
POEMS syndrome associated with Castleman disease: a case report and literature review

Abstract
Polyneuropathy, organomegaly, endocrinopathy, M proteins, and skin changes (POEMS) syndrome is a multisystemic disorder that clinically manifests as paraneoplastic and monoclonal plasma cell dyscrasia. Its acronym is derived from its principal characteristics: polyneuropathy, organomegaly, endocrinopathy, M proteins, and skin changes. Here, the authors reported a case of POEMS syndrome that was also associated with Castleman disease. A 53-year-old female patient was admitted to our hospital with limb weakness, numbness, edema, abdominal distention, and fever. Physical examination revealed tetraplegia, paraesthesia, and hyporeflexia in all four limbs, in addition to lymphadenectasis, splenomegaly, skin hyperpigmentation, hypertrichosis, and pitting edema. Laboratory tests and imaging revealed thrombocytosis, hypothyroidism, diabetes, hydropericardium, hydrothorax, splenomegaly, and lymphadenectasis. Electromyography showed the characteristic patterns of both demyelinating disease and axonal degeneration. Serum protein electrophoresis revealed monoclonal immunoglobulin G-lambda paraproteins. Histological examination clearly diagnosed the disease as the hyaline vascular subtype. The final diagnosis in this case was POEMS syndrome in association with Castleman disease.
Keywords
Introduction
Polyneuropathy, organomegaly, endocrinopathy, M proteins, and skin changes (POEMS) syndrome, also known as Crow-Fukase syndrome, osteosclerotic myeloma, and Takatsuki syndrome,[1-4] is the paraneoplastic clinical manifestation of monoclonal plasma cell dyscrasia. POEMS syndrome is a multisystemic disorder, and its acronym is derived from its principal characteristics: polyneuropathy, organomegaly, endocrinopathy, M proteins, and skin changes.[5,6] Other important clinical features include fever, papilledema, extravascular volume overload, sclerosis, bone lesions, thrombocytosis, erythrocytosis, elevated vascular endothelial growth factor (VEGF) levels, abnormal pulmonary function, predisposition toward thrombosis, etc.[2,3,6-13] Early diagnosis is a challenge because of the diverse clinical manifestations that are often accompanied with multiple organ injury. Here, we reported a patient with the Castleman disease variant of POEMS syndrome, which we hope will prompt the universal recognition of POEMS.
Case report
A 53-year-old Chinese woman was admitted to the Neurology Department of our hospital because of progressive limbs weakness, numbness, and edema.
Approximately 7 months before admission, she began to develop limb weakness and numbness. Twenty days before admission, the patient developed fever and edema. She had been diagnosed with diabetes and treated with insulin i.h. for 3 months prior to admission. Her past medical history was unremarkable, with no history of smoking, alcohol use, HIV infection, tuberculosis, or tumor.
On examination, the patient was suffering from progressive tetraplegia, paraesthesia, and edema. Physical examination revealed fever, skin hyperpigmentation, hypertrichosis, multiple small peripheral lymph nodes, splenomegaly, pitting edema in the lower extremities, tetraplegia, paraesthesia, and hyporeflexia in all four limbs. Routine blood examination revealed thrombocytosis (527 × 109 platelets/L; normal = (101-320) × 109 platelets/L). Blood biochemistry analysis revealed low albumin and globulin. Laboratory tests on admission were positive for hepatitis B surface antigen, hepatitis B core antibody, and hepatitis B e-antibody, but serum hepatitis B virus (HBV)-DNA was within normal limits. Fibrinogen was 4.72 g/L (normal = 1.8-3.5 g/L). Thyroid function test revealed hypothyroidism. C-reactive protein was 22.60 mg/L, and erythrocyte sedimentation rate was 40 mm/h. Lumbar puncture was performed on admission, and cerebrospinal fluid testing revealed increased protein and pressure levels, but the cell count was normal. Serum protein electrophoresis revealed monoclonal immunoglobulin G-lambda (IgG-λ) paraprotein, while serum immunoglobulin levels were within normal limits [Figure 1]. Ultrasonic examination indicated hydropericardium, right hydrothorax, splenomegaly, and lymphadenectasis (including the anterior cervical, axillary, and inguinal lymph nodes). At the same time, ultrasound revealed multiple hemangiomas and splenomegaly. Needle electromyography confirmed diffuse, symmetrical, demyelinating, and axonal lesions in the sensorimotor fibers that affected all four limbs. Meanwhile, a portion of the F-waves in the peripheral nerves had reduced and disappeared. Electromyography revealed a pattern that is characteristic both of demyelinating disease and axonal degeneration.
