.
75. Henderson AP, Barnett MH, Parratt JD, Prineas JW. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol 2009;66:739-53.
76. Maranzano J, Rudko DA, Nakamura K, Cook S, Cadavid D, et al. MRI evidence of acute inflammation in leukocortical lesions of patients with early multiple sclerosis. Neurology 2017;89:714-21.
77. Kuhlmann T, Ludwin S, Prat A, Antel J, Brück W, et al. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol 2017;133:13-24.
78. van Wageningen TA, Vlaar E, Kooij G, Jongenelen CAM, Geurts JJG, et al. Regulation of microglial TMEM119 and P2RY12 immunoreactivity in multiple sclerosis white and grey matter lesions is dependent on their inflammatory environment. Acta Neuropathol Commun 2019;7:206.
79. Peferoen LA, Gerritsen WH, Breur M, Ummenthum KM, Peferoen-Baert RM, et al. Small heat shock proteins are induced during multiple sclerosis lesion development in white but not grey matter. Acta Neuropathol Commun 2015;3:87.
80. Schirmer L, Velmeshev D, Holmqvist S, Kaufmann M, Werneburg S, et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019;573:75-82.
81. Popescu BF, Pirko I, Lucchinetti CF. Pathology of multiple sclerosis: where do we stand? Continuum (Minneap Minn) 2013;19:901-21.
82. Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 2012;3:1227.
83. Lee NJ, Ha SK, Sati P, Absinta M, Luciano NJ, et al. Spatiotemporal distribution of fibrinogen in marmoset and human inflammatory demyelination. Brain 2018;141:1637-49.
84. Ryu JK, Petersen MA, Murray SG, Baeten KM, Meyer-Franke A, et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun 2015;6:8164.
85. Yates RL, Esiri MM, Palace J, Jacobs B, Perera R, et al. Fibrin(ogen) and neurodegeneration in the progressive multiple sclerosis cortex. Ann Neurol 2017;82:259-70.
86. Magliozzi R, Hametner S, Facchiano F, Marastoni D, Rossi S, et al. Iron homeostasis, complement, and coagulation cascade as CSF signature of cortical lesions in early multiple sclerosis. Ann Clin Transl Neurol 2019;6:2150-63.
87. Davalos D, Mahajan KR, Trapp BD. Brain fibrinogen deposition plays a key role in MS pathophysiology - Yes. Mult Scler 2019;25:1434-5.
88. Bevan RJ, Evans R, Griffiths L, Watkins LM, Rees MI, et al. Meningeal inflammation and cortical demyelination in acute multiple sclerosis. Ann Neurol 2018;84:829-42.
89. Griffiths L, Reynolds R, Evans R, Bevan RJ, Rees MI, et al. Substantial subpial cortical demyelination in progressive multiple sclerosis: have we underestimated the extent of cortical pathology? Neuroimmunol Neuroinflammation 2020;7:51-67.
90. Calabrese M, Gallo P. Magnetic resonance evidence of cortical onset of multiple sclerosis. Mult Scler 2009;15:933-41.
91. Trapp BD, Vignos M, Dudman J, Chang A, Fisher E, et al. Cortical neuronal densities and cerebral white matter demyelination in multiple sclerosis: a retrospective study. Lancet Neurol 2018;17:870-84.
92. Magliozzi R, Howell OW, Durrenberger P, Aricò E, James R, et al. Meningeal inflammation changes the balance of TNF signalling in cortical grey matter in multiple sclerosis. J Neuroinflammation 2019;16:259.
93. Choi SR, Howell OW, Carassiti D, Magliozzi R, Gveric D, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 2012;135:2925-37.
94. Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011;134:2755-71.
95. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 2004;14:164-74.
96. Farina G, Magliozzi R, Pitteri M, Reynolds R, Rossi S, et al. Increased cortical lesion load and intrathecal inflammation is associated with oligoclonal bands in multiple sclerosis patients: a combined CSF and MRI study. J Neuroinflammation 2017;14:40.
97. Junker A, Wozniak J, Voigt D, Scheidt U, Antel J, et al. Extensive subpial cortical demyelination is specific to multiple sclerosis. Brain Pathol 2020;30:641-52.
98. Moll NM, Rietsch AM, Ransohoff AJ, Cossoy MB, Huang D, et al. Cortical demyelination in PML and MS: Similarities and differences. Neurology 2008;70:336-43.
99. Höftberger R, Guo Y, Flanagan EP, Lopez-Chiriboga AS, Endmayr V, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol 2020;139:875-92.
