
Neuroinflammation in cortical and meningeal pathology in multiple sclerosis: understanding from animal models
 0
0Abstract
Multiple sclerosis (MS) is a neurodegenerative and inflammatory disease usually presenting with acute demyelinating events that can start as, or progress to, chronic damage. The development of animal experimental models, specific for each stage of MS will aid in the design of new drugs specific for the different forms of the disease. Animal models of experimental autoimmune encephalomyelitis successfully reflect the pathophysiological mechanisms of the early phases of MS. However, few models resemble the features of the progressive forms of MS such as cortical demyelination and meningeal inflammation. Recently, a few auspicious animal models recapitulating many of the characteristics of progressive MS, aimed at a better understanding of the pathology of these forms of the disease, have been developed. In this review, we will summarize the latest developments in animal models reflecting the cortical and meningeal pathological features of progressive MS, as well as their response to drugs specifically targeting these forms.
Keywords
Introduction
Multiple sclerosis (MS) is a neurodegenerative and inflammatory disease of unknown etiology characterized by repeated inflammatory events, demyelination and axonal damage which cause loss of function in the central nervous system (CNS)[1]. MS displays different clinical patterns: recurrent episodes with periods of remission classified as relapsing-remitting MS (RRMS), which can progress to secondary progressive form (SPMS), or persistent progression from the onset of the disease classified as primary progressive MS (PPMS)[2].
Even though MS was considered a disease that exclusively involved the white matter[3], it has been described to present neurodegenerative and demyelinating grey matter (GM) lesions, mostly located in the cortex[4]. These lesions can occur at early stages of the disease and worsen over time, contributing to the clinical disability and cognitive impairment in PPMS and SPMS[5-7]. Two theories tried to explain the relationship between cortical damage and meningeal inflammation; it was suggested that meningeal inflammation may play a role in the pathogenesis of cortical pathology (“outside-in” theory), but it is also plausible that cortical damage could induce meningeal inflammation (“inside-out” theory)[8].
Neuroinflammation and neurodegeneration play a key part in every stage of MS in all three forms of the disease (RRMS, PPMS, and SPMS)[9], but their exact role in each one is still debated[10,11]. For instance, anti-inflammatory and immunomodulatory treatments that are beneficial in the early stages of RRMS are ineffective in PPMS and SPMS where cortical damage and neurodegeneration are more prominent[9,12,13].
It has been previously demonstrated that immune cell infiltration and inflammation within the meninges correlate with the degree of GM demyelination, microglial activation, axonal pathology, and neuronal loss[14-16]. Neuronal loss within the motor cortex was associated with worse clinical progression such as the need of a wheelchair, longer disease duration, and younger age at death[17].
Here, we will review the available animal models related to cortical lesions and its associated meningeal inflammation from the PubMed database. Data related to the spinal cord or other brain regions are out of the scope of this review. The animal models of cortical and meningeal pathology reviewed here are summarized in Table 1 and Figure 1.
Animal models of cortical and meningeal pathology
| Model | Cortical pathology | Follicle-like structure | Meningeal inflammatory cell types | Cytokines involved in meningeal pathology | Tested treatment |
|---|---|---|---|---|---|
| EAE induced SJL/J mice (Magliozzi et al.[44], 2004 and Columba-Cabezas et al.[42], 2006) | No | Yes | T cells
B cells Dendritic cells | CXCL13, CXCL10, BAFF | No |
| EAE induced SJL/J mice
(Pikor et al.[45], 2015) | Subpial demyelination and glial activation | Yes | T cells
B cells | CXCL13, IL-6, TGFβ, IL-23 | No |
| EAE induced marmoset plus intracortical TNF-α and IFN-γ (Stassart et al.[28], 2015) | Focal demyelinating lesion | No | No | - | Laquinimod |
| PLP-primed Th17 cells into SJL/J mice (Ward et al.[37], 2020) | Subpial demyelination and glial activation | Yes | T cells
B cells | IL-17, GM-CSF | Siponimod improves cortical and meningeal pathology |