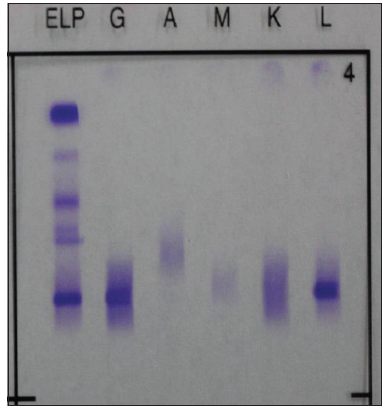
Figure 1. Immunofixation, a monoclonal immunoglobulin G-lambda paraprotein, showing deep dyeing belts (Band L)
Biopsy of the lymph nodes from the right cervical chain revealed vascular, follicular, and lymphoid hyperplasia, thickening of the mantle zone, and the formation of concentric lymphocytes surrounding the germinal center, which were hyalinized, atrophic, and surrounded by blood vessels. Vascular proliferation was found, in addition to sinus histiocytosis in the interfollicular parenchyma [Figure 2]. Meanwhile, immunohistochemical analysis revealed positive staining for CD3, CD4, CD8, CD20, CD21, CD138, kappa, lambda, Pax-5, and Ki-67. These histological findings are consistent with the hyaline vascular variant of Castleman disease. Patient improvement was not apparent after treatment with dexamethasone for 10 days. The patient was transferred to the Hematology Department for further treatment.
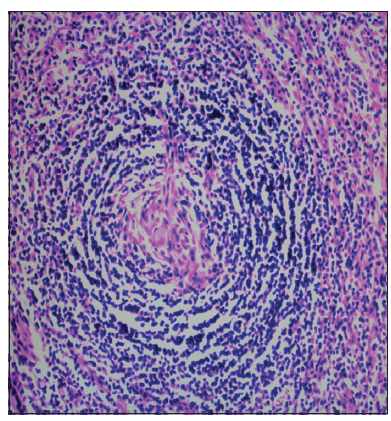
Figure 2. Right cervical lymph node biopsy showing hyaline vascular Castleman disease, follicular, lymphocytes, vascularity hyperplasia, and the formation of concentric of lymphocytes surrounding the germinal center. H and E staining (original magnification, ×200)
Discussion
POEMS syndrome was first reported by Scheinker in 1938.[2,5] The first Chinese case of POEMS syndrome was described in 1986.[14] POEMS syndrome is a rare multisystemic disorder that is related to underlying plasma cell dyscrasia. The important traits of POEMS syndrome including polyneuropathy, organomegaly, endocrinopathy, M proteins, and skin changes.[6] The other important features include Castleman disease, sclerotic bone lesions, VEGF elevation, etc. The diagnosis of POEMS syndrome is based on having both polyradiculoneuropathy and monoclonal plasma cell disorder, at least 1 of 3 other major criteria (Castleman disease, sclerotic bone lesions, or elevated VEGF), and at least 1 minor criterion [Table 1].
Criteria for the diagnosis of POEMS syndrome[11]
| Criteria/other symptoms and signs | Affected, %* |
|---|---|
| Mandatory major criteria (both required) | |
| Polyradiculoneuropathy (typically demyelinating) | 100 |
| Monoclonal plasma cell disorder (almost always λ) | 100† |
| Other major criteria (1 required) | |
| Castleman disease‡ | 11–25 |
| Sclerotic bone lesions | 27–97 |
| VEGF elevation§ | |
| Minor criteria (1 required) | |
| Organomegaly (splenomegaly, hepatomegaly, or lymphadenopathy) | 45–85 |
| Extravascular volume overload (edema, pleural effusion, or ascites) | 29–87 |
| Endocrinopathy (adrenal, thyroid, pituitary**, gonadal, parathyroid, pancreatic**) | 67–84 |
| Skin changes (hyperpigmentation, hypertrichosis, glomeruloid hemangiomata, plethora, acrocyanosis, flushing, white nails) | 68–89 |
| Papilledema | 29–64 |
| Thrombocytosis/polycythemia*** | 54–88 |
| Other symptoms and signs | |
| Clubbing, weight loss, hyperhidrosis, pulmonary hypertension/restrictive lung disease, thrombotic diatheses, diarrhea, low vitamin B12 values | |
Our patient had almost all the features of POEMS syndrome, including multiple peripheral neuropathy, splenomegaly, and multiple enlarged peripheral lymph nodes, diabetes, hypothyroidism, monoclonal IgG-λ paraprotein, skin hyperpigmentation, and hypertrichosis. Meanwhile, our patient also presented with systemic signs such as fever, abdominal distention, pitting edema of the lower extremities, hydropericardium, hydrothorax, thrombocytosis, hypoalbuminemia, and increased fibrinogen levels. In brief, this patient is consistent with the standard diagnostic standard of POEMS syndrome.