100. Androdias G, Reynolds R, Chanal M, Ritleng C, Confavreux C, et al. Meningeal T cells associate with diffuse axonal loss in multiple sclerosis spinal cords. Ann Neurol 2010;68:465-76.
101. Calabrese M, Agosta F, Rinaldi F, Mattisi I, Grossi P, et al. Cortical lesions and atrophy associated with cognitive impairment in relapsing-remitting multiple sclerosis. Arch Neurol 2009;66:1144-50.
102. Roosendaal SD, Moraal B, Pouwels PJ, Vrenken H, Castelijns JA, et al. Accumulation of cortical lesions in MS: relation with cognitive impairment. Mult Scler 2009;15:708-14.
103. Kooi EJ, Geurts JJ, van Horssen J, Bø L, van der Valk P. Meningeal inflammation is not associated with cortical demyelination in chronic multiple sclerosis. J Neuropathol Exp Neurol 2009;68:1021-8.
104. Pikor NB, Prat A, Bar-Or A, Gommerman JL. Meningeal tertiary lymphoid tissues and multiple sclerosis: a gathering place for diverse types of immune cells during CNS autoimmunity. Front Immunol 2015;6:657.
105. Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisäkk P, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 2006;129:200-11.
106. Krumbholz M, Theil D, Derfuss T, Rosenwald A, Schrader F, et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med 2005;201:195-200.
107. Magliozzi R, Columba-Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol 2004;148:11-23.
108. Ragheb S, Li Y, Simon K, Vanhaerents S, Galimberti D, et al. Multiple sclerosis: BAFF and CXCL13 in cerebrospinal fluid. Mult Scler 2011;17:819-29.
109. Gardner C, Magliozzi R, Durrenberger PF, Howell OW, Rundle J, et al. Cortical grey matter demyelination can be induced by elevated pro-inflammatory cytokines in the subarachnoid space of MOG-immunized rats. Brain 2013;136:3596-608.
110. Merkler D, Ernsting T, Kerschensteiner M, Brück W, Stadelmann C. A new focal EAE model of cortical demyelination: multiple sclerosis-like lesions with rapid resolution of inflammation and extensive remyelination. Brain 2006;129:1972-83.
111. Pikor NB, Astarita JL, Summers-Deluca L, Galicia G, Qu J, et al. Integration of Th17- and lymphotoxin-derived signals initiates meningeal-resident stromal cell remodeling to propagate neuroinflammation. Immunity 2015;43:1160-73.
112. Magliozzi R, Howell OW, Nicholas R, Cruciani C, Castellaro M, et al. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann Neurol 2018;83:739-55.
113. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity 2011;35:986-96.
114. Ward LA, Lee DS, Sharma A, Wang A, Naouar I, et al. Siponimod therapy implicates Th17 cells in a preclinical model of subpial cortical injury. JCI Insight 2020;5:132522.
115. Harrison DM, Wang KY, Fiol J, Naunton K, Royal W 3rd, et al. Leptomeningeal Enhancement at 7T in Multiple Sclerosis: Frequency, Morphology, and Relationship to Cortical Volume. J Neuroimaging 2017;27:461-8.
116. Ighani M, Jonas S, Izbudak I, Choi S, Lema-Dopico A, et al. No association between cortical lesions and leptomeningeal enhancement on 7-Tesla MRI in multiple sclerosis. Mult Scler 2020;26:165-76.
117. Zurawski J, Tauhid S, Chu R, Khalid F, Healy BC, et al. 7T MRI cerebral leptomeningeal enhancement is common in relapsing-remitting multiple sclerosis and is associated with cortical and thalamic lesions. Mult Scler 2020;26:177-87.
118. Absinta M, Vuolo L, Rao A, Nair G, Sati P, et al. Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis. Neurology 2015;85:18-28.
119. Zivadinov R, Ramasamy DP, Vaneckova M, Gandhi S, Chandra A, et al. Leptomeningeal contrast enhancement is associated with progression of cortical atrophy in MS: a retrospective, pilot, observational longitudinal study. Mult Scler 2017;23:1336-45.
120. Absinta M, Cortese IC, Vuolo L, Nair G, de Alwis MP, et al. Leptomeningeal gadolinium enhancement across the spectrum of chronic neuroinflammatory diseases. Neurology 2017;88:1439-44.
121. James RE, Schalks R, Browne E, Eleftheriadou I, Munoz CP, et al. Persistent elevation of intrathecal pro-inflammatory cytokines leads to multiple sclerosis-like cortical demyelination and neurodegeneration. Acta Neuropathol Commun 2020;8:66.