| EAE induced transgenic mice plus TNF-α and IFN-γ in the cortex
EAE induced marmoset (Lagumersindez-Denis et al.[29], 2017) | Demyelinating lesions | No | No | - | No |
| EAE induced agouti rat plus TNF-α and IFN-γ in the cortex (James et al.[17], 2020) | Subpial demyelination, microglial activation and neurodegeneration | No | T cells
B cells | TNF signalling genes | No |
| Cortical IL-1β injection
(Silva et al.[33], 2018) | Pancortical demyelination, neurodegeneration and glial activation | Yes | T cells
B cells Dendritic cells | IL-1β, TNF-α, IL-6 | Environmental enrichment improves cortical pathology |
| EAE induced agouti rat plus TNF-α and IFN-γ in the subarachnoid space
(Gardner et al.[31] 2013) | Subpial demyelination and microglial activation | No | T cells
B cells Macrophages | no | |
| EAE induced agouti rat plus TNF-α and IFN-γ in the cortex (Ucal et al.[32] 2017) | Subpial demyelination and glial activation. Neuronal death | No | Macrophages
T cells | No |
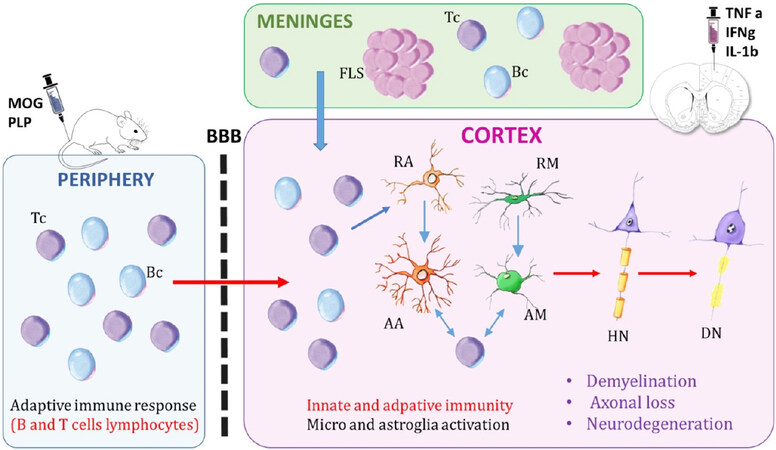
Figure 1. Schematic representation of cortical damage and meningeal inflammation in animal models. The meninges contain the cerebrospinal fluid, which includes T cells and B cells. Meningeal inflammation can appear either diffuse or forming follicle-like structures, which are associated with B cells. The animal models are mostly based on peripheral immunization with MOG or PLP peptides, which activate the innate and adaptive immune response, coupled with the intracortical injection of a cocktail of pro-inflammatory cytokines (TNF-α and IFN-γ). Also, cortical damage can be developed by a single injection of IL-1β into the cortex. The peripheral immune cells cross the blood brain barrier into the central nervous system. Activated microglia and astroglia, induced by the pro-inflammatory environment within the cortex, induce demyelination and neurodegeneration. HN: healthy neuron; RA: resting astrocyte; RM: resting microglia; Tc: T cells; Bc: B cells; FLS: follicle-like structures; BBB: blood brain barrier; AM: activated microglia; AA: activated astroglia; DN: neurodegeneration; MOG: myelin oligodendrocyte glycoprotein; PLP: proteolipid protein
Animal models of cortical pathology
The development of experimental animal models of MS is a key step towards clarifying the immune and pathological mechanisms of the progressive forms of this pathology and, in turn, advance the development new therapeutic approaches. Even though no single model represents all the pathophysiological characteristic of PPMS or SPMS, each one allows the study of different aspects of the disease. The ideal pre-clinical animal model that represents the cortical damage of progressive MS should include GM pathology with chronic demyelination, neurodegeneration, neuroinflammation with innate and adaptive immune infiltrate, and glial activation[18,19].
Cortical inflammatory demyelinating lesions are considered a key feature of both PPMS and SPMS patients but are sparse in RRMS[20]. The mechanisms involved in cortical demyelination and neurodegeneration are still unknown. Therefore, the main challenge in MS therapeutics so far has been the difficulty of replicating the chronicity and injury pattern of SPMS and PPMS which are rather different from RRMS[18].
Experimental autoimmune encephalomyelitis (EAE) was developed 70 years ago and since then, it has greatly contributed to the study of MS pathophysiology, especially of RRMS. EAE was induced in rodents and monkeys, providing models of acute monophasic relapsing-remitting and chronic progressive CNS inflammation (reviewed in[21]). Depending on the immunization agent, EAE can result in either a monophasic, relapsing-remitting, or even chronic disease. Therefore, based on the selected rodent strain, the immunogen, and the adjuvant, EAE can model different aspect of the disease. Spinal cord homogenate or purified antigens, including myelin basic protein, proteolipid protein (PLP), and myelin oligodendrocyte glycoprotein (MOG), induced acute monophasic EAE in Lewis and Dark Agouti rats[22-24]. Moreover, the immunization of SJL/J mice with PLP39-155 and PLP180-199 induced a form of RRMS. Additionally, immunization of Dark Agouti rats with MOG1-125, as well as of C57BL/6 mice with MOG35-55 peptide and BALB/c and C57BL/6 mice with PLP180-199 resulted in a chronic disease course, emulating the progressive forms of MS (reviewed in[24,25]).