Castleman disease (CD, or angiofollicular lymph node hyperplasia) is also a rare lymphoproliferative disorder.[15] Castleman disease was first described by Castleman et al.[16] in 1956. According to previous studies, the pathological feature of Castleman disease is reactive proliferation in the lymphoid tissues.[17] The clinical features of Castleman disease are classified into two categories: localized and multicentric.[18] There are also three histological forms of CD: (1) hyaline vascular form, (2) plasma cell form, and (3) mixed. Multicentric Castleman disease (MCD) is generally the plasma cell type, but the hyaline vascular type has been described in some patients.[19] Localized Castleman disease usually presents as masses in young adults (20-30 years of age). Systemic symptoms are rare in localized Castleman disease patients. In contrast, MCD develops in old patients (40-50 years of age). The involvement of multiple lymph nodes and organs is frequent.[20] Our older patient presented with systemic symptoms and multiple enlarged lymph nodes. The histological findings in this case are consistent with the hyaline vascular form of Castleman disease.
Castleman disease and POEMS syndrome are closely related. An association with MCD was reported in about 50% of patients with POEMS.[6,19,20] Of 113 patients with MCD, 32% presented with criteria sufficient for a diagnosis of POEMS syndrome.[21] Here, our patient presented with POEMS syndrome in association with Castleman disease.
The pathogeny of POEMS syndrome remains unclear. It is assumed that hepatitis B antigen may play a role in the etiology of this lymphatic disorder.[22] Our patient was positive for HBV. A previous study confirmed that increased levels of tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and VEGF in patients of POEMS syndrome are correlated with disease activity.[23] However, we did not assess TNF-α, IL-6, or VEGF. Therapy for POEMS syndrome should include radiation, chemotherapy, peripheral blood stem cell transplant, targeting therapy, intravenous gamma-globulin therapy, plasmapheresis, corticosteroids, etc.[11] The clinical course of POEMS syndrome is also chronic. A previous study revealed that the median survival time of patients with POEMS syndrome is 165 months.[6] Another study reported that the prognosis of MCD patients was poor, demonstrating a median survival time of 30 months.[20]
The diagnosis of POEMS syndrome is often delayed because the syndrome is rare and can be mistaken for other neurological disorders. Thus, we hope our patient with the Castleman disease variant of POEMS syndrome will prompt the universal recognition of this disease.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
2. Nakanishi T, Sobue I, Toyokura Y, Nishitani H, Kuroiwa Y, Satoyoshi E, Tsubaki T, Igata A, Ozaki Y. The Crow-Fukase syndrome.a study of 102 cases in Japan. Neurology 1984;34:712-20.
3. Takatsuki K, Sanada I. Plasma cell dyscrasia with polyneuropathy and endocrine disorder: clinical and laboratory features of 109 reported cases. Jpn J Clin Oncol 1983;13:543-55.
4. Fukase M, Kakimatsu T, Nishitani H. Report of a case of solitary plasmacytoma in the abdomen presenting with polyneuropathy and endocrinological disorders. Clin Neurol 1969;9:657.
5. Bardwick PA, Zvaifler NJ, Gill GN, Newman D, Greenway GD, Resnick DL. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine (Baltimore) 1980;59:311-22.
6. Dispenzieri A, Kyle RA, Lacy MQ, Rajkumar SV, Therneau TM, Larson DR, Greipp PR, Witzig TE, Basu R, Suarez GA, Fonseca R, Lust JA, Gertz MA. POEMS syndrome: definitions and long-term outcome. Blood 2003;101:2496-506.