122. Lisak RP, Nedelkoska L, Studzinski D, Bealmear B, Xu W, et al. Cytokines regulate neuronal gene expression: differential effects of Th1, Th2 and monocyte/macrophage cytokines. J Neuroimmunol 2011;238:19-33.
123. Üçal M, Haindl MT, Adzemovic MZ, Strasser J, Theisl L, et al. Widespread cortical demyelination of both hemispheres can be induced by injection of pro-inflammatory cytokines via an implanted catheter in the cortex of MOG-immunized rats. Exp Neurol 2017;294:32-44.
124. Silva BA, Leal MC, Farías MI, Avalos JC, Besada CH, et al. A new focal model resembling features of cortical pathology of the progressive forms of multiple sclerosis: Influence of innate immunity. Brain Behav Immun 2018;69:515-31.
125. Correale J. The role of microglial activation in disease progression. Mult Scler 2014;20:1288-95.
126. Correale J, Farez MF. The role of astrocytes in multiple sclerosis progression. Front Neurol 2015;6:180.
127. Lisak RP, Benjamins JA, Nedelkoska L, Barger JL, Ragheb S, et al. Secretory products of multiple sclerosis B cells are cytotoxic to oligodendroglia in vitro. J Neuroimmunol 2012;246:85-95.
128. Lisak RP, Nedelkoska L, Benjamins JA, Schalk D, Bealmear B, et al. B cells from patients with multiple sclerosis induce cell death via apoptosis in neurons in vitro. J Neuroimmunol 2017;309:88-99.
129. Benjamins JA, Nedelkoska L, Touil H, Stemmer PM, Carruthers NJ, et al. Exosome-enriched fractions from MS B cells induce oligodendrocyte death. Neurol Neuroimmunol Neuroinflamm 2019;6:e550.
130. Selmaj I, Cichalewska M, Namiecinska M, Galazka G, Horzelski W, et al. Global exosome transcriptome profiling reveals biomarkers for multiple sclerosis. Ann Neurol 2017;81:703-17.
131. Basso M, Bonetto V. Extracellular vesicles and a novel form of communication in the brain. Front Neurosci 2016;10:127.
132. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 2014;14:195-208.
133. Ebrahimkhani S, Vafaee F, Young PE, Hur SSJ, Hawke S, et al. Exosomal microRNA signatures in multiple sclerosis reflect disease status. Sci Rep 2017;7:14293.
134. Pieragostino D, Cicalini I, Lanuti P, Ercolino E, di Ioia M, et al. Enhanced release of acid sphingomyelinase-enriched exosomes generates a lipidomics signature in CSF of Multiple Sclerosis patients. Sci Rep 2018;8:3071.
135. Vidaurre OG, Haines JD, Katz Sand I, Adula KP, Huynh JL, et al. Cerebrospinal fluid ceramides from patients with multiple sclerosis impair neuronal bioenergetics. Brain 2014;137:2271-86.
136. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008;319:1244-7.
137. Serafini B, Rosicarelli B, Veroni C, Mazzola GA, Aloisi F. Epstein-Barr virus-specific CD8 T cells selectively infiltrate the brain in multiple sclerosis and interact locally with virus-infected cells: clue for a virus-driven immunopathological mechanism. J Virol 2019;93:e00980-19.
138. Kaskow BJ, Baecher-Allan C. Effector T cells in multiple sclerosis. Cold Spring Harb Perspect Med 2018;8:a029025.
139. Lisak RP, Benjamins JA, Bealmear B, Nedelkoska L, Studzinski D, et al. Differential effects of Th1, monocyte/macrophage and Th2 cytokine mixtures on early gene expression for molecules associated with metabolism, signaling and regulation in central nervous system mixed glial cell cultures. J Neuroinflammation 2009;6:4.
140. Lisak RP, Benjamins JA, Bealmear B, Nedelkoska L, Yao B, et al. Differential effects of Th1, monocyte/macrophage and Th2 cytokine mixtures on early gene expression for glial and neural-related molecules in central nervous system mixed glial cell cultures: neurotrophins, growth factors and structural proteins. J Neuroinflammation 2007;4:30.
141. Lisak RP, Benjamins JA, Bealmear B, Yao B, Land S, et al. Differential effects of Th1, monocyte/macrophage and Th2 cytokine mixtures on early gene expression for immune-related molecules by central nervous system mixed glial cell cultures. Mult Scler 2006;12:149-68.
142. Haider L, Fischer MT, Frischer JM, Bauer J, Höftberger R, et al. Oxidative damage in multiple sclerosis lesions. Brain 2011;134:1914-24.