Despite its usefulness for studying RRMS, EAE has poorly contributed to the analysis of cortical lesions, demonstrating its limitations for studying the progressive forms of MS. Due to this situation, several animal models of focal cortical pathology of MS have emerged in recent years in order to address this gap in the knowledge.
EAE is a model based on immune stimulation with myelin proteins, such as MOG or PLP, which induce CNS autoimmunity with infiltration of CD4+ and CD8+ B cells as well as monocytes[26]. Some EAE-based models were adapted to mimic focal cortical lesions. For example, one study induced chronic EAE in several strains of Lewis rats by active immunization with recombinant rat MOG1-125. In this study, cortical demyelination was dependent on particular combinations of MHC class I and II isotypes and alleles. These cortical lesions presented demyelination, microglial activation, and the deposition of immunoglobulins on myelin sheaths[27]. Other EAE-based models were modified to mimic cortical lesions by injecting a cocktail of cytokines, tumor necrosis factor-alpha (TNF-α) and interferon gamma (IFN-γ) in the cortex of MOG immunized animals. These lesions presented demyelination, neuroinflammation, glial activation, and neuronal degeneration[28-30]. However, each model exhibited some differences. In particular, Merkler et al.[30] developed a focal cortical model based on the stereotactical injection of TNF-α and IFN-γ in the cortex of rat MOG1-125 immunized Lewis rats. Thus, the authors generated demyelinated intracortical and subpial lesions with inflammatory cell infiltration (CD4+ T cells, CD8+ T cells, and ED1+ macrophages), deposition of complement proteins, axonal damage and neurodegeneration which were all resolved within 14 days[30]. In another model, the cocktail of TNF-α and IFN-γ was injected in the intrathecal space of Dark Agouti rats immunized with a subclinical dose recombinant mouse MOG in incomplete Freund’s adjuvant. This design induced meningeal inflammation which is associated with cortical subpial demyelination and microglia/macrophage activation in a gradient from the pial surface to the deeper cortical layers, with no neurodegeneration, lasting for 14 days[31]. The meningeal infiltration included lymphocytes CD4+ and CD8+ T cells and CD79+ B cells along with extensive microglial/macrophage activation[31].
Other authors achieved a chronic cortical pathology by placing a catheter in the rodent’s cortex which allowed the reinjection of cytokines. Dark Agouti rats were subclinically immunized with a low dose of MOG1-125 dissolved in incomplete Freund’s Adjuvant, then subsequent injections of TNF-α + IFN-γ were administered in the cortex using a catheter. The cortical and subcortical lesions presented inflammatory infiltrate (macrophages and T-cells), demyelination, microglia activation, neurodegeneration, and meningeal inflammation[32]. These demyelinated cortical lesions reached maximum size on days 9-15 and lasted for 30 days. Additionally, the cortical tissue showed upregulation of genes related to TNF-α signaling pathways involved in apoptosis/necrosis. In particular, the presence of neuronal loss and the upregulation of necroptosis signaling was demonstrated by the presence pRIPK3 and MLKM in cortical neurons[17].
The contribution of the immune mechanisms to cortical demyelination was also studied in several lines of transgenic mice, which showed that CCR2+ monocytes play a role in this process. One of these models was based on the intracerebral injection of a cocktail of cytokines (TNF-α and IFN-γ) in MOG-immunized transgenic mice. The authors demonstrated that transgenic mice with high titers of MOG-specific immunoglobulins in the serum (Th+) crossbreeding with CC chemokine receptor 2 (CCR2) deficient (Th/+ CCR2-/-) mice have less activated microglia (Mac-3+) perivascular cuffing, diminished mRNA levels of TNF-α and iNOS (monocytes-related genes) and reduced subpial and cortical demyelination compared to controls. In addition, the authors studied the depletion of CCR2+ monocytes in the common marmoset by administrating DOC-2 Fr-2 (a marmoset IgG1-chimeric humanized mouse anti-human CCR2 antibody) twice a week. The depletion was performed 14 days after the immunization of the marmoset with rat MOG1-125. Monocyte-depleted animals exhibited less cortical and perivascular demyelination along with an improved clinical disease course. Thus, the author proposed that CCR2+ monocytes are required for triggering cortical demyelination[29].