7. Soubrier MJ, Dubost JJ, Sauvezie BJ. POEMS syndrome: a study of 25 cases and a review of the literature. French Study Group on POEMS Syndrome. Am J Med 1994;97:543-53.
8. Li J, Zhou DB, Huang Z, Jiao L, Duan MH, Zhang W, Zhao YQ, Shen T. Clinical characteristics and long-term outcome of patients with POEMS syndrome in China. Ann Hematol 2011;90:819-26.
9. Gandhi GY, Basu R, Dispenzieri A, Basu A, Montori VM, Brennan MD. Endocrinopathy in POEMS syndrome: the Mayo Clinic experience. Mayo Clin Proc 2007;82:836-42.
10. Jeunon T, Sampaio AL, Caminha RC, Reis CU, Dib C. Glomeruloid hemangioma in POEMS syndrome: a report on two cases and a review of the literature. An Bras Dermatol 2011;86:1167-73.
12. D'Souza A, Hayman SR, Buadi F, Mauermann M, Lacy MQ, Gertz MA, Kyle RA, Kumar S, Greipp PR, Lust JA, Russell SJ, Zeldenrust S, Dingli D, Witzig TE, Rajkumar SV, Dispenzieri A. The utility of plasma vascular endothelial growth factor levels in the diagnosis and follow-up of patients with POEMS syndrome. Blood 2011;118:4663-5.
13. Dispenzieri A. POEMS syndrome: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol 2011;86:591-601.
14. Bo Q, Wen B. Polyneuropathy accompanied by impotence, edema and lymphadenopathy. Chin J Intern Med 1986;25:570-2.
15. Dispenzieri A. POEMS syndrome: 2014 update on diagnosis, risk-s tratification, and management. A m J Hematol 2014;89:214-23.
16. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer 1956;9:822-30.
17. McCarty MJ, Vukelja SJ, Banks PM, Weiss RB. Angiofollicular lymph node hyperplasia (Castleman's disease). Cancer Treat Rev 1995;21:291-310.
18. Aguilar-Rodriguez R, Milea SL, Demirci I, Herold S, Flasshove M, Klosterhalfen B, Kinkel H, Janßen H. Localized retroperitoneal Castleman's disease: a case report and review of the literature. J Med Case Rep 2014;8:93.
20. Perdaens C, De Raeve H, Goossens A, Sennesael J. POEMS syndrome characterized by glomeruloid angioma, osteosclerosis and multicentric Castleman disease. J Eur Acad Dermatol Venereol 2006;20:480-1.
21. Dispenzieri A, Armitage JO, Loe MJ, Geyer SM, Allred J, Camoriano JK, Menke DM, Weisenburger DD, Ristow K, Dogan A, Habermann TM. The clinical spectrum of Castleman's disease. Am J Hematol 2012;87:997-1002.
22. Ben-Chetrit E, Flusser D, Okon E, Ackerman Z, Rubinow A. Multicentric Castleman's disease associated with rheumatoid arthritis: a possible role of hepatitis B antigen. Ann Rheum Dis 1989;48:326-30.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Kang J, Yang F, Zhang HY, Hu MM, Xia F, Wang JC, Deng YC, Zhao G. POEMS syndrome associated with Castleman disease: a case report and literature review. Neurosciences 2014;1:40-3. http://dx.doi.org/10.4103/2347-8659.135577
AMA Style
Kang J, Yang F, Zhang HY, Hu MM, Xia F, Wang JC, Deng YC, Zhao G. POEMS syndrome associated with Castleman disease: a case report and literature review. Neuroimmunology and Neuroinflammation. 2014; 1: 40-3. http://dx.doi.org/10.4103/2347-8659.135577
Chicago/Turabian Style
Kang, Juan, Fang Yang, Hong-Ya Zhang, Meng-Meng Hu, Feng Xia, Jin-Cun Wang, Yan-Chun Deng, Gang Zhao. 2014. "POEMS syndrome associated with Castleman disease: a case report and literature review" Neuroimmunology and Neuroinflammation. 1: 40-3. http://dx.doi.org/10.4103/2347-8659.135577
ACS Style
Kang, J.; Yang F.; Zhang H.Y.; Hu M.M.; Xia F.; Wang J.C.; Deng Y.C.; Zhao G. POEMS syndrome associated with Castleman disease: a case report and literature review. Neurosciences. 2014, 1, 40-3. http://dx.doi.org/10.4103/2347-8659.135577
About This Article
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.