143. Gray E, Thomas TL, Betmouni S, Scolding N, Love S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol 2008;18:86-95.
144. Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 2011;17:495-9.
145. Vercellino M, Merola A, Piacentino C, Votta B, Capello E, et al. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol 2007;66:732-9.
146. Mayo L, Trauger SA, Blain M, Nadeau M, Patel B, et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med 2014;20:1147-56.
147. Quintana FJ. Astrocytes play a crucial role in the formation and evolution of MS lesions - Commentary. Mult Scler 2019;25:19-20.
148. Chao CC, Gutiérrez-Vázquez C, Rothhammer V, Mayo L, Wheeler MA, et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 2019;179:1483-98.e22.
149. Wheeler MA, Clark IC, Tjon EC, Li Z, Zandee SEJ, et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020;578:593-9.
150. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998;338:278-85.
151. Carassiti D, Altmann DR, Petrova N, Pakkenberg B, Scaravilli F, et al. Neuronal loss, demyelination and volume change in the multiple sclerosis neocortex. Neuropathol Appl Neurobiol 2018;44:377-90.
152. Jurkiewicz MT, Crawley AP, Verrier MC, Fehlings MG, Mikulis DJ. Somatosensory cortical atrophy after spinal cord injury: a voxel-based morphometry study. Neurology 2006;66:762-4.
153. Sepulcre J, Goñi J, Masdeu JC, Bejarano B, Vélez de Mendizábal N, et al. Contribution of white matter lesions to gray matter atrophy in multiple sclerosis: evidence from voxel-based analysis of T1 lesions in the visual pathway. Arch Neurol 2009;66:173-9.
154. Fisher E, Lee JC, Nakamura K, Rudick RA. Gray matter atrophy in multiple sclerosis: a longitudinal study. Ann Neurol 2008;64:255-65.
156. Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci 2008;31:535-61.
157. Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol 2009;8:280-91.
158. Watkins LM, Neal JW, Loveless S, Michailidou I, Ramaglia V, et al. Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J Neuroinflammation 2016;13:161.
159. Brink BP, Veerhuis R, Breij EC, van der Valk P, Dijkstra CD, et al. The pathology of multiple sclerosis is location-dependent: no significant complement activation is detected in purely cortical lesions. J Neuropathol Exp Neurol 2005;64:147-55.
160. Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 2011;69:481-92.
161. Cristofanilli M, Rosenthal H, Cymring B, Gratch D, Pagano B, et al. Progressive multiple sclerosis cerebrospinal fluid induces inflammatory demyelination, axonal loss, and astrogliosis in mice. Exp Neurol 2014;261:620-32.
162. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, et al. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017;140:1900-13.
163. Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012;135:886-99.
164. Correale J, Gaitán MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain 2017;140:527-46.
166. Campbell GR, Kraytsberg Y, Krishnan KJ, Ohno N, Ziabreva I, et al. Clonally expanded mitochondrial DNA deletions within the choroid plexus in multiple sclerosis. Acta Neuropathol 2012;124:209-20.
167. Campbell GR, Worrall JT, Mahad DJ. The central role of mitochondria in axonal degeneration in multiple sclerosis. Mult Scler 2014;20:1806-13.
168. Touil H, Kobert A, Lebeurrier N, Rieger A, Saikali P, et al; Canadian B Cell Team in MS. Human central nervous system astrocytes support survival and activation of B cells: implications for MS pathogenesis. J Neuroinflammation 2018;15:114.
169. Michel L, Touil H, Pikor NB, Gommerman JL, Prat A, et al. B cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS-compartmentalized inflammation. Front Immunol 2015;6:636.
170. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, et al; OPERA I and OPERA II Clinical Investigators. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2017;376:221-34.
171. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, et al; ORATORIO Clinical Investigators. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017;376:209-20.
172. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol 2006;180:63-70.
173. Petereit HF, Moeller-Hartmann W, Reske D, Rubbert A. Rituximab in a patient with multiple sclerosis--effect on B cells, plasma cells and intrathecal IgG synthesis. Acta Neurol Scand 2008;117:399-403.
174. Bhargava P, Wicken C, Smith MD, Strowd RE, Cortese I, et al. Trial of intrathecal rituximab in progressive multiple sclerosis patients with evidence of leptomeningeal contrast enhancement. Mult Scler Relat Disord 2019;30:136-40.
175. Greenfield AL, Hauser SL. B-cell therapy for multiple sclerosis: entering an era. Ann Neurol 2018;83:13-26.
176. Kappos L, Bar-or A, Cree BAC, Fox RJ, Giovannoni G, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet 2018;391:1263-73.