We have developed a new animal model of cortical pathology based on the innate immune response achieved by the long-term expression of interleukin 1beta (IL-1β) in the rat cortex, along with peripheral inflammation. IL-1β injected rats presented neuroinflammation (with neutrophil and monocyte infiltration), demyelination, neurodegeneration, meningeal inflammation and upregulation of pro-inflammatory cytokine mRNAs for IL-1β, TNF-α, and IL-6. These pathological features induced cognitive impairment and anxiety-like symptoms in IL-1β injected animals compared to controls. The cortical lesions could be visualized by MRI and the symptoms remained for almost 50 days, demonstrating that it is a chronic model of MS[33].
An EAE model induced by recombinant rat MOG-immunized marmosets along with stereotactic injection of cytokines (TNF-α and IFN-γ) was proposed as an intermediate step between pre-clinical and clinical trials[28,34]. This experimental design induced two types of lesions: (1) large demyelinating lesions like those found in human MS patients; and (2) perivascular foci of demyelination. These lesions contained CD3+ T cells and reactive GFAP+ astroglia. The macrophages expressed the myeloid-related protein-14 as well as myelin proteins such as 2’,3’-cyclic-nucleotide-3’-phosphodiesterase and MOG, which are observed in active human MS lesions. Numerous OLIG2+ cells were observed in the demyelinating areas, thus demonstrating the presence of oligodendrocyte precursor cells. Additionally, APP+ degenerating axons could be observed within the demyelinating lesions. These cortical lesions could be observed by MRI two weeks after the injection of the cytokines. Together with previous data, the authors concluded that the histopathology of the lesions developed in marmoset resembles early active human MS[28]. Another model induced cortical grey matter demyelinating lesions by immunizing marmosets with human MOG34-56 in incomplete Freund’s adjuvant. This model is based on the activation of adaptive immune mechanisms, the antigen-experienced T and/or B cells response. The pathology of the lesions in the cortex included demyelination, microglial activation, oxidative stress, and redistribution of iron[35,36]. Similar results were obtained if the marmosets were immunized using rat MOG1-125, which induce Type II MS pattern subcortical white matter lesions[30].
As it was previously stated, the mechanisms that mediate the pathogenesis of subpial lesions remain uncertain. However, it has been proposed that the inflammation of the overlying meninges could be linked to subpial injury. Based on this hypothesis, Ward et al.[37] showed in a non-focal EAE model that adoptive transfer of PLP-primed Th17+ cells into recipient SJL/J mice induced subpial demyelination associated with microglial/macrophage activation, disruption of the glia limitans as well as an oxidative stress response. This pathology was topologically associated with foci of immune cells in the meninges and occurred in the absence of measurable anti-MOG IgM or IgG antibodies.
Animal models of meningeal inflammation
The meningeal inflammation found in MS patients is a hallmark of the progressive forms of MS which contributes to the chronification of the cortical pathology. This knowledge required the development of specific animal models.
Cortical lesions have been associated with increased meningeal inflammation, which can be either diffuse or, more often, be organized in lymphoid-like tissue associated to grey matter pathology, leading to a more severe and rapid disease progression in MS patients[14-16,38]. Follicular-like structures (FLS) have recently been found in the meninges, specifically in the subarachnoid space, of SPMS patients and several animal models. FLS appear at different stages of development, from clusters of cells to highly organized follicles[14]. These structures contain proliferating B lymphocytes, plasma cells, mesenquimal cells, T lymphocytes and a network of follicular dendritic cells[39]. Chemokines and lymphotoxins, including CXCL13, CCL19, CCL21, CXCL12 and B cell activating factor (BAFF), influence the organization of B cells inside the follicles[40]. These follicular structures were areas of B-cell reactivation and antibody production, which suggests that soluble factors might diffuse into the cortex and induce cortical damage[14,15,41,42].
However, the exact relationship between these follicular meningeal structures and disease progression is not yet fully understood[43]. A very early study demonstrated the presence of similar FLS containing B cells, T cells and dendritic cells in the meninges of SJL/J mice with EAE[44]. Following that seminal study, several researchers started investigating the presence of follicle-like structures in different types of EAE immunization[42,45-47]. We will review the most outstanding data that correlates the presence of meningeal inflammation with cortical damage.
SJL/J mice immunized with PLP139-151 peptide developed meningeal B cell follicles. The expression of lymphotoxins was essential for lymphoid tissue organization in this model of relapsing-remitting EAE, while the blockage of the lymphotoxin pathway inhibited the formation of FLS, reduced T and B cell infiltration and prevented the expression of chemokines such as CXCL10 and CXCL13[42]. Moreover, the presence of IL-17, IL-22, and lymphotoxin beta was described in the FLS of SJL/J mice immunized with PLP139-151 in an emulsion of complete Freund’s adjuvant[45]. On the other hand, a model of non-obese diabetic mouse strain demonstrated the occurrence of meningeal inflammation, mostly composed of B cells, without the presence of FLS[46]. Similarly, meningeal inflammation in mice with MOG-induced EAE was characterized by sporadic distribution of cells expressing CD45 and CD19, clusters of Iba1+ macrophages and lesions that could be visualized by a 9.4 tesla MRI scanner[48].
Meningeal and subpial brain pathology were also described in a model of subclinical MOG-induced EAE, driven by adoptive transfer of PLP-primed Th17+ lymphocytes, which induced subpial demyelination, microglial/macrophage activation and oxidative stress[37]. In a Dark Agouti rat model of subclinical MOG-induced EAE with persistent infusion of a lentiviral vector expressing TNF-α and IFN-γ in the meninges, the authors demonstrated the presence of cortical pathology together with activation of TNF-α signaling pathways in cortical cells, leading to neuronal death and subpial demyelination. The meningeal inflammatory infiltrate was mostly composed of densely packed CD4+ T cells, CD8+ T cells and CD79+ B cells filling the subarachnoid space, which lasted for 56 days[16]. CD4+ and CD8+ T cells were roughly evenly distributed throughout the subarachnoid space, but CD79a+ B cells appeared to form discrete clusters. However, data demonstrated no features of lymphoid-like tissues in these clusters[17].
Even though most animal models for the study of meningeal FLS were based on EAE induction, our novel rat model of focal cortical inflammation, generated by the long-term expression of IL-1β in the cortex, also induced meningeal inflammation with FLS. These structures contained CD4+, CD8+, CD20+, CD23+ and CD39+ cells and were associated with cortical demyelination and neurodegeneration along with anxiety-like symptoms and short-term memory deterioration. Additionally, these cortico-meningeal structures could be visualized by MRI with gadolinium contrast[33].
Until now, the animal models described previously have represented the first step towards modelling the meningeal pathology associated with cortical damage. The development of a variety of animal models that mimic most of the characteristics of meningeal inflammation may help answer questions on the effect of the meninges in cortical tissue damage and, in turn, the effect of cortical damage on the inflammatory events occurring in the meninges.
Effect of therapeutic agents on animal models of cortical and meningeal animal models
Although cortical pathology is important for the prognosis of patients, no new treatments have been specifically designed to target this aspect of disease. Thus, the evidence that some currently available drugs can also target cortical pathology provides valuable aid to the treatment of MS patients while waiting for the development of specific therapies targeting cortical pathology. A few previously approved drugs have already been tested in animal models of cortical pathology with the aim of showing their efficacy in cortical MS pathology, among them siponimod, glatiramer acetate and laquinimod.
Siponimod (BAF312) is a novel sphingosine-1-phosphate receptor modulator demonstrated to delay progression in PMS. The drug was recently approved by the United States Food and Drug Administration and the European Medicines Agency[37,49]. It was proposed that siponimod could reduce inflammation by sequestering lymphocytes in lymphoid tissues and could cross the blood-brain barrier by binding to its receptors on neurons, astrocytes and oligodendrocytes[50]. Siponimod was tested in a cortical model of MOG35-55EAE C57BL/6J mice with the addition of intracortical administration of a cocktail of cytokines (TNF-α and IFN-γ). The authors demonstrated that demonstrated that oral, but not intracerebral, administration of siponimod diminished the infiltration of immune cells within both grey and white matter lesions. Siponimod administration partially restored cortical neuronal circuit function by exerting a neuroprotective effect after crossing the blood-brain barrier[49]. Moreover, the administration of siponimod significantly ameliorated subclinical MOG-induced EAE symptoms and improved subpial pathology concomitantly with selective reduction in the capacity of transferred T cells to induce Th17 cytokines[37]. In addition, they demonstrated that siponimod impaired the formation of meningeal tertiary lymphoid tissue, although it did not completely prevent the associated clinical symptoms of EAE. Siponimod diminished fibronectin network formation in the meninges by about 47% compared to control mice. The accumulation of B220+ and CD3+ cells was also significantly decreased compared to control animals, possibly due to the lack of fibronectin matrix support which is necessary for tertiary lymphoid tissue formation[37].
Glatiramer acetate is an immunomodulatory drug for RRMS that was demonstrated to have neuroprotective effects in two different EAE models, relapsing-remitting PLP-induced EAE disease and chronic MOG-induced EAE disease. The administration of glatiramer acetate alleviated cognitive impairment and cortico/hippocampal damage in these animals by downregulating pro-inflammatory pathways[51]. In addition, glatiramer acetate treatment in MOG35-55-induced EAE mice potentiated neuroprotection by incrementing neurotrophic factors (BDNF, NT-3, and NT-4)[51-53]. Additionally, BDNF expression in glatiramer acetate treated mice induced the differentiation/proliferation of neuronal progenitors that migrated into lesions in injured regions. These results indicate that the immunomodulator glatiramer acetate exerts anti-inflammatory, neuroprotective and regenerative effects in the diseased brain[52].