177. Gentile A, Musella A, Bullitta S, Fresegna D, De Vito F, et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J Neuroinflammation 2016;13:207.
178. Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, et al; CLARITY Study Group. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med 2010;362:416-26.
179. Rice GP, Filippi M, Comi G. Cladribine and progressive MS: clinical and MRI outcomes of a multicenter controlled trial. Cladribine MRI Study Group. Neurology 2000;54:1145-55.
180. Jørgensen LØ, Hyrlov KH, Elkjaer ML, Weber AB, Pedersen AE, et al. Cladribine modifies functional properties of microglia. Clin Exp Immunol 2020;201:328-40.
181. Musella A, Mandolesi G, Gentile A, Rossi S, Studer V, et al. Cladribine interferes with IL-1β synaptic effects in experimental multiple sclerosis. J Neuroimmunol 2013;264:8-13.
182. Beutler E, Sipe JC, Romine JS, Koziol JA, McMillan R, et al. The treatment of chronic progressive multiple sclerosis with cladribine. Proc Natl Acad Sci U S A 1996;93:1716-20.
183. Sipe JC, Romine JS, Koziol JA, McMillan R, Zyroff J, et al. Cladribine in treatment of chronic progressive multiple sclerosis. Lancet 1994;344:9-13.
184. Lubetzki C, Zalc B, Williams A, Stadelmann C, Stankoff B. Remyelination in multiple sclerosis: from basic science to clinical translation. Lancet Neurol 2020;19:678-88.
185. Villoslada P, Steinman L. New targets and therapeutics for neuroprotection, remyelination and repair in multiple sclerosis. Expert Opin Investig Drugs 2020;29:443-59.
186. Calabrese M, De Stefano N, Atzori M, Bernardi V, Mattisi I, et al. Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch Neurol 2007;64:1416-22.
187. Calabrese M, Rocca MA, Atzori M, Mattisi I, Bernardi V, et al. Cortical lesions in primary progressive multiple sclerosis: a 2-year longitudinal MR study. Neurology 2009;72:1330-6.
188. Calabrese M, Rocca MA, Atzori M, Mattisi I, Favaretto A, et al. A 3-year magnetic resonance imaging study of cortical lesions in relapse-onset multiple sclerosis. Ann Neurol 2010;67:376-83.
189. Bagnato F, Salman Z, Kane R, Auh S, Cantor FK, et al. T1 cortical hypointensities and their association with cognitive disability in multiple sclerosis. Mult Scler 2010;16:1203-12.
190. Mike A, Glanz BI, Hildenbrand P, Meier D, Bolden K, et al. Identification and clinical impact of multiple sclerosis cortical lesions as assessed by routine 3T MR imaging. AJNR Am J Neuroradiol 2011;32:515-21.
191. Kolber P, Montag S, Fleischer V, Luessi F, Wilting J, et al. Identification of cortical lesions using DIR and FLAIR in early stages of multiple sclerosis. J Neurol 2015;262:1473-82.
192. Geisseler O, Pflugshaupt T, Bezzola L, Reuter K, Weller D, et al. The relevance of cortical lesions in patients with multiple sclerosis. BMC Neurol 2016;16:204.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Zuroff LR, Benjamins JA, Bar-Or A, Lisak RP. Inflammatory mechanisms underlying cortical injury in progressive multiple sclerosis. Neurosciences 2021;8:111. http://dx.doi.org/10.20517/2347-8659.2020.35
AMA Style
Zuroff LR, Benjamins JA, Bar-Or A, Lisak RP. Inflammatory mechanisms underlying cortical injury in progressive multiple sclerosis. Neuroimmunology and Neuroinflammation. 2021; 8: 111. http://dx.doi.org/10.20517/2347-8659.2020.35
Chicago/Turabian Style
Zuroff, Leah R., Joyce A. Benjamins, Amit Bar-Or, Robert P. Lisak. 2021. "Inflammatory mechanisms underlying cortical injury in progressive multiple sclerosis" Neuroimmunology and Neuroinflammation. 8: 111. http://dx.doi.org/10.20517/2347-8659.2020.35
ACS Style
Zuroff, LR.; Benjamins JA.; Bar-Or A.; Lisak RP. Inflammatory mechanisms underlying cortical injury in progressive multiple sclerosis. Neurosciences. 2021, 8, 111. http://dx.doi.org/10.20517/2347-8659.2020.35
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 49 clicks
Cite This Article 49 clicks

 Like This Article 15
likes
Like This Article 15
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.