Laquinimod is a quinoline-3-carboxamide derivative that was demonstrated to inhibit the development of disease in inflammatory mouse models for MS such as EAE[54-56]. A focal model based on the combination of EAE induced with rat recombinant MOG, together with the injection of TNF-α and IFN-γ in the cortex and corpus callosum of the common marmoset, presented leukocortical demyelinating lesions at the site of the injections in both white and gray matter, as previously described. The prophylactic treatment with laquinimod completely prevented the leuko-cortical lesion formation in this model. The cortex of animals treated with laquinimod presented a lack of demyelinating lesions which could not be detected by MRI, and minor astrocytic and microglial reaction at the injection site. Thus, prophylactic modulation of the immune system by laquinimod prevents the development of focal EAE in marmoset[28]. Additionally, laquinimod demonstrated immunosuppressive and neuroprotective properties in a model based on a combination of neurodegeneration induced by cuprizone feeding and MOG35-55-induced EAE in mice, which provokes an infiltration of myelin autoreactive T cells from peripheral lymphoid organs. The treatment with laquinimod was suspended two weeks prior to active EAE induction. Laquinimod ameliorated cuprizone pathology reduced demyelination, axonal loss, and microglial and astroglial activation but, at the same time, it decreased peripheral immune cell recruitment, particularly of lymphocytes[54-56]. This study demonstrated that ameliorating a primary degenerative CNS process might reduce the secondary inflammatory lesion development[56].
Conclusion
Grey matter lesions and meningeal inflammation are two of the most outstanding features of the progressive forms of MS and of RRMS, resulting in higher disease activity and poorer outcome. Animal models provide valuable insight into the knowledge of the role of cortical damage and its association with meningeal inflammation in MS. They also collaborated with the identification of the immune cells and molecules such as specific cytokines and chemokines involved in the development of cortical damage and meningeal inflammation pathologic processes. In addition, animal models helped to understand how immune molecules, either from cells within the cortex or from the inflammatory events in the meninges, impact glial activation, axonal degeneration, and oligodendrocyte death. Even though it was suggested that meningeal inflammation may play a role in the pathogenesis of cortical pathology (“outside-in” theory), it is also plausible that cortical damage induces meningeal inflammation (“inside-out” theory). In order to address this dichotomy, we need new information from animal models.
The importance of good preclinical studies is highlighted by the fact that research with animal models has already led to the development of three approved therapies for use in MS. However, most new therapeutic drugs showing promise in the preclinical studies end up failing in the clinical practice. One of the reasons that might explain this is the lack of development or evaluation of suitable animal models of MS. Still, there is no specific MS model that allows to study the entire pathology of the disease; several models reflecting the different aspects of the disease are available. The election of the right model to answer a specific question should be done in accordance with the specific aims of the study.
Declarations
Authors’ contributionsConceptualized and wrote the review: Silva BA, Ferrari CC
Wrote and corrected the manuscript: Miglietta E
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Brück W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J Neurol 2005;252 Suppl 5:v3-9.
2. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014;83:278-86.
3. Kidd D, Barkhof F, McConnell R, et al. Cortical lesions in multiple sclerosis. Brain 1999;122:17-26.
4. Calabrese M, Favaretto A, Martini V, Gallo P. Grey matter lesions in MS: from histology to clinical implications. Prion 2013;7:20-7.
5. Nelson F, Datta S, Garcia N, et al. Intracortical lesions by 3T magnetic resonance imaging and correlation with cognitive impairment in multiple sclerosis. Mult Scler 2011;17:1122-9.
6. Calabrese M, Poretto V, Favaretto A, et al. Cortical lesion load associates with progression of disability in multiple sclerosis. Brain 2012;135:2952-61.
7. Roosendaal SD, Moraal B, Pouwels PJ, et al. Accumulation of cortical lesions in MS: relation with cognitive impairment. Mult Scler 2009;15:708-14.
8. Lassmann H. Pathophysiology of inflammation and tissue injury in multiple sclerosis: what are the targets for therapy. J Neurol Sci 2011;306:167-9.
10. Louapre C, Lubetzki C. Neurodegeneration in multiple sclerosis is a process separate from inflammation: Yes. Mult Scler 2015;21:1626-8.
11. Hutchinson M. Neurodegeneration in multiple sclerosis is a process separate from inflammation: No. Mult Scler 2015;21:1628-31.
12. Comi G. Disease-modifying treatments for progressive multiple sclerosis. Mult Scler 2013;19:1428-36.
13. Lorscheider J, Jokubaitis VG, Spelman T, et al. Anti-inflammatory disease-modifying treatment and short-term disability progression in SPMS. Neurology 2017;89:1050-9.
14. Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011;134:2755-71.
15. Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007;130:1089-104.
16. Magliozzi R, Howell OW, Reeves C, et al. A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 2010;68:477-93.
17. James RE, Schalks R, Browne E, et al. Persistent elevation of intrathecal pro-inflammatory cytokines leads to multiple sclerosis-like cortical demyelination and neurodegeneration. Acta Neuropathol Commun 2020;8:66.
18. Fox RJ, Thompson A, Baker D, et al. Setting a research agenda for progressive multiple sclerosis: the international collaborative on progressive MS. Mult Scler 2012;18:1534-40.
19. Denic A, Johnson AJ, Bieber AJ, et al. The relevance of animal models in multiple sclerosis research. Pathophysiology 2011;18:21-9.
20. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005;128:2705-12.
21. Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol 2011;164:1079-106.
22. Polman CH, Matthaei I, de Groot CJ, et al. Low-dose cyclosporin A induces relapsing remitting experimental allergic encephalomyelitis in the Lewis rat. J Neuroimmunol 1988;17:209-16.
23. Shin T, Ahn M, Matsumoto Y. Mechanism of experimental autoimmune encephalomyelitis in Lewis rats: recent insights from macrophages. Anat Cell Biol 2012;45:141-8.
24. Weissert R. Actively induced experimental autoimmune encephalomyelitis in rats. Methods Mol Biol 2016;1304:161-9.
25. Terry RL, Ifergan I, Miller SD. Experimental autoimmune encephalomyelitis in mice. Methods Mol Biol 2016;1304:145-60.
26. Procaccini C, De Rosa V, Pucino V, Formisano L, Matarese G. Animal models of Multiple Sclerosis. Eur J Pharmacol 2015;759:182-91.
27. Storch MK, Bauer J, Linington C, et al. Cortical demyelination can be modeled in specific rat models of autoimmune encephalomyelitis and is major histocompatibility complex (MHC) haplotype-related. J Neuropathol Exp Neurol 2006;65:1137-42.
28. Stassart RM, Helms G, Garea-Rodriguez E, et al. A new targeted model of experimental autoimmune encephalomyelitis in the common marmoset. Brain Pathol 2015;26:452-64.
29. Lagumersindez-Denis N, Wrzos C, Mack M, et al. Differential contribution of immune effector mechanisms to cortical demyelination in multiple sclerosis. Acta Neuropathol 2017;134:15-34.
30. Merkler D, Ernsting T, Kerschensteiner M, Bruck W, Stadelmann C. A new focal EAE model of cortical demyelination: multiple sclerosis-like lesions with rapid resolution of inflammation and extensive remyelination. Brain 2006;129:1972-83.
31. Gardner C, Magliozzi R, Durrenberger PF, et al. Cortical grey matter demyelination can be induced by elevated pro-inflammatory cytokines in the subarachnoid space of MOG-immunized rats. Brain 2013;136:3596-608.
32. Ucal M, Haindl MT, Adzemovic MZ, et al. Widespread cortical demyelination of both hemispheres can be induced by injection of pro-inflammatory cytokines via an implanted catheter in the cortex of MOG-immunized rats. Exp Neurol 2017;294:32-44.
33. Silva BA, Leal MC, Farias MI, et al. A new focal model resembling features of cortical pathology of the progressive forms of multiple sclerosis: Influence of innate immunity. Brain Behav Immun 2018;69:515-31.
34. Mangiardi M, Crawford DK, Xia X, et al. An animal model of cortical and callosal pathology in multiple sclerosis. Brain Pathol 2011;21:263-78.
35. Jagessar SA, Heijmans N, Bauer J, et al. B-cell depletion abrogates T cell-mediated demyelination in an antibody-nondependent common marmoset experimental autoimmune encephalomyelitis model. J Neuropathol Exp Neurol 2012;71:716-28.
36. ‘t Hart BA, Dunham J, Faber BW, et al. A B cell-driven autoimmune pathway leading to pathological hallmarks of progressive multiple sclerosis in the marmoset experimental autoimmune encephalomyelitis model. Front Immunol 2017;8:804.
37. Ward LA, Lee DS, Sharma A, et al. Siponimod therapy implicates Th17 cells in a preclinical model of subpial cortical injury. JCI Insight 2020;5:132522.
38. Magliozzi R, Howell OW, Durrenberger P, et al. Meningeal inflammation changes the balance of TNF signalling in cortical grey matter in multiple sclerosis. J Neuroinflammation 2019;16:259.
39. Silva BA, Ferrari CC. Cortical and meningeal pathology in progressive multiple sclerosis: a new therapeutic target? Rev Neurosci 2019;30:221-32.
40. Haugen M, Frederiksen JL, Degn M. B cell follicle-like structures in multiple sclerosis-with focus on the role of B cell activating factor. J Neuroimmunol 2014;273:1-7.
41. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 2004;14:164-74.
42. Columba-Cabezas S, Griguoli M, Rosicarelli B, et al. Suppression of established experimental autoimmune encephalomyelitis and formation of meningeal lymphoid follicles by lymphotoxin beta receptor-Ig fusion protein. J Neuroimmunol 2006;179:76-86.
43. Absinta M, Vuolo L, Rao A, et al. Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis. Neurology 2015;85:18-28.
44. Magliozzi R, Columba-Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol 2004;148:11-23.
45. Pikor NB, Prat A, Bar-Or A, Gommerman JL. Meningeal tertiary lymphoid tissues and multiple sclerosis: a gathering place for diverse types of immune cells during CNS autoimmunity. Front Immunol 2016;6:657.
46. Dang AK, Tesfagiorgis Y, Jain RW, Craig HC, Kerfoot SM. Meningeal infiltration of the spinal cord by non-classically activated B cells is associated with chronic disease course in a spontaneous B cell-dependent model of CNS autoimmune disease. Front Immunol 2015;6:470.
47. Peters A, Pitcher LA, Sullivan JM, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity 2011;35:986-96.
48. Pol S, Schweser F, Bertolino N, et al. Characterization of leptomeningeal inflammation in rodent experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis. Exp Neurol 2019;314:82-90.
49. Hundehege P, Cerina M, Eichler S, et al. The next-generation sphingosine-1 receptor modulator BAF312 (siponimod) improves cortical network functionality in focal autoimmune encephalomyelitis. Neural Regen Res 2019;14:1950-60.
50. Choi JW, Gardell SE, Herr DR, et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A 2011;108:751-6.
51. Aharoni R, Vainshtein A, Stock A, et al. Distinct pathological patterns in relapsing-remitting and chronic models of experimental autoimmune enchephalomyelitis and the neuroprotective effect of glatiramer acetate. J Autoimmun 2011;37:228-41.
52. Aharoni R, Eilam R, Domev H, et al. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc Natl Acad Sci U S A 2005;102:19045-50.
53. Azoulay D, Vachapova V, Shihman B, Miler A, Karni A. Lower brain-derived neurotrophic factor in serum of relapsing remitting MS: reversal by glatiramer acetate. J Neuroimmunol 2005;167:215-8.
54. Bruck W, Popescu B, Lucchinetti CF, et al. Neuromyelitis optica lesions may inform multiple sclerosis heterogeneity debate. Ann Neurol 2012;72:385-94.
55. Kramann N, Menken L, Hayardeny L, Hanisch UK, Brück W. Laquinimod prevents cuprizone-induced demyelination independent of Toll-like receptor signaling. Neurol Neuroimmunol Neuroinflamm 2016;3:e233.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Silva BA, Miglietta E, Ferrari CC. Neuroinflammation in cortical and meningeal pathology in multiple sclerosis: understanding from animal models. Neurosciences 2021;8:174-84. http://dx.doi.org/10.20517/2347-8659.2020.47
AMA Style
Silva BA, Miglietta E, Ferrari CC. Neuroinflammation in cortical and meningeal pathology in multiple sclerosis: understanding from animal models. Neuroimmunology and Neuroinflammation. 2021; 8(3): 174-84. http://dx.doi.org/10.20517/2347-8659.2020.47
Chicago/Turabian Style
Silva, Berenice Anabel, Esteban Miglietta, Carina Cintia Ferrari. 2021. "Neuroinflammation in cortical and meningeal pathology in multiple sclerosis: understanding from animal models" Neuroimmunology and Neuroinflammation. 8, no.3: 174-84. http://dx.doi.org/10.20517/2347-8659.2020.47
ACS Style
Silva, BA.; Miglietta E.; Ferrari CC. Neuroinflammation in cortical and meningeal pathology in multiple sclerosis: understanding from animal models. Neurosciences. 2021, 8, 174-84. http://dx.doi.org/10.20517/2347-8659.2020.47
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 15
likes
Like This Article 15
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.